
Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors


 ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
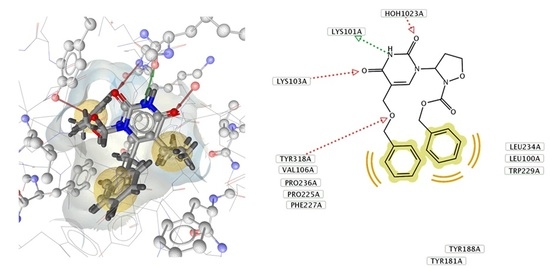
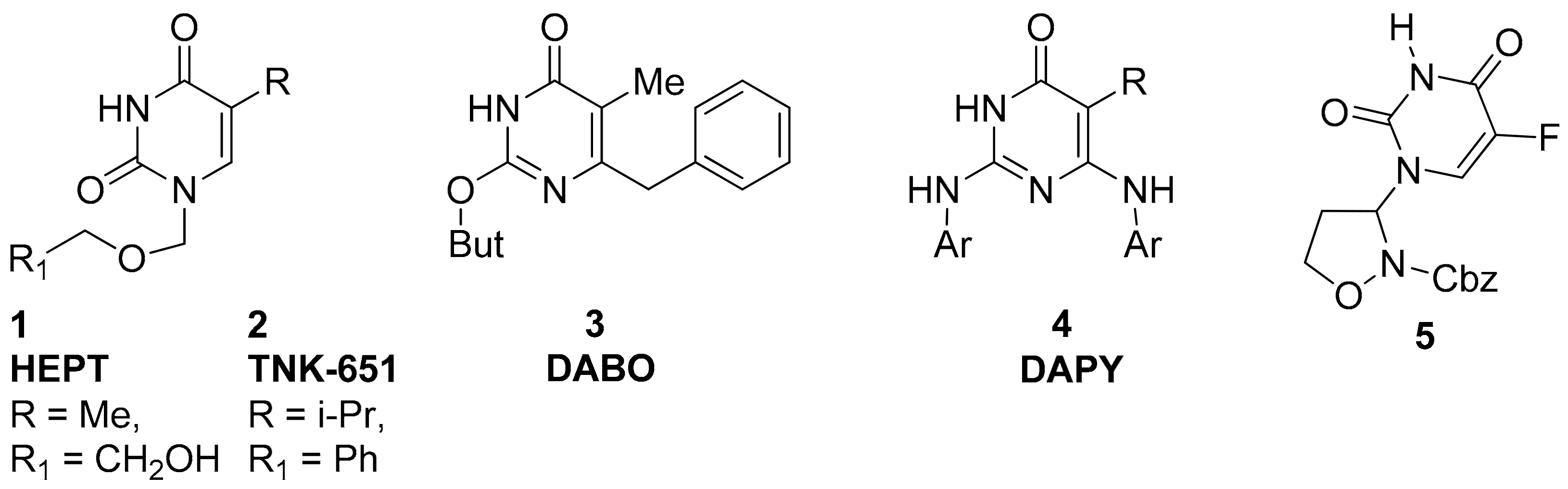
2. Results and Discussion
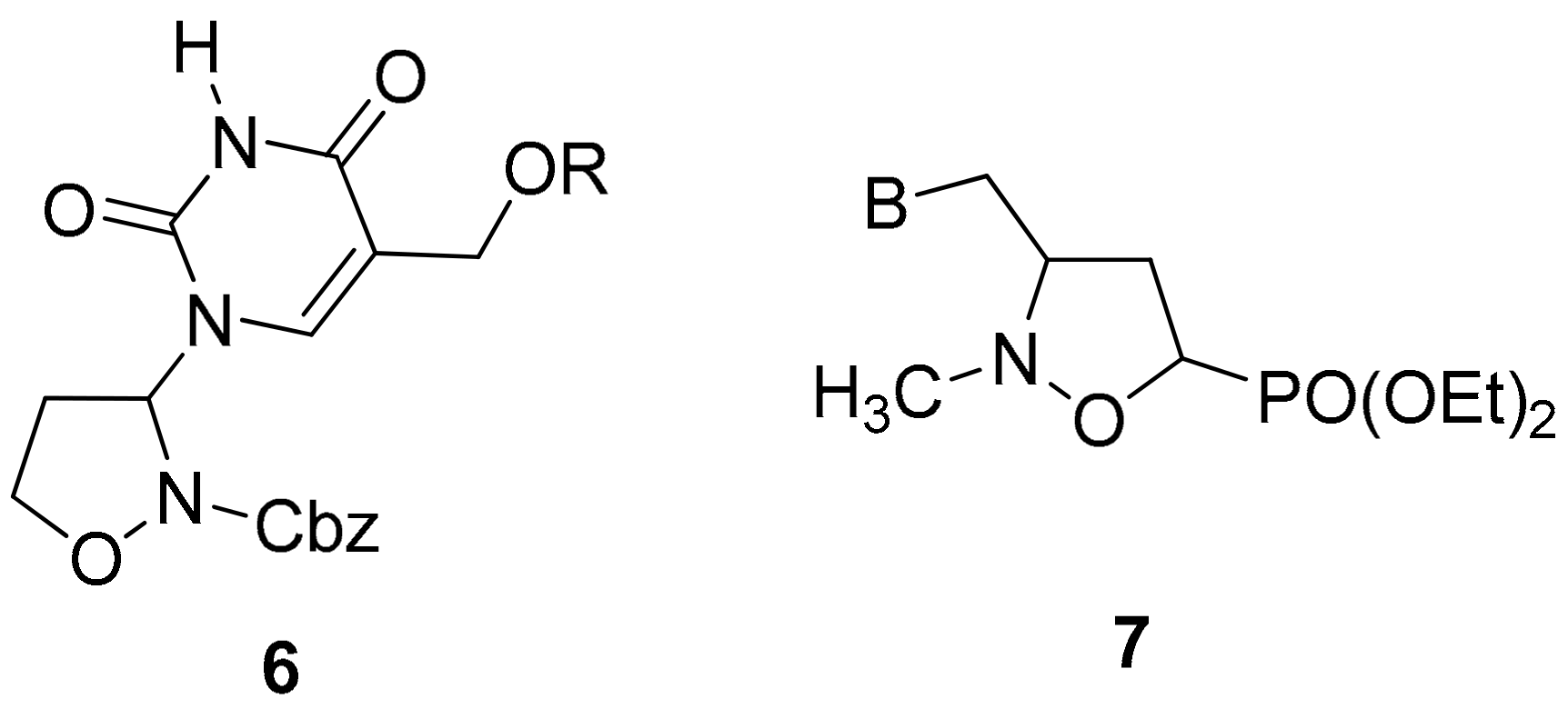
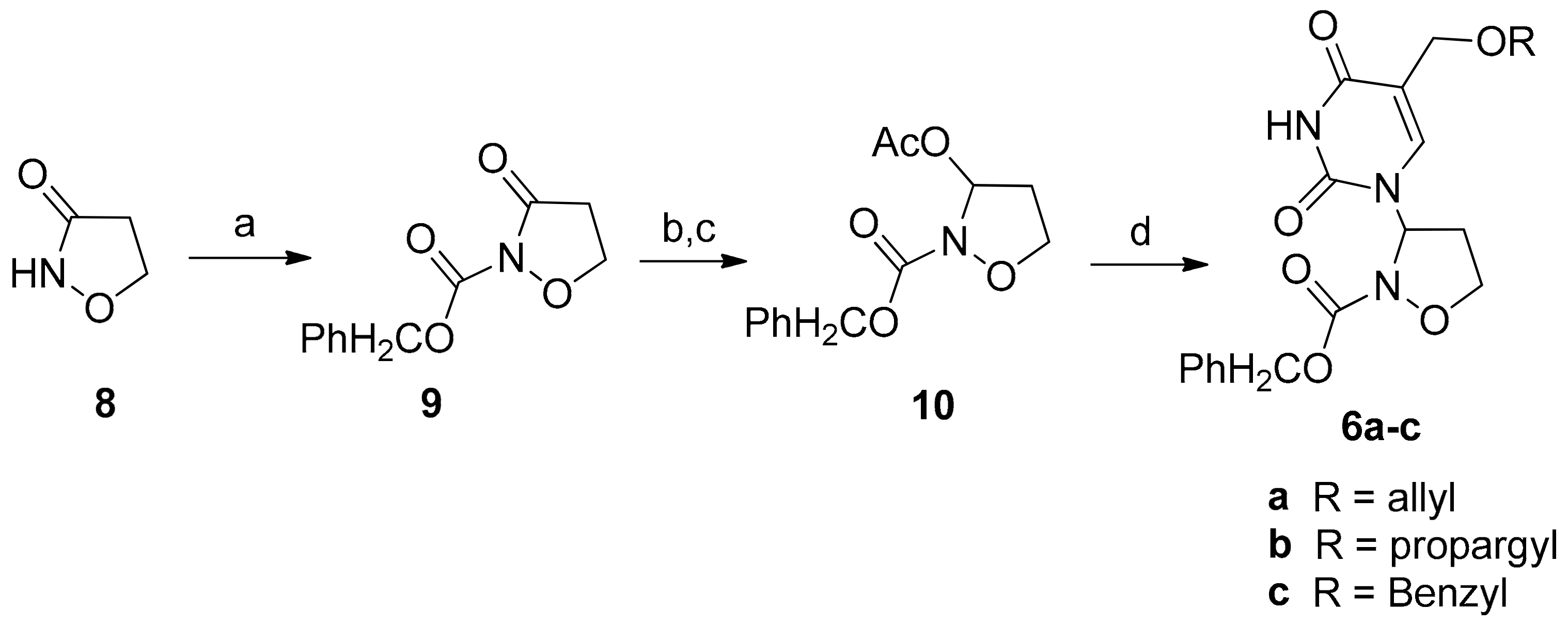
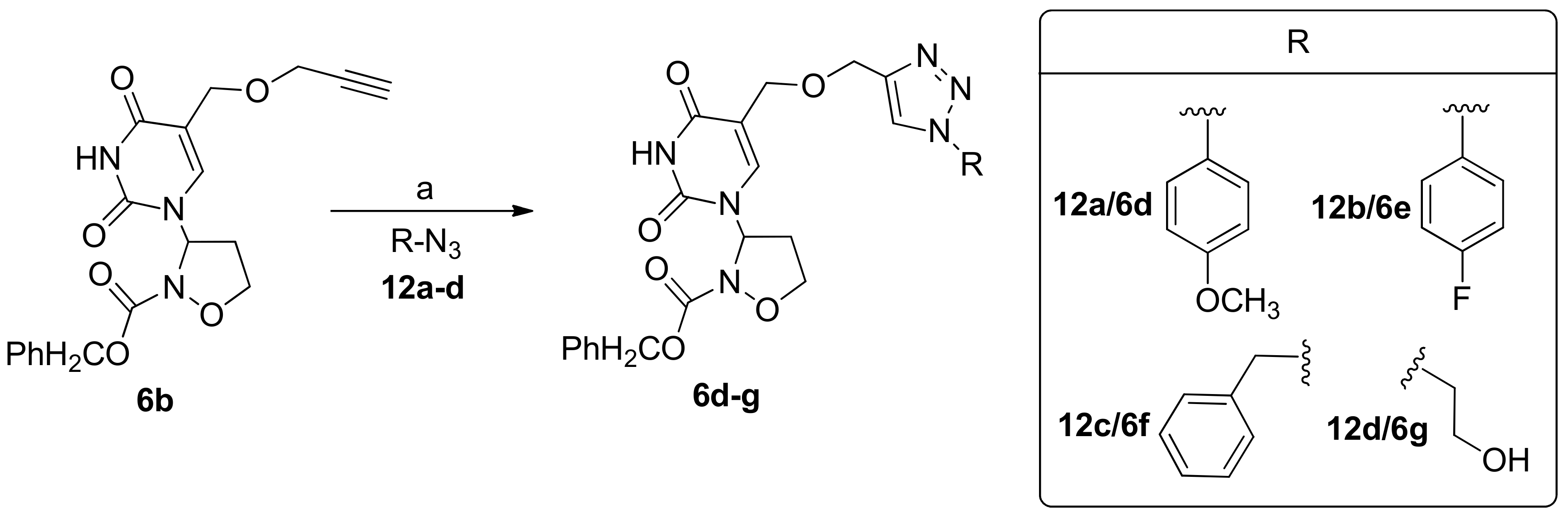
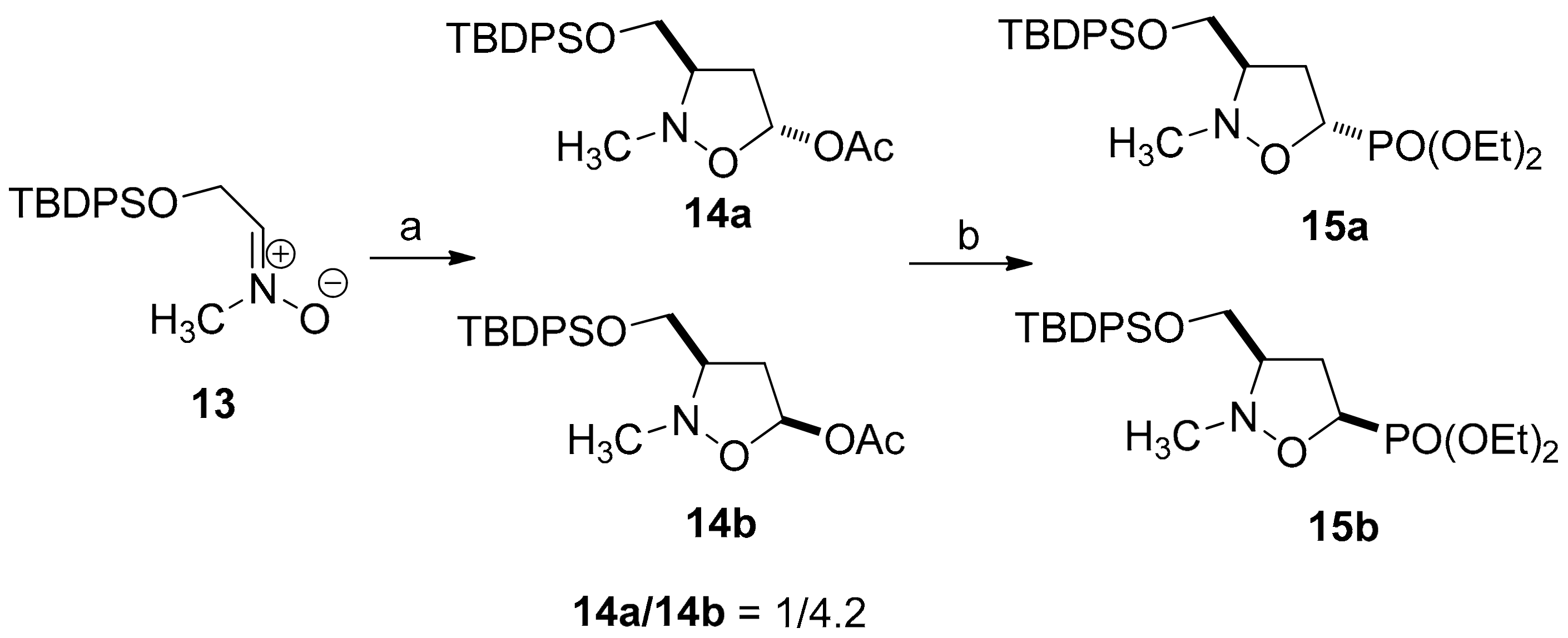
2.1. Chemistry
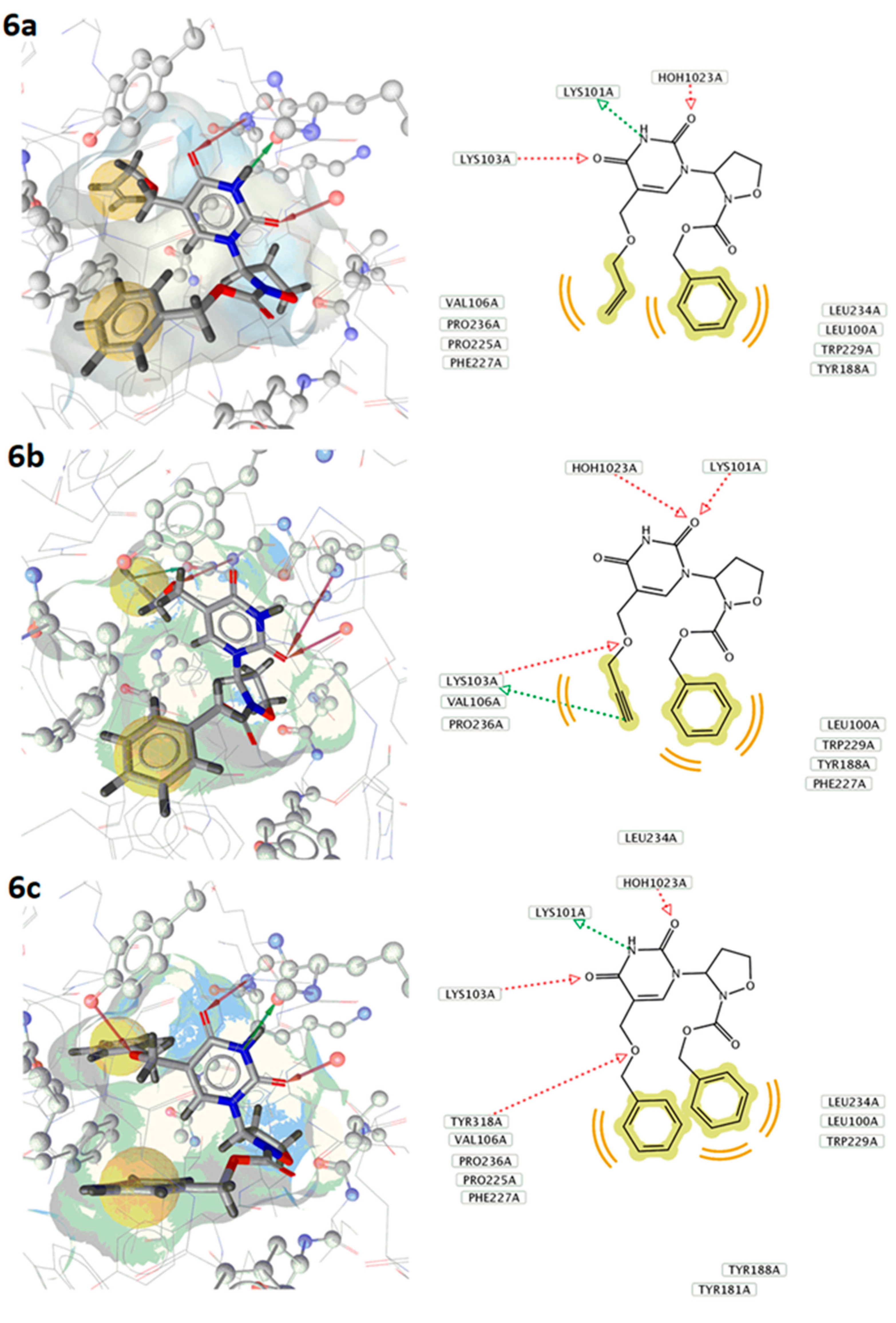
2.2. Biological Activity
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Preparation of 3-Pyrimidinyl-Isoxazolidines 6a–c
3.3. General Procedure for the Preparation of 3-Pyrimidinyl-Isoxazolidines 6d–g
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weber, R.; Ruppik, M.; Rickenbach, M.; Spoerri, A.; Furrer, H.; Battegay, M.; Cavassini, M.; Calmy, A.; Bernasconi, E.; Schmid, P.; et al. Decreasing mortality and changing patterns of causes of death in the Swiss HIV Cohort Study. HIV Med. 2013, 14, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Ryom, L.; Weber, R.; Morlat, P.; Pradier, G.; Reiss, P.; Kowalska, J.; de Wit, S.; Law, M.; El Sadr, W.; et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): A multicohort collaboration. Lancet 2014, 384, 241–248. [Google Scholar] [CrossRef]
- McManus, H.; O’Connor, C.C.; Boyd, M.; Broom, J.; Russell, D.; Watson, K.; Roth, N.; Read, P.J.; Petoumenos, K.; Law, M.G. Long-Term Survival in HIV Positive Patients with up to 15 Years of Antiretroviral Therapy. PLoS ONE 2012, 7, e48839. [Google Scholar] [CrossRef]
- Costagliola, D. Demographics of HIV and aging. Curr. Opin. HIV AIDS 2014, 9, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.N.; Grey, P.; Shaik, A.; Cooper, D.A.; Kelleher, A.D.; Petoumenos, K. Early Treatment of Primary HIV Infection Is Associated with Decreased Mortality. AIDS Res. Hum. Retrovir. 2018, 34, 936–946. [Google Scholar] [CrossRef]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 life cycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2003, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV β-chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef]
- Nicoletti, F.; Lapenta, C.; Donati, S.; Spada, M.; Ranazzi, A.; Cacopardo, B.; Mangano, K.; Belardelli, F.; Perno, C.; Aquaro, S. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin. Exp. Immunol. 2009, 155, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; McCubrey, J.A.; Bendtzen, K.; Nicoletti, F. Potential use of rapamycin in HIV infection. Br. J. Clin. Pharmacol. 2010, 70, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Fagone, P.; Meroni, P.; McCubrey, J.; Bendtzen, K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov. Today 2011, 16, 715–721. [Google Scholar] [CrossRef]
- Agnello, S.; Brand, M.; Chellat, M.F.; Gazzola, S.; Riedl, R. A structural view on medicinal chemistry strategy against drug resistance. Angew. Chem. Int. Ed. 2019, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, C.; Grelli, S.; Balestrieri, E.; Minutolo, A.; Argaw-Denboba, A.; Macchi, B.; Sinibaldi-Vallebona, P.; Perno, C.F.; Mastino, A.; Garaci, E. Thymosin alpha 1 and HIV-1: Recent advances and future perspectives. Future Microbiol. 2017, 12, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Beyrer, C.; Pozniak, A. HIV drug resistance—An emerging threat to epidemic control. N. Engl. J. Med. 2017, 377, 1605–1607. [Google Scholar] [CrossRef]
- Lehman, D.A.; Wamelwa, D.C.; McCoy, C.O.; Matsen, F.A.; Langat, A.; Chohan, B.H.; Benki-Nugent, S.; Custers-Allen, R.; Bushman, F.D.; John-Stewart, G.C.; et al. Low-frequency Nevirapine resistance at multiple sites may predict treatment failure in infants on nevirapine-based treatment. J. Acq. Immun. Def. Synd. 2012, 60, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Geronikaki, A.; Eleftheriou, P.; Poroikov, V. Anti-HIV Agents: Current status and recent trends. In Communicable Diseases of the Developing World; Springer: Berlin, Germany, 2018; pp. 37–95. [Google Scholar]
- D’Ettorre, G.; Ceccarelli, G.; Pavone, P.; Vittozzi, P.; De Girolamo, G.; Schietroma, I.; Serafino, S.; Giustini, N.; Vullo, V. What happens to cardiovascular system behind the undetectablelevel of HIV viremia? AIDS Res. Ther. 2016, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, N.S.; Chow, F.C. Neurologic Complications in Treated HIV-1 Infection. Curr. Neurol. Neurosci. Rep. 2016, 16, 62. [Google Scholar] [CrossRef]
- Haas, D.W.; Tarr, P.E. Perspectives on pharmacogenomics of antiretroviral medications and HIV-associated comorbidities. Curr. Opin. HIV AIDS 2015, 10, 116–122. [Google Scholar] [CrossRef]
- De Clercq, E. A 40-year journey in search of selective antiviral chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 1–24. [Google Scholar] [CrossRef]
- Ghosh, R.K.; Ghosh, S.M.; Chawla, S. Recent advances in antiretroviral drugs. Expert Opin. Pharmacother. 2011, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Deeks, S.G. When to Start Antiretroviral Therapy. Curr. Hiv Aids Rep. 2010, 7, 60–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Clercq, E. Antiviral drug discovery: Ten more compounds, and ten more stories (part B). Med. Res. Rev. 2009, 29, 611–645. [Google Scholar] [CrossRef]
- De Clercq, E. The discovery of antiviral agents: Ten different compounds, ten different stories. Med. Res. Rev. 2008, 28, 929–953. [Google Scholar] [CrossRef]
- Dal Pozzo, F.; Andrei, G.; Lebeau, I.; Beadle, J.R.; Hostetler, K.Y.; De Clercq, E.; Snoeck, R. In vitro evaluation of the anti-orf virus activity of alkoxyalkyl esters of CDV, cCDV and (S)-HPMPA. Antivir. Res. 2007, 75, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Holy, A. Antiviral acyclic nucleoside phosphonates structure activity studies. Antivir. Res. 2006, 71, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Dash, C.; Ahmadibeni, Y.; Hanley, M.J.; Pandhare, J.; Gotte, M.; Le Grice, S.F.J.; Parang, K. Inhibition of multi-drug resistant HIV-1 reverse transcriptase by nucleoside β-triphosphates. Bioorg. Med. Chem. Lett. 2011, 21, 3519–3522. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Himmel, D.M.; Jiang, J.-K.; Wojtak, K.; Bauman, J.D.; Rausch, J.W.; Wilson, J.A.; Beutler, J.A.; Thomas, C.J.; Arnold, E.; et al. Synthesis, Activity, and Structural Analysis of Novel α-Hydroxytropolone Inhibitors of Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease, H. J. Med. Chem. 2011, 54, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Koczor, C.A.; Lewis, W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Exp. Opin. Drug Metab. Toxicol. 2010, 6, 1493–1504. [Google Scholar] [CrossRef]
- Martin, J.C.; Hitchcock, M.J.M.; De Clercq, E.; Prusoff, W.H. Early nucleoside reverse transcriptase inhibitors for the treatment of HIV: A brief history of stavudine (D4T) and its comparison with other dideoxynucleosides. Antivir. Res. 2010, 85, 34–38. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs): Past, Present, and Future. Chem. Biodivers. 2004, 1, 44–64. [Google Scholar] [CrossRef]
- De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antivir. Res. 1998, 38, 153–179. [Google Scholar] [CrossRef]
- Ding, J.; Das, K.; Moereels, H.; Koymans, L.; Andries, K.; Janssen, P.A.; Hughes, S.H.; Arnold, E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Lewi, P.; Hughes, S.H.; Arnold, E. Crystallography and the design of anti-AIDS drugs: Conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 2005, 88, 209–231. [Google Scholar] [CrossRef]
- Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. ChemicalSynthesis of Heterocyclic−Sugar Nucleoside Analogues. Chem. Rev. 2010, 110, 3337–3370. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Carnovale, C.; Giofrè, S.V.; Macchi, B.; Frezza, A.; Marino-Merlo, C.; Pistarà, V.; Chiacchio, U. Truncatedphosphonated C-1′-branched N,O-nucleosides: A new class of antiviral agents. Bioorg. Med. Chem. 2012, 20, 3652–3657. [Google Scholar] [CrossRef]
- Balestrieri, E.; Matteucci, C.; Ascolani, A.; Piperno, A.; Romeo, R.; Romeo, G.; Chiacchio, U.; Mastino, A.; Macchi, B. Effect of Phosphonated Carbocyclic 2′-Oxa-3′-Aza-Nucleoside on Human T-Cell Leukemia Virus Type 1 Infection In Vitro. Antimicrob. Agents Chemother. 2008, 52, 54–64. [Google Scholar] [CrossRef]
- Chiacchio, U.; Iannazzo, D.; Piperno, A.; Romeo, R.; Romeo, G.; Rescifina, A.; Saglimbeni, M. Synthesis and biological evaluation of phosphonated carbocyclic 2′-oxa-3′-aza-nucleosides. Bioorg. Med. Chem. 2006, 14, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Guillemont, G.; Pasquier, E.; Palandjian, P.; Vernier, D.; Gaurrand, S.; Lewi, P.J.; Heeres, J.; de Jonge, L.M.; Koymans, F.F.; Daeyaert, M.H.; et al. Synthesis of Novel Diarylpyrimidine Analogues and Their Antiviral Activity against Human Immunodeficiency Virus Type 1. J. Med. Chem. 2005, 48, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhan, P.; Li, D.; De Clercq, E.; Liu, X. Recent advances in DAPYs and related analogues as HIV-1 NNRTIs. Curr. Med. Chem. 2011, 18, 359–376. [Google Scholar] [CrossRef]
- Schrijvers, R. Etravirine for the treatment of HIV/AIDS. Expert Opin. Pharmacother. 2013, 2014, 1087–1096. [Google Scholar] [CrossRef]
- Minudo, J.J.; Haubrich, R. Etravirine: A second-generation NNRTI for treatment-experienced adults with resistant HIV-1 infection. Futur. HIV Ther. 2008, 2, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhan, P.; De Clercq, E.; Liu, X. Strategies for the Design of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Lessons from the Development of Seven Representative Paradigms. J. Med. Chem. 2012, 55, 3595–3613. [Google Scholar] [CrossRef]
- Marino-Merlo, F.; Macchi, B.; Armenia, D.; Bellocchi, M.C.; Ceccherini-Silberstein, F.; Mastino, A.; Grelli, S. Focus on recently developed assays for detection of resistance/sensitivity to reverse transcriptase inhibitors. Appl. Microbiol. Biotechnol. 2018, 102, 9925–9936. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, P.; Huang, B.; Zhou, Z.; Yu, Z.; Yuan, Z.; Liu, H.; Pannecouque, C.; Daelemans, D.; De Clercq, E.; et al. Discovery of novel piperidine-substituted indolylarylsulfones as potent HIV NNRTIs via structure-guided scaffold morphing and fragment rearrangement. Eur. J. Med. Chem. 2017, 126, 190–201. [Google Scholar] [CrossRef]
- Li, X.; Zhang, L.; Tien, Y.; Song, Y.; Zhan, P.; Liu, X. Novel HIV-1 non-nucleoside reverse transcriptaseinhibitors: A patentreview (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1199–1227. [Google Scholar] [CrossRef]
- Zhan, P.; Chen, X.; Li, D.; Fang, Z.; De Clercq, E.; Liu, X. HIV NNRTIs: Structural diversity, pharmacophore similarity, and implications for drug design. Med. Res. Rev. 2013, 33, E1–E72. [Google Scholar] [CrossRef]
- De Bethune, M.P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in treatment of HIV infections: A review of the last 20 years (1989–2009). Antivir. Res. 2010, 85, 75–90. [Google Scholar] [CrossRef]
- Mordant, C.; Schmitt, B.; Pasquier, E.; Demestre, C.; Queguiner, L.; Masungi, C.; Peeters, A.; Smeulders, L.; Bettern, E.; Hertogs, K.; et al. Synthesis of novel diarylpyrimidine analogues of TMC278 and their antiviral activity against HIV-1 wild-type and mutant strains. Eur. J. Med. Chem. 2007, 42, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-Q.; Liang, Y.-H.; Zeng, Z.-S.; Chen, F.-E.; Balzarini, J.; Pannecouque, C.; De Clercq, E. Structural modifications of DAPY analogues with potent HIV activity. Chem. Med. Chem. 2009, 4, 219–224. [Google Scholar] [CrossRef]
- Liang, Y.-H.; He, Q.-Q.; Zeng, Z.-S.; Liu, Z.-Q.; Feng, X.-Q.; Chen, F.-E.; Pannecouque, C.; De Clercq, E. Synthesis and anti-HIV activity of 2-naphtyl substituted DAPY analogues as non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2010, 18, 4601–4605. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Tarantino, D.; Artico, M.; Nawrozkij, M.B.; Gonzalez-Ortega, E.; Clotet, B.; Samuele, A.; EstØ, J.A.; Maga, G.; Mai, A. Diarylpyrimidine-dihydrobenzyloxopyrimidine hybrids: New, wide-spectrumanti-HIV-1 agents active at (sub)nanomolar level. J. Med. Chem. 2011, 54, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Nawrozkij, M.B.; Rotili, D.; Tarantino, D.; Botta, G.; Eremiychuk, A.S.; Musmuca, I.; Ragno, R.; Samuele, A.; Zanoli, S.; Armand-Ugòn, M.; et al. 5-Alkyl-6-benzyl-2-(2-oxo-2-phenylethylsulfonyl)pyrimidine-4(3H)-ones, a series of anti-HIV-1 agents of the dihydro-alkoxy-benzyl-oxopyrimidine family with peculiar structure-activity relationship profile. J. Med. Chem. 2008, 51, 4641–4652. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Y.; Guo, Y.; Liu, Z.; Shi, X.; Xu, Y.; Wang, X.; Zhang, Z.; Liu, J. The design and synthesis of N-1-alkylated-5-aminoarylalkylsubstituted-6-methyluracils as potential non-nucleoside HIV-1 RT inhibitors. Bioorg. Med. Chem. 2007, 15, 7399–7407. [Google Scholar] [CrossRef]
- Danel, K.; Larsen, L.M.; Pedersen, E.B.; Sanna, G.; La Colla, P.; Loddo, R. Synthesis and antiviral activity of new dimeric inhibitors against HIV-1. Bioorg. Med. Chem. 2008, 16, 511–517. [Google Scholar] [CrossRef]
- Campiani, G.; Ramunno, A.; Maga, G.; Nacci, V.; Fattorusso, C.; Catalanotti, B.; Morelli, E.; Novellino, E. Non-nucleoside HIV-1 reverse transcriptase (RT) inhibitors: Past, present, and future perspectives. Curr. Pharm. Des. 2002, 8, 615–657. [Google Scholar] [CrossRef]
- Romeo, R.; Giofrè, S.V.; Macchi, B.; Balestrieri, E.; Mastino, A.; Merino, P.; Romeo, G.; Chiacchio, U. Truncated reverse isoxazolidinyl nucleosides: A new class of allosteric HIV-1 reverse transcriptase inhibitors. Chem. Med. Chem. 2012, 7, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Macchi, B.; Romeo, G.; Chiacchio, U.; Frezza, C.; Marino Merlo, F.; Mastino, A. Phosphonated nucleoside analoguesasantiviral agents. In Topics in Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2015; Volume 9, pp. 53–92. [Google Scholar]
- Reddy, A.S.; Kumar, M.S.; Reddy, G.R. A convenient method for the preparation of hydroxamic acids. Tetrahedron Lett. 2000, 41, 6285–6288. [Google Scholar] [CrossRef]
- Piperno, A.; Chiacchio, U.; Iannazzo, D.; Giofrè, S.V.; Romeo, G.; Romeo, R. First example of direct RuO4-catalyzed oxidation of isoxazolidines to 3-oxazolidones. J. Org. Chem. 2007, 72, 3958–3960. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Balzarini, J.; Andrei, G.; Schols, D.; Snoeck, R.; Wroblewski, A.E.; Gatkowska, J. Novel isoxazolidine analogues of homonuclemosides and homonucleotides. Tetrahedron 2016, 72, 8294–8308. [Google Scholar] [CrossRef]
- Romeo, R.; Giofrè, S.V.; Iaria, D.; Sciortino, M.T.; Ronsisvalle, S.; Chiacchio, M.A.; Scala, A. Synthesis of 5-alkynyl isoxazolidinyl nucleosides. Eur. J. Org. Chem. 2011, 28, 5690–5695. [Google Scholar] [CrossRef]
- Romeo, R.; Giofrè, S.V.; Garozzo, A.; Bisignano, B.; Corsaro, A.; Chiacchio, M.A. Synthesis and biological evaluation of furopyrimidine N,O-nucleosides. Bioorg. Chem. Med. 2013, 21, 5688–5693. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Balestrieri, E.; Marino-Merlo, F.; Mastino, A.; Macchi, B. A novel, cell-free PCR-based assay for evaluating the inhibitor activity of antiretroviral compounds against HIV reverse transcriptase. J. Med. Virol. 2014, 86, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Grelli, S.; Federico, M.; Marino-Merlo, F.; Mastino, A.; Macchi, B. Testing anti-HIV activity of antiretroviral agents in vitro using flow cytometry analysis of CEM-GFP cells infected with transfection-derived HIV-1 NL4-3. J. Med. Virol. 2016, 88, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Medici, M.A.; Sciortino, M.T.; Perri, D.; Amici, C.; Avitabile, E.; Ciotti, M.; Balestrieri, E.; De Smaele, E.; Franzoso, G.; Mastino, A. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: Role of nuclear factor kappaB. J. Biol. Chem. 2003, 19, 36059–36067. [Google Scholar] [CrossRef]
- Romeo, R.; Giofrè, S.V.; Carnovale, C.; Chiacchio, M.A.; Campisi, A.; Mancuso, R.; Cirmi, S.; Navarra, M. Synthesis and Biological Activity of Triazole-Appended N,O-Nucleosides. Eur. J. Org. Chem. 2014, 25, 5442–5447. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 6a–g and 7a,b are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT Inhibition Assay 1 MRTIC (μM) | Cytotoxicity CC50 ± SD (μM) 2 | ||
|---|---|---|---|
| Compound | HIV-RT | MOLT-3 | Pearson’s 3 |
| 6a | 0.1 | >1000 | 0.94 |
| 6b | 1 | >1000 | 0.95 |
| 6c | 0.01 | >1000 | 0.97 |
| 6d | >100 | 594 ± 0.8 | - |
| 6e | >100 | 579 ± 2 | - |
| 6f | >100 | 552 ± 3 | - |
| 6g | 100 | 670 ± 1 | - |
| 7a | n.d. | >1000 | - |
| 7b | 90 4 | >1000 | - |
| NVP | 0.001 | >1000 | 0.97 |
| RPV | 0.038 | n.d. | n.d. |
| EVF | 0.0000094 | n.d. | n.d. |
| IC50 ± SD (µM) 1 | Cytotoxicity CC50 ± SD (µM) 2 | Selectivity Index 3 | |
|---|---|---|---|
| Compound | % GFP + cells | CEM/GFP | SI |
| 6a | 10.1 | 747 ± 10.7 | 73.9 |
| 6b | 0.69 | 1053 ± 8.2 | 1526 |
| 6c | 15.7 | 1127 ± 6.6 | 71.8 |
| 6g | 103 | 427 ± 0.1 | 4 |
| NVP | 0.14 | 980 ± 9.9 | 7000 |
| Compound | ΔG Kcal/mol |
|---|---|
| 6a | −8.51 |
| 6b | −7.45 |
| 6c | −8.62 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, R.; Iannazzo, D.; Veltri, L.; Gabriele, B.; Macchi, B.; Frezza, C.; Marino-Merlo, F.; Giofrè, S.V. Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors. Molecules 2019, 24, 1718. https://doi.org/10.3390/molecules24091718
Romeo R, Iannazzo D, Veltri L, Gabriele B, Macchi B, Frezza C, Marino-Merlo F, Giofrè SV. Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors. Molecules. 2019; 24(9):1718. https://doi.org/10.3390/molecules24091718
Chicago/Turabian StyleRomeo, Roberto, Daniela Iannazzo, Lucia Veltri, Bartolo Gabriele, Beatrice Macchi, Caterina Frezza, Francesca Marino-Merlo, and Salvatore V. Giofrè. 2019. "Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors" Molecules 24, no. 9: 1718. https://doi.org/10.3390/molecules24091718
APA StyleRomeo, R., Iannazzo, D., Veltri, L., Gabriele, B., Macchi, B., Frezza, C., Marino-Merlo, F., & Giofrè, S. V. (2019). Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors. Molecules, 24(9), 1718. https://doi.org/10.3390/molecules24091718