Al(Salen) Metal Complexes in Stereoselective Catalysis


Abstract
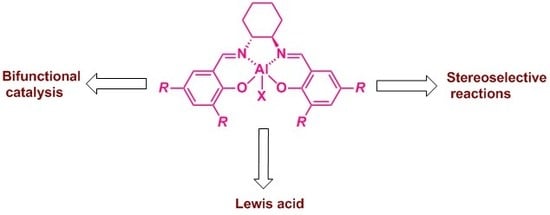
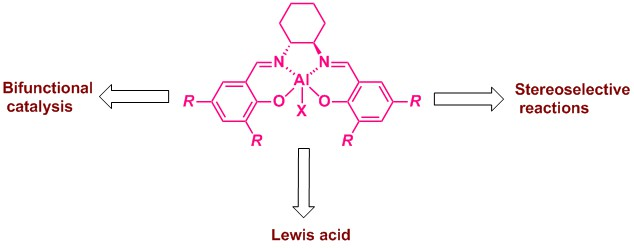
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
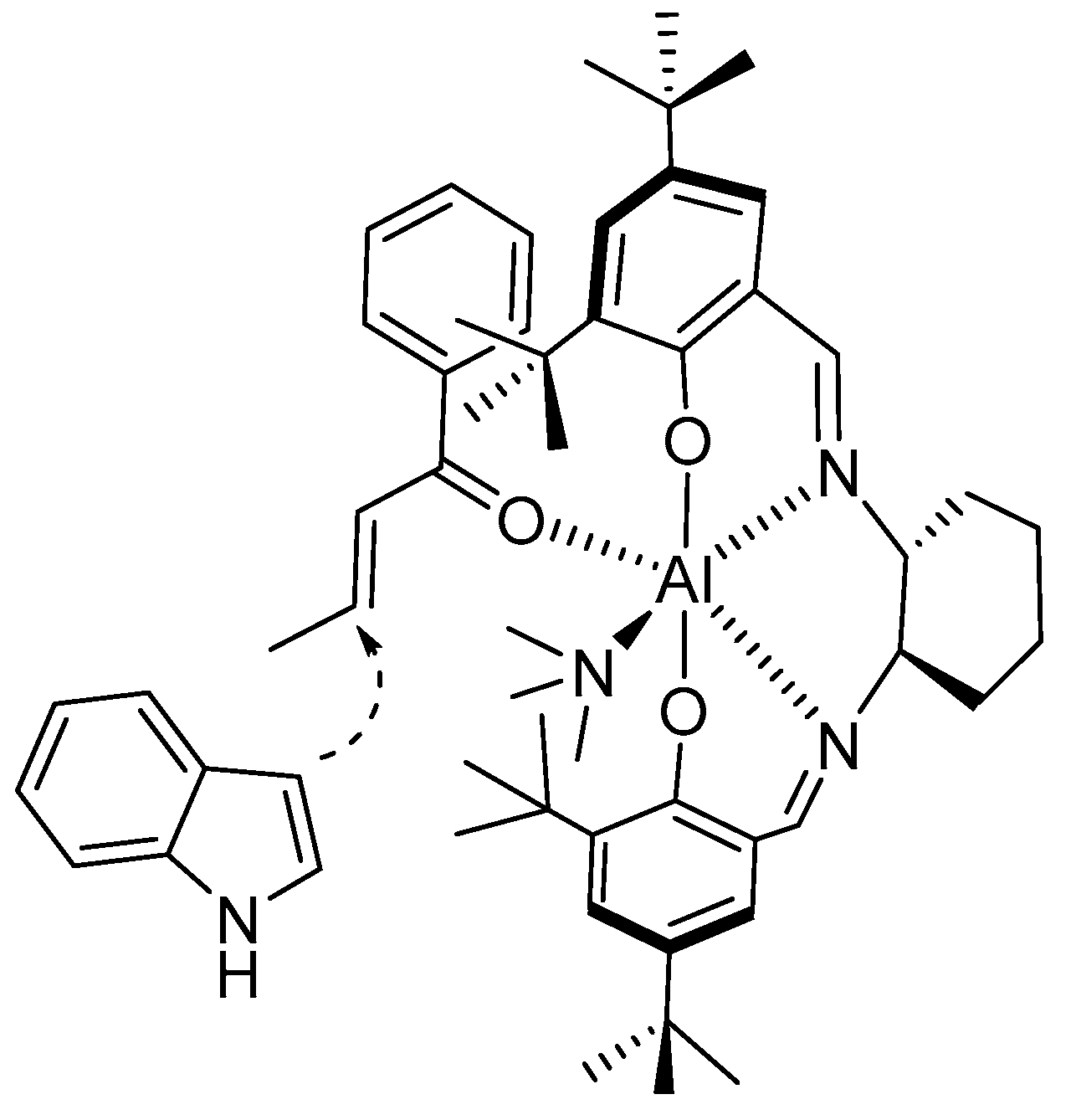{kind=link}
{kind=link}
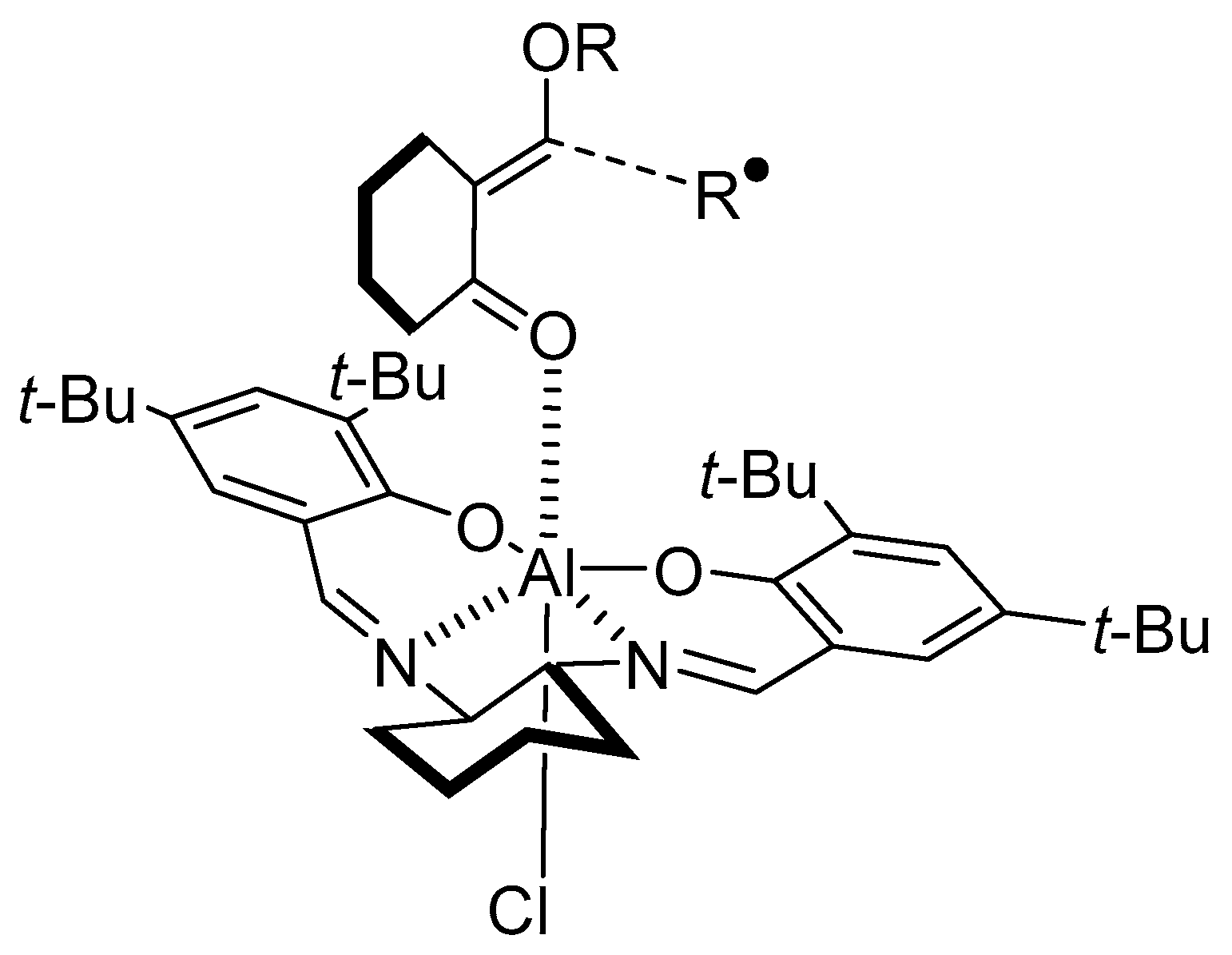{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
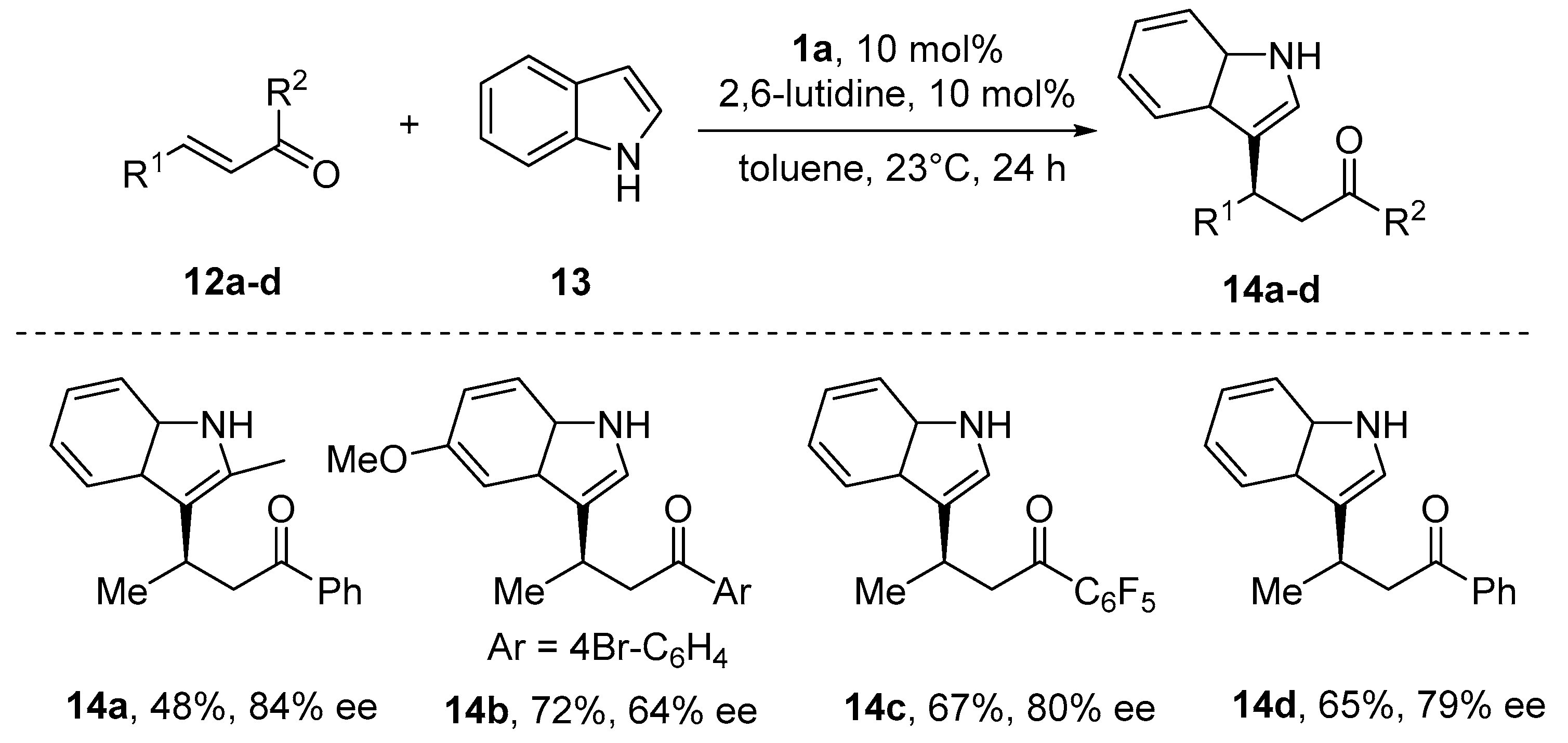{kind=link}
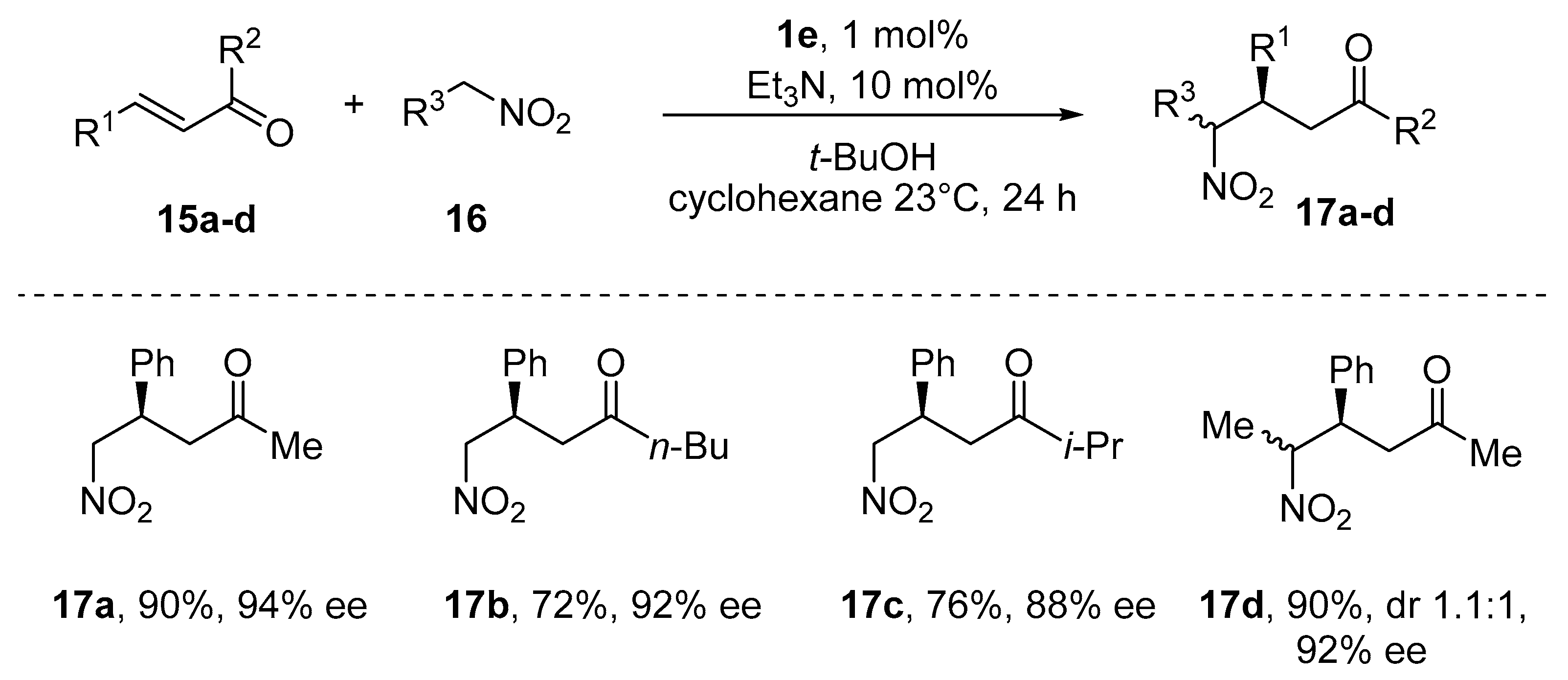{kind=link}
{kind=link}
{kind=link}
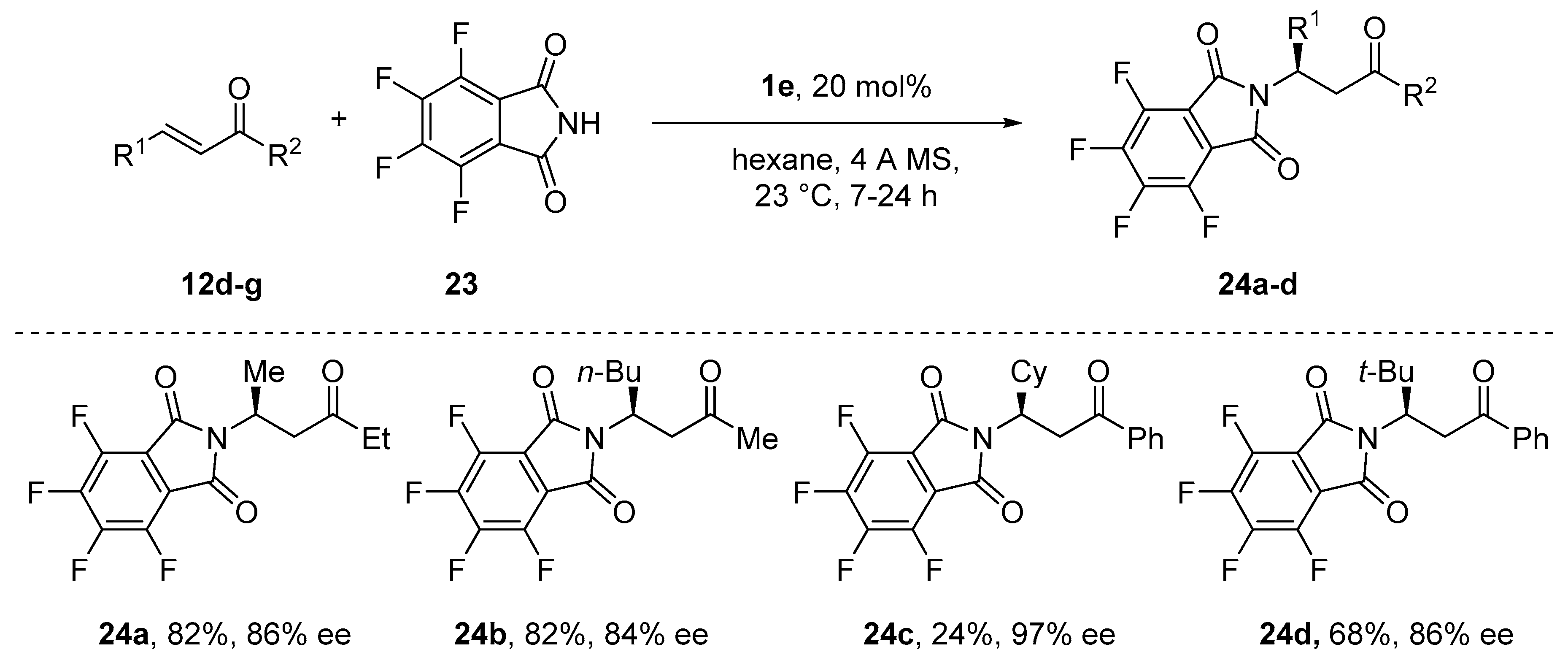{kind=link}
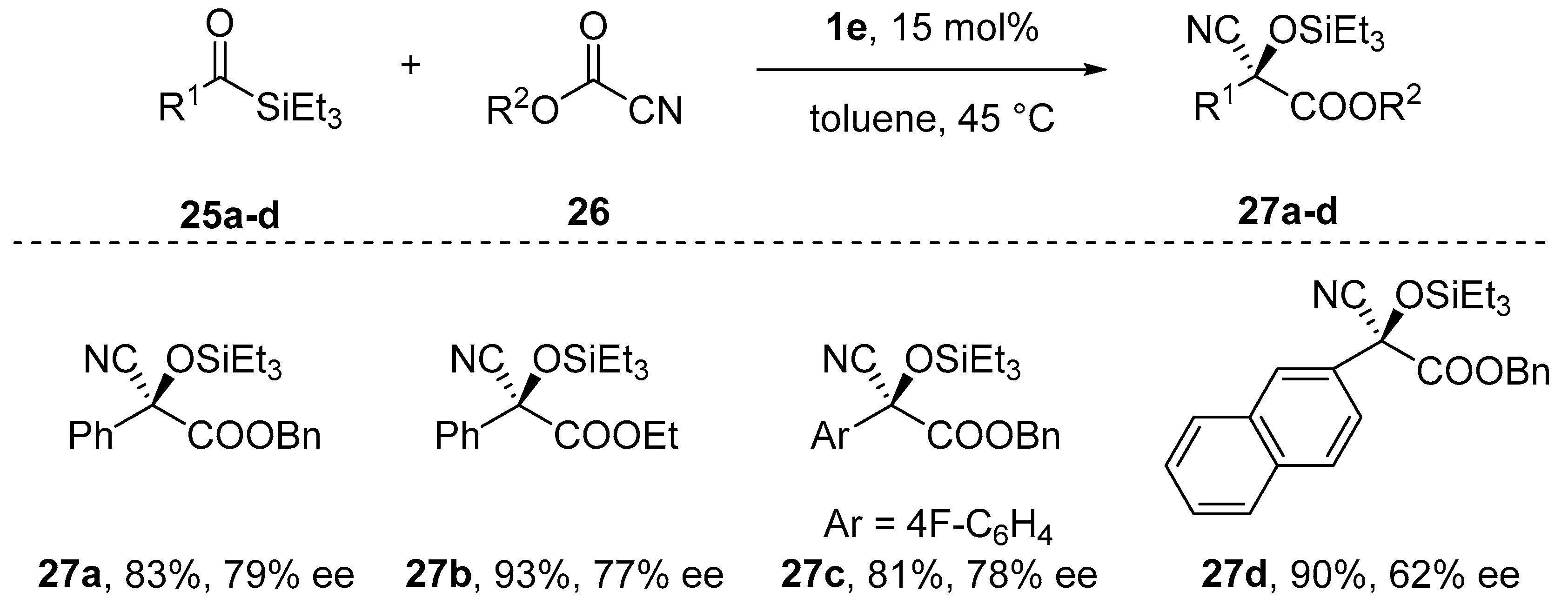{kind=link}
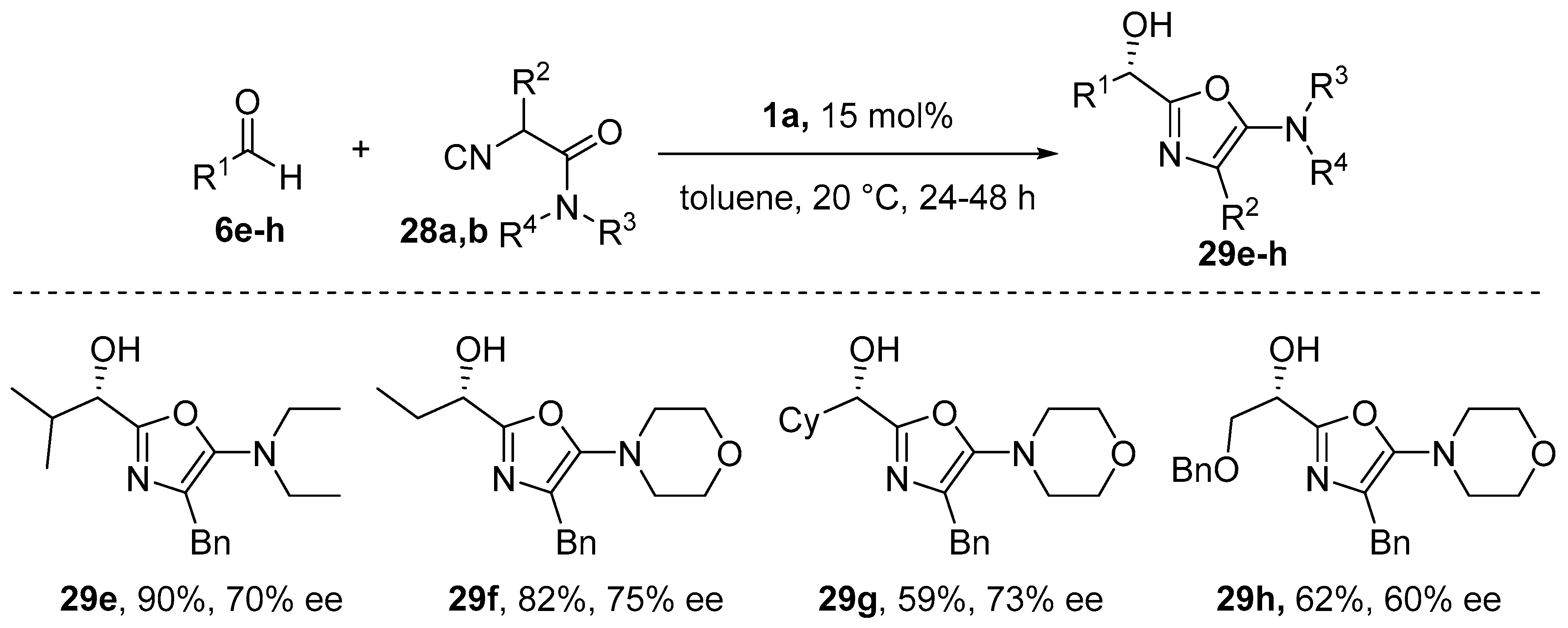{kind=link}
{kind=link}
{kind=link}
{kind=link}
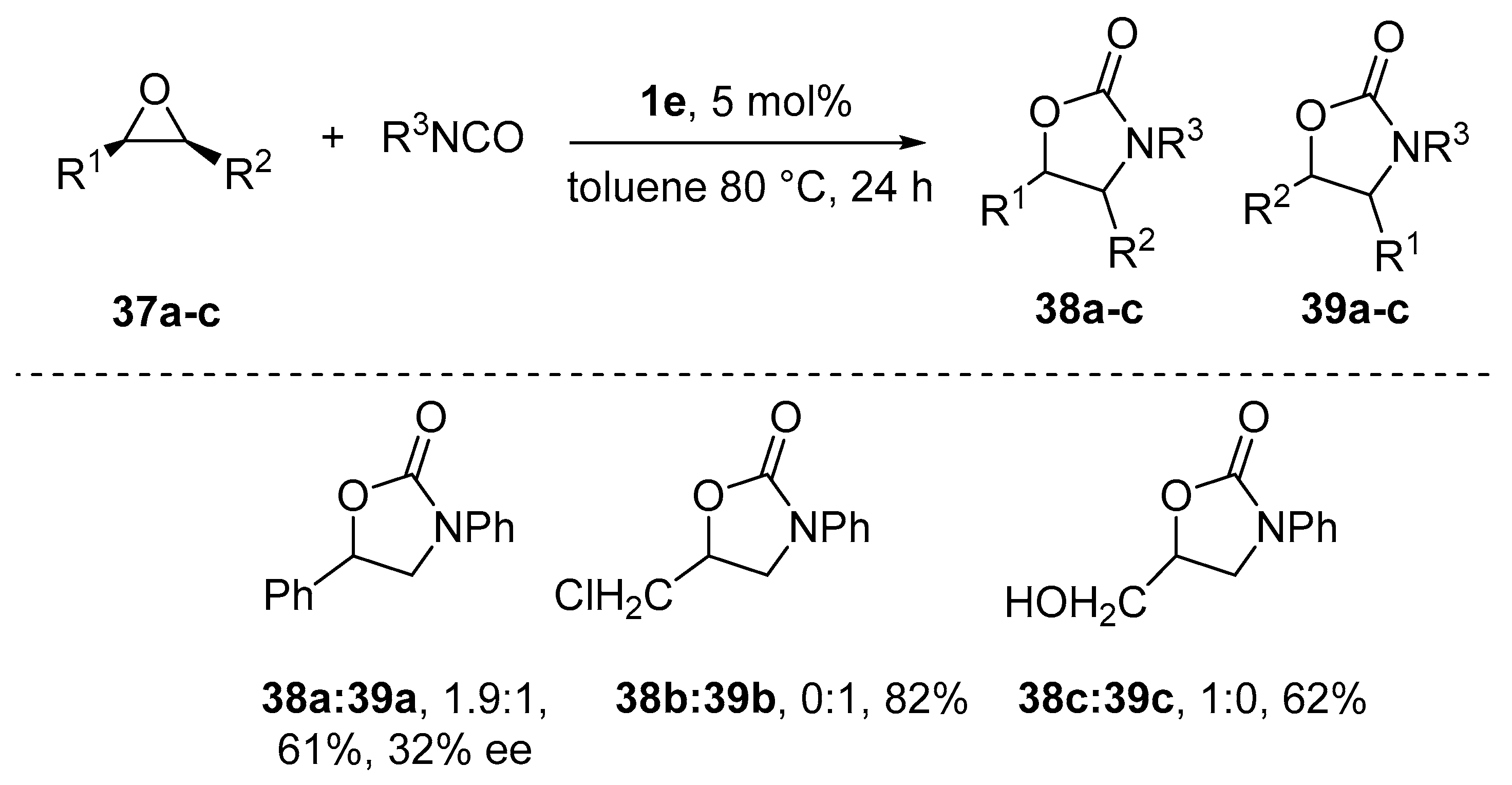{kind=link}
{kind=link}
1. Introduction
2. Aluminum: General Properties and Character
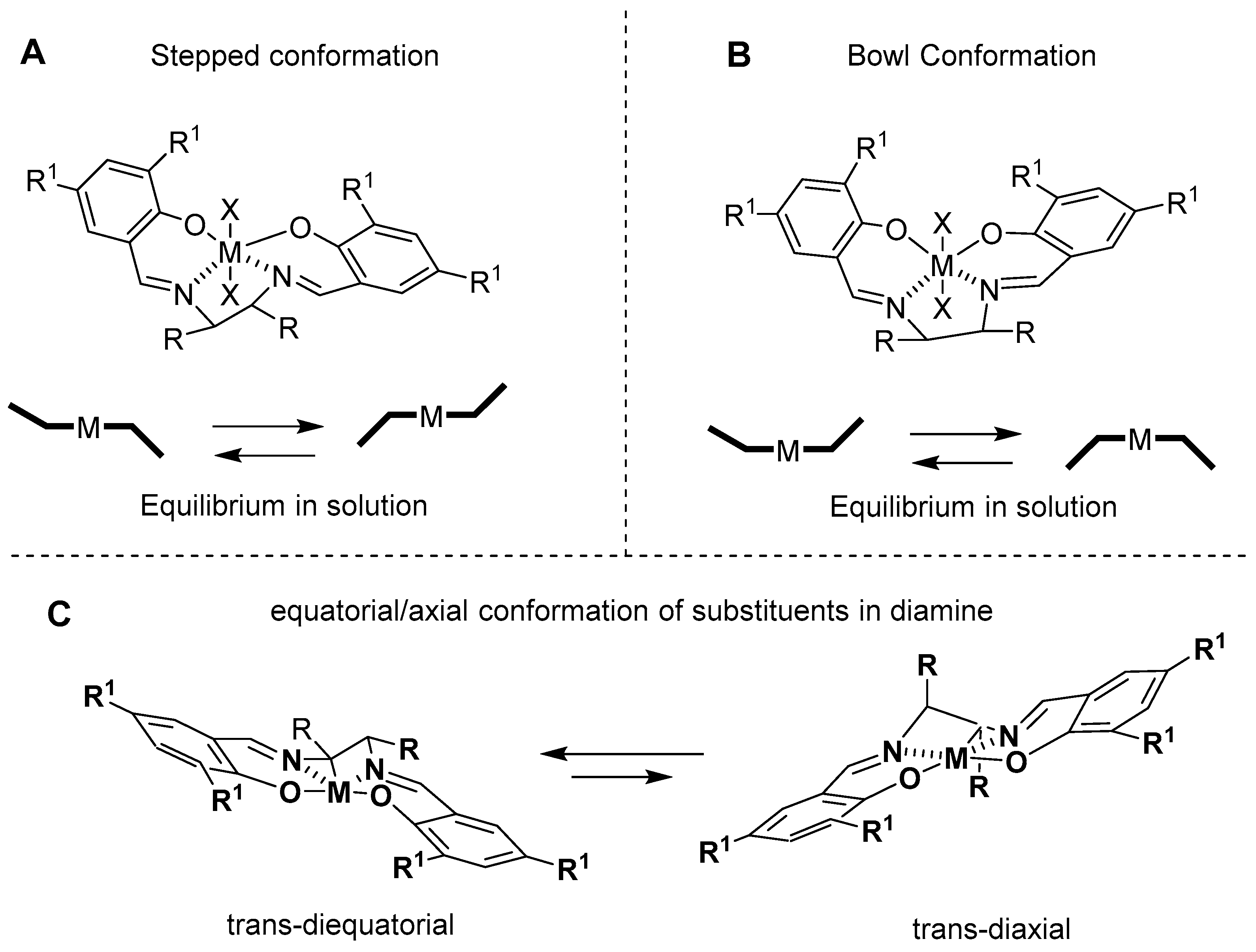
3. Al(Salen): Synthesis, Properties and Coordination Chemistry
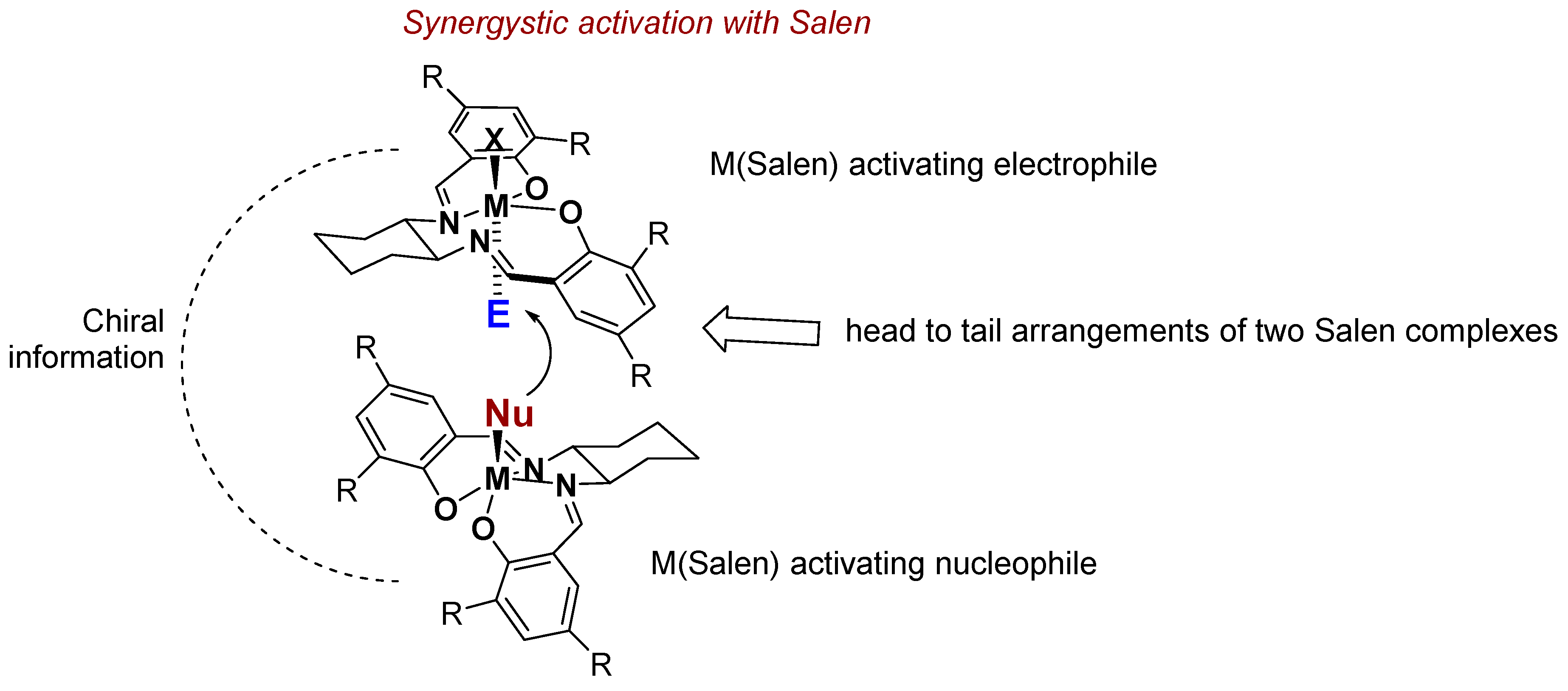
4. Chiral Al(salen) Complexes Used in Stereoselective Reactions
5. Addition of CN to Electrophiles Promoted by Chiral Al(Salen) Complexes
6. Michael-Type Reactions Promoted by Chiral Al(salen) Complexes
7. Reaction of Nucleophiles/Enolates Promoted by Chiral Al(salen) Complexes
8. Cycloaddition and Multicomponent Stereocontrolled Reactions Promoted by Chiral Al(Salen) Complexes
9. Chiral Al(salen) Complexes, CO2 and Related Electrophiles
10. Polymerization Reactions Promoted by Chiral Al(Salen) Complexes
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wei, Y.; Zhang, B.-L.; Liu, P.; He, W.; Zhang, S.-Y. The Application of Chiral Schiff Base in Asymmetric Catalysis. Mini-Rev. Org. Chem. 2011, 8, 66–90. [Google Scholar] [CrossRef]
- Canali, L.; Sherrington, D.C. Utilisation of Homogeneous and Supported Chiral Metal(salen) Complexes in Asymmetric Catalysis. Chem. Soc. Rev. 1999, 28, 85–93. [Google Scholar] [CrossRef]
- Katsuki, T. Some Recent Advances in Metallosalen Chemistry. Synlett 2003, 3, 281–297. [Google Scholar] [CrossRef]
- Che, C-M.; Huang, J-S. Metal complexes of chiral binaphthyl Schiff-base ligands and their application in stereoselective organic transformations. Coord. Chem. Rev. 2003, 242, 97–113. [Google Scholar] [CrossRef]
- Cozzi, P.G. Metal-Salen Schiff base complexes in catalysis: practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Larrow, J.F.; Jacobsen, E.N. Asymmetric Processes Catalyzed by Chiral (Salen)Metal Complexes. Topics Organomet. Chem. 2004, 6, 123–152. [Google Scholar] [CrossRef]
- Baleizão, C.; Garcia, H. Chiral Salen Complexes: An Overview to Recoverable and Reusable Homogeneous and Heterogeneous Catalysts. Chem. Rev. 2006, 106, 3987–4043. [Google Scholar] [CrossRef]
- Matsumoto, K.; Saito, B.; Katsuki, T. Asymmetric catalysis of metal complexes with non-planar ONNO ligands: salen, salalen and salan. Chem. Commun. 2007, 3619–3627. [Google Scholar] [CrossRef] [PubMed]
- Clever, G.H.; Sötl, Y.; Spahl, W.; Carell, T. Metal-Salen-Based Pair Complex Inside DNA: Complexation Overrides Sequence Information. Chem. Eur. J. 2006, 12, 8708–8718. [Google Scholar] [CrossRef]
- Kleij, A.W. Zinc-centred salen complexes: Versatile and accessible supramolecular building motifs. Dalton Trans. 2009, 4635–4639. [Google Scholar] [CrossRef] [PubMed]
- Plattner, D.A.; Feichtinger, D.; El-Bahraoui, J.; Wiest, O. Coordination chemistry of manganese–salen complexes studied by electrospray tandem mass spectrometry: The significance of axial ligands. Int. J. Mass. Spectr. 2000, 195/196, 351–362. [Google Scholar] [CrossRef]
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds: Principles and Applications; Royal Society of Chemistry: Cambridge, UK, 2007; ISBN 978-0-85404-246-3. [Google Scholar]
- Ford, D.D.; Nielsen, L.P.C.; Zuend, S.J.; Musgrave, C.B.; Jacobsen, E.N. Mechanistic Basis for High Stereoselectivity and Broad Substrate Scope in the (salen)Co(III)-Catalyzed Hydrolytic Kinetic Resolution. J. Am. Chem. Soc. 2013, 135, 15595–15608. [Google Scholar] [CrossRef]
- Liu, Y.S.; Li, J.; Ma, X.L.; Yang, Z.; Roesky, H.W. The chemistry of aluminum(I) with β-diketiminate ligands and pentamethylcyclopentadienyl-substituents: Synthesis, reactivity and applications. Coord. Chem. Rev. 2018, 374, 387–415. [Google Scholar] [CrossRef]
- Woodward, S.; Dagorne, S. Modern Organoaluminum Reagents: Preparation, Structure, Reactivity and Use; Woodward, S., Dagorne, S., Eds.; Springer: Heidelberg, Germany, 2013; Volume 41, ISBN 978-3-642-33672-0. [Google Scholar] [CrossRef]
- Goldmisth, C.R. Aluminum and gallium complexes as homogeneous catalysts for reduction/oxidation reactions. Coord. Chem. Rev. 2018, 377, 209–224. [Google Scholar] [CrossRef]
- Ooi, T.; Miura, T.; Maruoka, K. Highly Efficient, Catalytic Meerwein-Ponndorf-Verley Reduction with a Novel Bidentate Aluminum Catalyst. Angew. Chem. Int. Ed. 1998, 37, 2347–2349. [Google Scholar] [CrossRef]
- Klaue, A.; Kruck, M.; Friederichs, N.; Bertola, F.; Wu, W.; Morbidelli, M. Insight into the synthesis process of an industrial Ziegler−Natta catalyst. Ind. Eng. Chem. Res. 2019, 58, 886–896. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Dolci, L.S.; Garelli, A.; Montalti, M.; Prodi, L.; Zaccheroni, N. Photophysical properties of Schiff-base metal complexes. New J. Chem. 2003, 27, 692–697. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Hockin, B.M.; Li, C.; Robertson, N.; Zysman-Colman, E. Photoredox catalysts based on earth-abundant metal complexes. Catal. Sci. Technol. 2019, 9, 889–915. [Google Scholar] [CrossRef]
- Gualandi, A.; Ceroni, P.; Cozzi, P.G. ClAl(Salen) as active photoredox catalysts. Manuscript in preparation. Manuscript in preparation.
- Atwood, D.A.; Harvey, M.J. Group 13 Compounds Incorporating Salen Ligands. Chem. Rev. 2001, 101, 37–52. [Google Scholar] [CrossRef]
- Dzugan, S.J.; Goedken, V.L. Factors affecting aluminum-carbon bond reactivity of tetradentate Schiff-based organoaluminum complexes. Inorg. Chem. 1986, 25, 2858–2864. [Google Scholar] [CrossRef]
- Atwood, D.A.; Hill, M.S.; Jegier, J.A.; Rutherford, D. The Use of Five-Coordinate Aluminum Alkyls to Prepare Molecules Containing a Single Al−O−Si Linkage. Organometallics 1997, 16, 2659–2664. [Google Scholar] [CrossRef]
- Rutherford, D.; Atwood, D.A. Five-Coordinate Aluminum Amides. Organometallics 1996, 15, 4417–4422. [Google Scholar] [CrossRef]
- Le Borgne, A.; Vincens, V.; Jouglard, M.; Spassky, N. Ring-opening oligomerization reactions using aluminium complexes of Schiff’s bases as initiators. Makromol. Chem. Macromol. Symp. 1993, 73, 37–46. [Google Scholar] [CrossRef]
- Duxbury, J.P.; Cawley, A.; Thornton-Pett, M.; Wantz, L.; Warne, J.N.D.; Greatrex, R.; Brown, D.; Kee, T.P. Chiral aluminum complexes as phospho-transfer catalysts. Tetrahedron Lett. 1999, 40, 4403–4406. [Google Scholar] [CrossRef]
- Atwood, D.A.; Jegier, J.A.; Rutherford, D. A New Class of Aluminum Cations Based upon Tetradentate (N2O2) Chelating Ligands. Inorg. Chem. 1996, 35, 63–70. [Google Scholar] [CrossRef]
- Munoz-Hernandez, M.-A.; Yearwood, B.; Wei, P.; Atwood, D.A.J. Chiral five- and six- coordinated aluminum. J. Chem. Crystallogr. 2000, 30, 215–218. [Google Scholar] [CrossRef]
- Mengozzi, L.; El Garah, M.; Gualandi, A.; Iurlo, M.; Fiorani, A.; Ciesielski, A.; Marcaccio, M.; Paolucci, F.; Samorì, P.; Cozzi, P.G. Phenoxyaluminum(salophen) Scaffolds: Synthesis, Electrochemical Properties, and Self-Assembly at Surfaces of Multifunctional Systems. Chem. Eur. J. 2018, 24, 11954–11960. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K.; Lin, C.-C. Polymer-supported Schiff base complexes in oxidation reactions. Coord. Chem. Rev. 2009, 253, 1926–1946. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Zhang, W.; Muci, R.A.; Ecker, J.M.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Feng, X. Asymmetric Strecker Reactions. Chem. Rev. 2011, 111, 6947–6983. [Google Scholar] [CrossRef]
- Williams, R.M.; Hendrix, J.A. Asymmetric synthesis of arylglycines. Chem. Rev. 1992, 92, 889–917. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Enantioselective Addition of Hydrogen Cyanide to Imines Catalyzed by a Chiral (Salen)Al(III) Complex. J. Am. Chem. Soc. 1998, 120, 5315–5316. [Google Scholar] [CrossRef]
- North, M. Synthesis and applications of non-racemic cyanohydrins. Tetrahedron: Asymmetry 2003, 14, 147–176. [Google Scholar] [CrossRef]
- Chen, F.-X.; Zhou, H.; Liu, X.-H.; Qin, B.; Feng, X.-M. Enantioselective Cyanosilylation of Ketones by a Catalytic Double-Activation Method with an Aluminium Complex and an N-Oxide. Chem. Eur. J. 2004, 10, 4790–4797. [Google Scholar] [CrossRef]
- Alaaeddine, A.; Roisnel, T.; Thomas, C.M.; Carpentier, J.-F. Discrete versus In Situ-Generated Aluminum-Salen Catalysts in Enantioselective Cyanosilylation of Ketones: Role of Achiral Ligands. Adv. Synth. Catal. 2008, 350, 731–740. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhao, G.; Zhou, Z.; Tang, C. A Novel Chiral (salen)AlIII Complex Catalyzed Asymmetric Cyanosilylation of Aldehydes. Eur. J. Org. Chem. 2008, 1615–1618. [Google Scholar] [CrossRef]
- Esteves, M.A.; Gigante, B.; Santos, C.; Guerreiro, A.M.; Baleizão, C. New heterogeneous catalysts for the synthesis of chiral amino acids: Functionalization of organic resins with chiral salen complexes. Cat. Today 2013, 218–219, 65–69. [Google Scholar] [CrossRef]
- Brodbeck, D.; Broghammer, F.; Meisner, J.; Klepp, J.; Garnier, D.; Frey, W.; Johannes Kästner, J.; Peters, R. An Aluminum Fluoride Complex with an Appended Ammonium Salt as an Exceptionally Active Cooperative Catalyst for the Asymmetric Carboxycyanation of Aldehydes. Angew. Chem. Int. Ed. 2017, 56, 4056–4060. [Google Scholar] [CrossRef]
- Brodbeck, D.; Àlvarez-Barcia, S.; Meisner, J.; Broghammer, F.; Klepp, J.; Garnier, D.; Frey, W.; Johannes Kästner, J.; Peters, R. Asymmetric Carbocyanation of Aldehydes by Cooperative AlF/Onium Salt Catalysis: From Cyanoformate to KCN as Cyanide Source. Chem. Eur. J. 2019, 25, 1515–1524. [Google Scholar] [CrossRef]
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis, 1st ed.; Baldwin, E.J., Ed.; Elsevier: Pergamon, Turkey, 2013; Volume 9, ISBN 9781483293783. [Google Scholar]
- Zheng, K.; Liu, X.; Feng, X. Recent Advances in Metal-Catalyzed Asymmetric 1,4-Conjugate Addition (ACA) of Nonorganometallic Nucleophiles. Chem. Rev. 2018, 118, 7586–7656. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
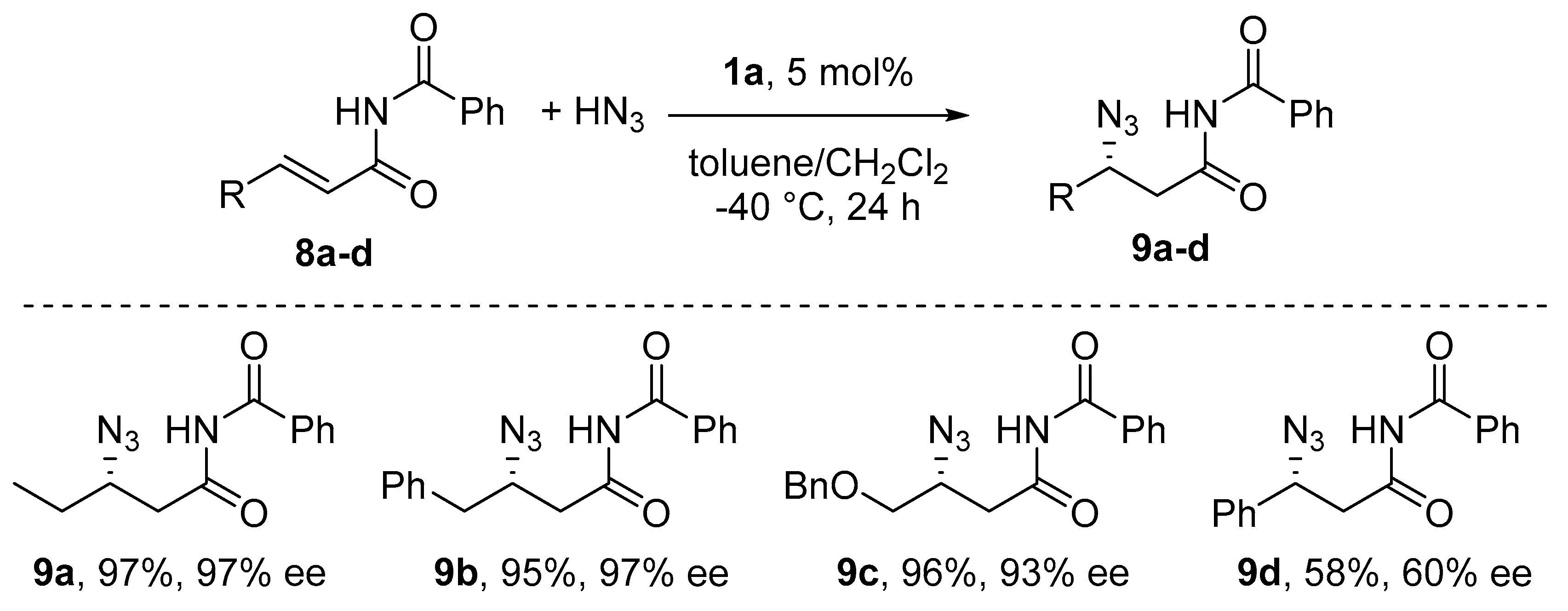
- Myers, J.K.; Jacobsen, E.N. Asymmetric Synthesis of β-Amino Acid Derivatives via Catalytic Conjugate Addition of Hydrazoic Acid to Unsaturated Imides. J. Am. Chem. Soc. 1999, 121, 8959–8960. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Enantioselective Michael Additions to α,β-Unsaturated Imides Catalyzed by a Salen-Al Complex. J. Am. Chm. Soc. 2003, 125, 11204–11205. [Google Scholar] [CrossRef]
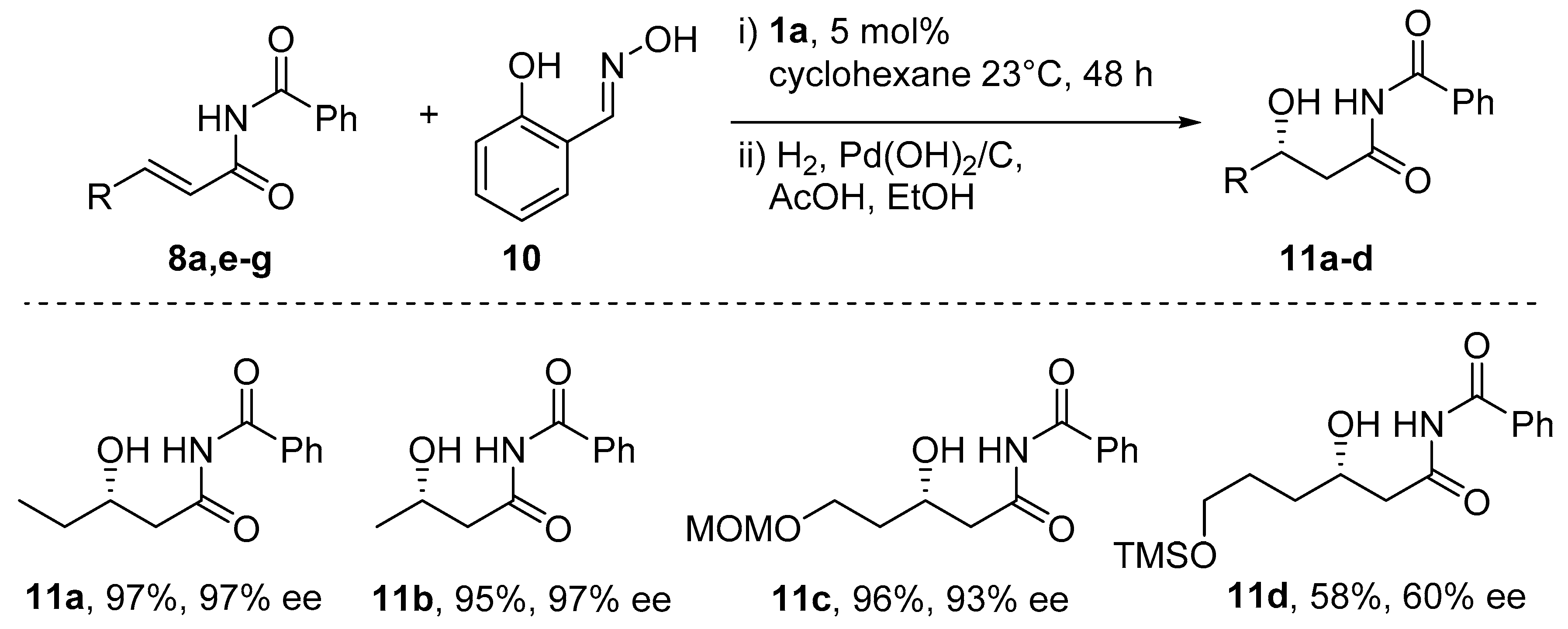
- Vanderwal, C.D.; Jacobsen, E.N. Enantioselective Formal Hydration of α,β-Unsaturated Imides by Al-Catalyzed Conjugate Addition of Oxime Nucleophiles. J. Am. Chem. Soc. 2004, 126, 14724–14725. [Google Scholar] [CrossRef]
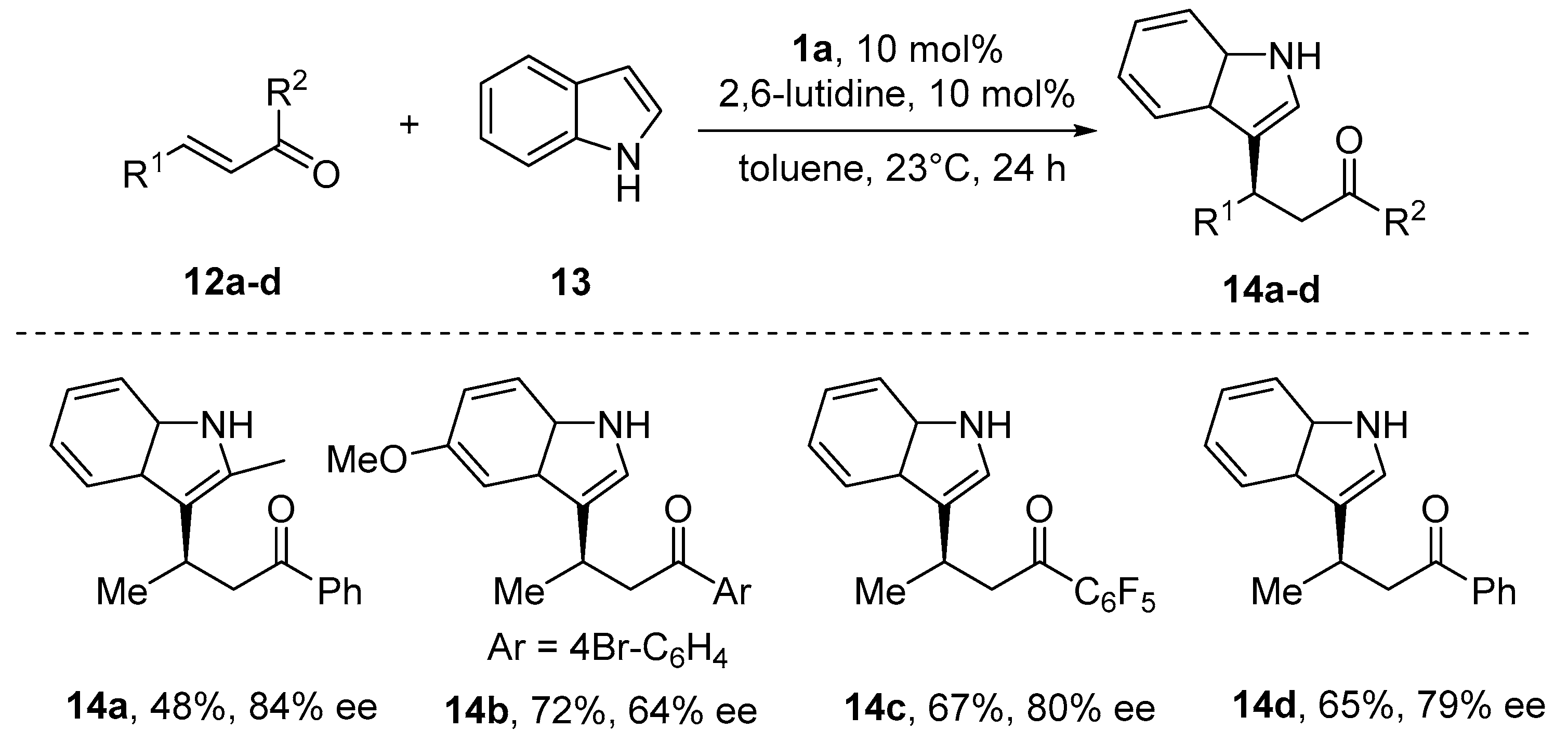
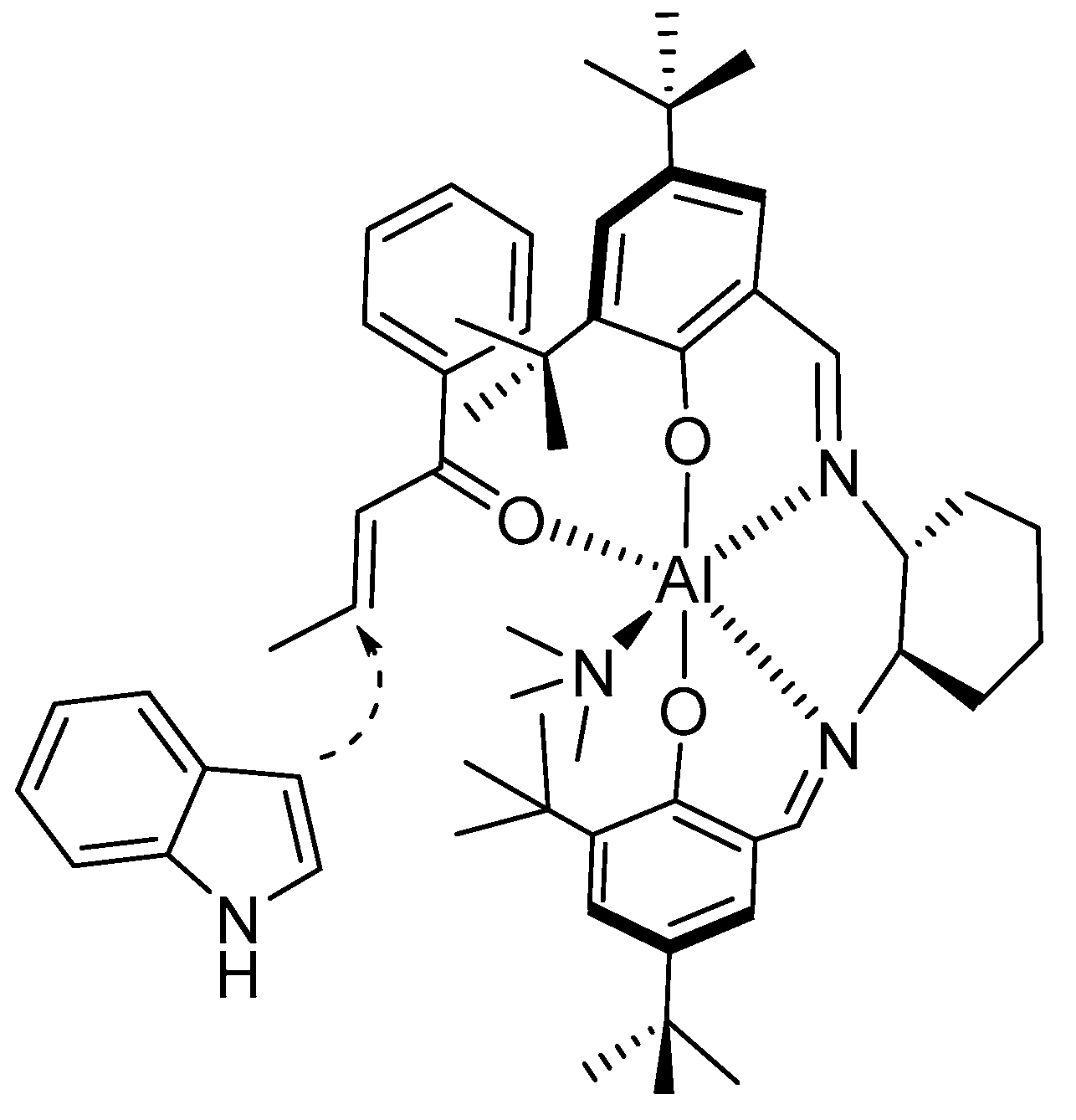
- Bandini, M.; Fagioli, M.; Melchiorre, P.; Melloni, A.; Umani-Ronchi, A. Catalytic enantioselective conjugate addition of indoles to simple α,β-unsaturated ketones. Tetrahedron Lett. 2003, 44, 5843–5846. [Google Scholar] [CrossRef]
- Bandini, B.; Fagioli, M.; Garavelli, M.; Melloni, A.; Trigari, V.; Umani-Ronchi, A. Can Simple Enones Be Useful Partners for the Catalytic Stereoselective Alkylation of Indoles? J. Org. Chem. 2004, 69, 7511–7518. [Google Scholar] [CrossRef]
- Gandelman, C.; Jacobsen, E.N. Highly Enantioselective Catalytic Conjugate Addition of N-Heterocycles to α,β-Unsaturated Ketones and Imides. Angew. Chem. Int. Ed. 2005, 44, 2393–2397. [Google Scholar] [CrossRef]
- Taylor, M.S.; Zalatan, D.N.; Lerchner, A.M.; Jacobsen, E.N. Highly Enantioselective Conjugate Additions to α,β-Unsaturated Ketones Catalyzed by a (Salen)Al Complex. J. Am. Chem. Soc. 2005, 127, 1313–1317. [Google Scholar] [CrossRef]
- Balskus, E.P.; Jacobsen, E.N. α,β-Unsaturated β-Silyl Imide Substrates for Catalytic, Enantioselective Conjugate Additions: A Total Synthesis of (+)-Lactacystin and the Discovery of a New Proteasome Inhibitor. J. Am. Chem. Soc. 2006, 128, 6811–6812. [Google Scholar] [CrossRef]
- Mazet, C.; Jacobsen, E.N. Dinuclear {(salen)Al} Complexes Display Expanded Scope in the Conjugate Cyanation of α,β-Unsaturated Imides. Angew. Chem. Int. Ed. 2008, 47, 1762–1765. [Google Scholar] [CrossRef] [PubMed]
- Sammis, G.M.; Danjo, H.; Jacobsen, E.N. Cooperative Dual Catalysis: Application to the Highly Enantioselective Conjugate Cyanation of Unsaturated Imides. J. Am. Chem. Soc. 2004, 126, 9928–9929. [Google Scholar] [CrossRef]
- Sibi, M.P.; Nad, S. Enantioselective Radical Reactions: Stereoselective Aldol Synthesis from Cyclic Ketones. Angew. Chem. Int. Ed. 2007, 46, 9231–9234. [Google Scholar] [CrossRef] [PubMed]
- Bar, G.; Parsons, A.F. Stereoselective radical reactions. Chem. Soc. Rev. 2003, 32, 251–263. [Google Scholar] [CrossRef]
- Silvi, M.; Melchiorre, P. Enhancing the potential of enantioselective organocatalysis with light. Nature 2018, 554, 41–49. [Google Scholar] [CrossRef]
- Madhvan, N.; Weck, M. Highly Active Polymer-Supported (Salen)Al Catalysts for the Enantioselective Addition of Cyanide to α,β-Unsaturated Imides. Adv. Synth. Catal. 2008, 350, 419–425. [Google Scholar] [CrossRef]
- Avidan-Shlomovich, S.; Ghosh, H.; Szpilman, A.M. Synthetic and Mechanistic Study of the Catalytic Enantioselective Preparation of Primary β-Amino Ketones from Enones and a Fluorinated Gabriel Reagent. ACS Catal. 2015, 5, 336–342. [Google Scholar] [CrossRef]
- Nicewicz, D.A.; Yates, C.M.; Johnson J., S. Enantioselective Cyanation/Brook Rearrangement/C-Acylation Reactions of Acylsilanes Catalyzed by Chiral Metal Alkoxides. J. Org. Chem. 2004, 69, 6548–6555. [Google Scholar] [CrossRef] [PubMed]
- Váradi, A.; Palmer, T.C.; Notis Dardashti, R.; Majumdar, S. Isocyanide-Based Multicomponent Reactions for the Synthesis of Heterocycles. Molecules 2016, 21, 19. [Google Scholar] [CrossRef]
- Kazemizadeh, A.R.; Ramazani, A. Synthetic Applications of Passerini Reaction. Curr. Org. Chem. 2012, 16, 418–450. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. Multicomponent Reactions with Isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Biggs-Houck, J.E.; Younai, A.; Shaw, J.T. Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr. Op. Chem. Biol. 2010, 14, 371–382. [Google Scholar] [CrossRef]
- van Berkel, S.S.; Bögels, B.G.M.; Wijdeven, M.A.; Westermann, B.; Rutjes, F.P.J.T. Recent Advances in Asymmetric Isocyanide-Based Multicomponent Reactions. Eur. J. Org. Chem. 2012, 3543–3559. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, P.; Li, S.-Y.; Sun, H.; Xiang, S.-H.; Wang, J.J.; Houk, K.N.; Tan, B. Asymmetric phosphoric acid–catalyzed four-component Ugi reaction. Science 2018, 361, eaas8707. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Chiral Salen-Aluminum Complex as a Catalyst for Enantioselective α-Addition of Isocyanides to Aldehydes: Asymmetric Synthesis of 2-(1-Hydroxyalkyl)-5-aminooxazoles. Org. Lett. 2007, 9, 3615–3618. [Google Scholar] [CrossRef]
- Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Catalytic Enantioselective Passerini Three-Component Reaction. Angew. Chem. Int. Ed. 2008, 47, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Asymmetric Synthesis of 5-(1-Hydroxyalkyl)tetrazoles by Catalytic Enantioselective Passerini-Type Reactions. Ang. Chem. Int. Ed. 2008, 47, 9454–9457. [Google Scholar] [CrossRef] [PubMed]
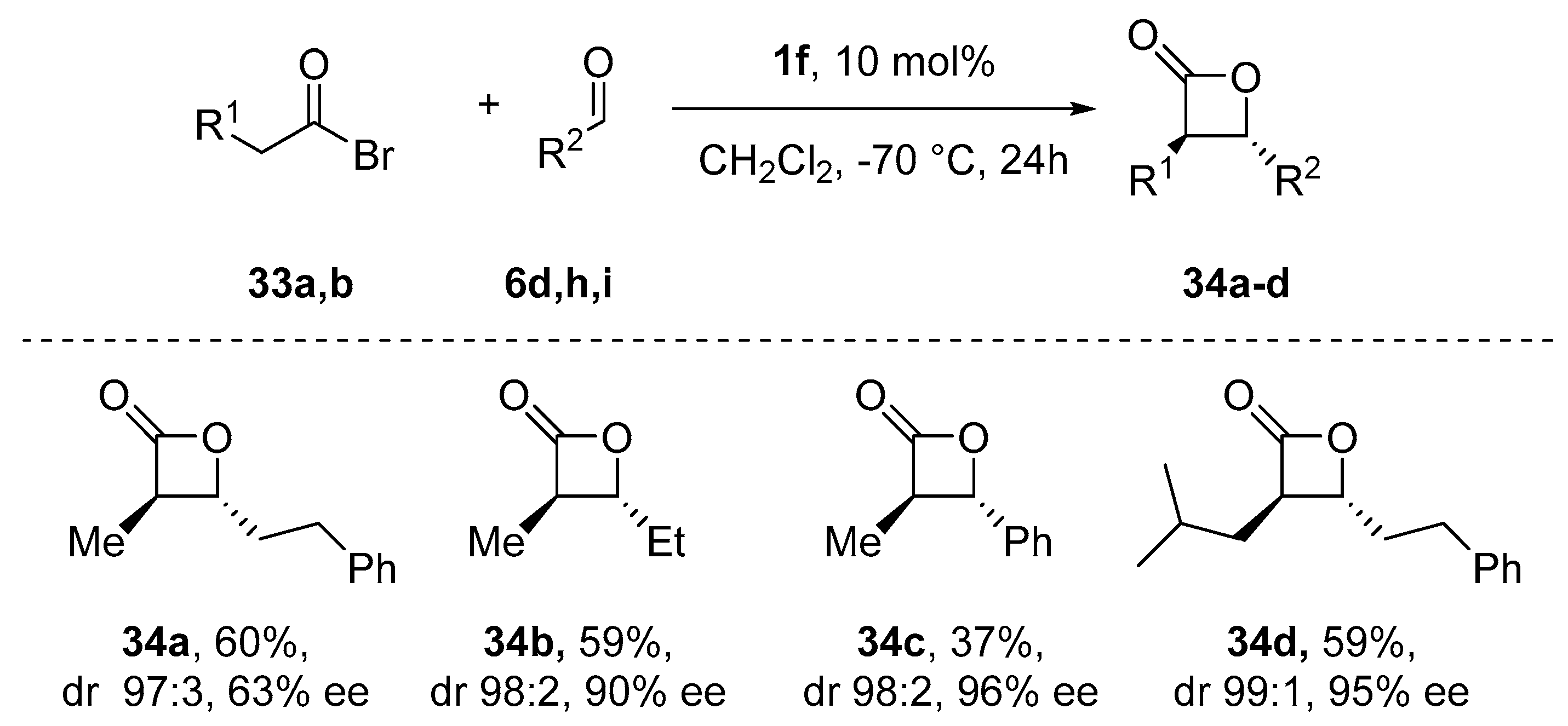
- Meier, P.; Broghammer, F.; Latendorf, K.; Rauhut, G.; Peters, R. Cooperative Al(Salen)-Pyridinium Catalysts for the Asymmetric Synthesis of trans-Configured β-Lactones by [2+2]-Cyclocondensation of Acylbromides and Aldehydes: Investigation of Pyridinium Substituent Effects. Molecules 2012, 17, 7121–7150. [Google Scholar] [CrossRef]
- Arakawa, H.; Aresta, M.; Armor, J.N.; Barteau, M.A.; Beckman, E.J.; Bell, A.T.; Bercaw, J.E.; Creutz, C.; Dinjus, E.; Dixon, D.A.; et al. Catalysis Research of Relevance to Carbon Management: Progress, Challenges, and Opportunities. Chem. Rev. 2001, 101, 953–996. [Google Scholar] [CrossRef]
- Taherimehr, M.; Pescarmona, P.P. Green Polycarbonates Prepared by the Copolymerization of CO2 with Epoxides. J. Appl. Polym. Sci. 2014, 131, 41141/1–41141/17. [Google Scholar] [CrossRef]
- Wang, C.; Yamamoto, H. Asymmetric Epoxidation Using Hydrogen Peroxide as Oxidant. Chem. Asian J. 2015, 10, 2056–2068. [Google Scholar] [CrossRef]
- Lu, X.-B.; Zhang, Y.-J.; Jin, K.; Luo, L.-M.; Wang, H. Highly active electrophile–nucleophile catalyst system for the cycloaddition of CO2 to epoxides at ambient temperature. J. Catal. 2004, 227, 537–541. [Google Scholar] [CrossRef]
- Alvaro, M.; Baleizao, C.; Carbonell, E.; El Ghoul, M.; García, H.; Gigante, B. Polymer-bound aluminium salen complex as reusable catalysts for CO2 insertion into epoxides. Tetrahedron 2005, 61, 12131–12139. [Google Scholar] [CrossRef]
- Luinstra, G.A.; Haas, G.R.; Molnar, F.; Bernhart, V.; Eberhardt, R.; Rieger, B. On the Formation of Aliphatic Polycarbonates from Epoxides with Chromium(III) and Aluminum(III) Metal–Salen Complexes. Chem. Eur. J. 2005, 11, 6298–6314. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, J.; North, M.; Pasquale, R. Synthesis of Cyclic Carbonates from Atmospheric Pressure Carbon Dioxide Using Exceptionally Active Aluminium(Salen) Complexes as Catalysts. Eur. J. Inorg. Chem. 2007, 3323–3326. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic Carbonate Synthesis Catalysed by Bimetallic Aluminium–Salen Complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R. Mechanism of Cyclic Carbonate Synthesis from Epoxides and CO2. Angew. Chem. Int. Ed. 2009, 48, 2946–2948. [Google Scholar] [CrossRef]
- North, M.; Young, C. Reducing the Cost of Production of Bimetallic Aluminium Catalysts for the Synthesis of Cyclic Carbonates. ChemSusChem. 2011, 4, 1685–1693. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Villuendas, P. A Bimetallic Aluminum(Salen) Complex for the Synthesis of 1,3-Oxathiolane-2-thiones and 1,3-Dithiolane-2-thiones. J. Org. Chem. 2010, 75, 6201–6207. [Google Scholar] [CrossRef]
- Baronsky, T.; Beattie, C.; Harrington, R.W.; Irfan, R.; North, M.; Osende, J.G.; Young, C. Bimetallic Aluminum(salen) Catalyzed Synthesis of Oxazolidinones from Epoxides and Isocyanates. ACS Catal. 2013, 3, 790–797. [Google Scholar] [CrossRef]
- Keith, J.M.; Larrow, J.F.; Jacobsen, E.N. Practical Considerations in Kinetic Resolution Reactions. Adv. Synth. Catal. 2001, 343, 5–26. [Google Scholar] [CrossRef]
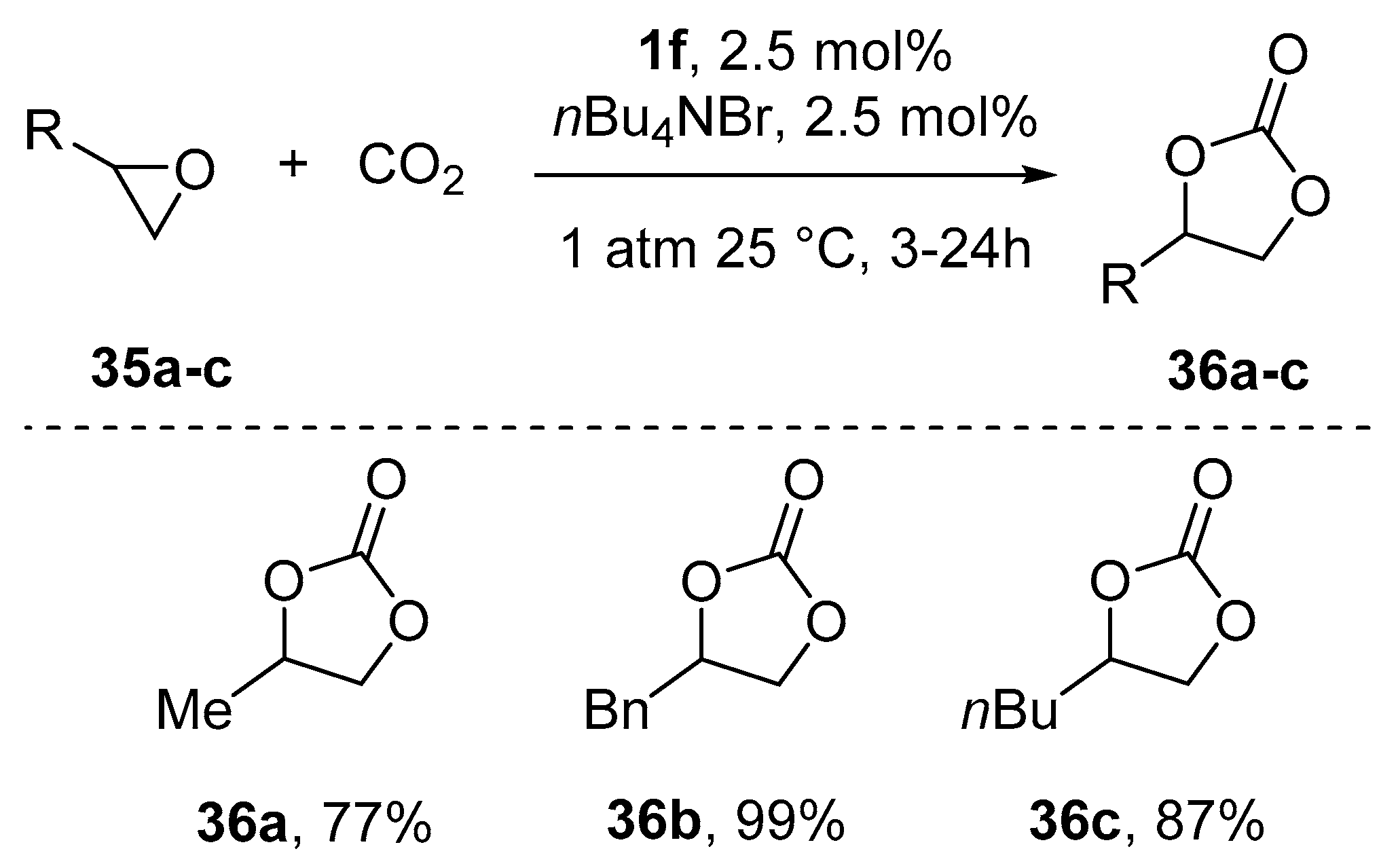
- North, M.; Quek, S.C.Z.; Pridmore, N.E.; Whitwood, A.C.; Wu, X. Aluminum(salen) Complexes as Catalysts for the Kinetic Resolution of Terminal Epoxides via CO2 Coupling. ACS Catal. 2015, 5, 3398–3402. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Spassky, N.; Wisniewski, M.; Pluta, C.; Borgne, A.L. Highly stereoelective polymerization of rac-(D,L)-lactide with a chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereochemistry of Lactide Polymerization with Chiral Catalysts: New Opportunities for Stereocontrol Using Polymer Exchange Mechanisms. J. Am. Chem. Soc. 2002, 124, 1316–1326. [Google Scholar] [CrossRef]
- Radano, C.P.; Baker, G.L.; Smith, M.R. Stereoselective Polymerization of a Racemic Monomer with a Racemic Catalyst: Direct Preparation of the Polylactic Acid Stereocomplex from Racemic Lactide. J. Am. Chem. Soc. 2000, 122, 1552–1553. [Google Scholar] [CrossRef]
- Zhong, Z.Y.; Dijkstra, P.J.; Feijen, J. [(salen)Al]-Mediated, Controlled and Stereoselective Ring-Opening Polymerization of Lactide in Solution and without Solvent: Synthesis of Highly Isotactic Polylactide Stereocopolymers from Racemic D,L-Lactide. Angew. Chem. Int. Ed. 2002, 41, 4510–4513. [Google Scholar] [CrossRef]
- Zhong, Z.Y.; Dijkstra, P.J.; Feijen, J. Controlled and Stereoselective Polymerization of Lactide: Kinetics, Selectivity, and Microstructures. J. Am. Chem. Soc. 2003, 125, 11291–11298. [Google Scholar] [CrossRef]
- Gao, B.; Duan, R.; Pang, R.; Li, X.; Qu, Z.; Tang, Z.; Zhuang, X.; Chen, X. Stereoselective Ring-Opening Polymerization of rac-Lactides Catalyzed by Aluminum Hemi-Salen Complexes. Organometallics 2013, 32, 5435–5444. [Google Scholar] [CrossRef]
- Kan, C.; Ge, J.; Ma, H. Aluminum methyl, alkoxide and α-alkoxy ester complexes supported by 6,6’-dimethylbiphenyl-bridged salen ligands: synthesis, characterization and catalysis for rac-lactide polymerization. Dalton Trans. 2016, 45, 6682–6695. [Google Scholar] [CrossRef]
- Robert, C.; Schmid, T.E.; Richard, V.; Haquette, P.; Raman, S.K.; Rager, M.-N.; Gauvin, R.M.; Morin, Y.; Trivelli, X.; Gueérineau, V.; et al. Mechanistic Aspects of the Polymerization of Lactide Using a Highly Efficient Aluminum(III) Catalytic System. J. Am. Chem. Soc. 2017, 139, 6217–6222. [Google Scholar] [CrossRef]
- Pang, X.; Duan, R.; Li, X.; Hu, C.; Wang, X.; Chen, C. Breaking the Paradox between Catalytic Activity and Stereoselectivity: rac-Lactide Polymerization by Trinuclear Salen−Al Complexes. Macromolecules 2018, 51, 906–913. [Google Scholar] [CrossRef]
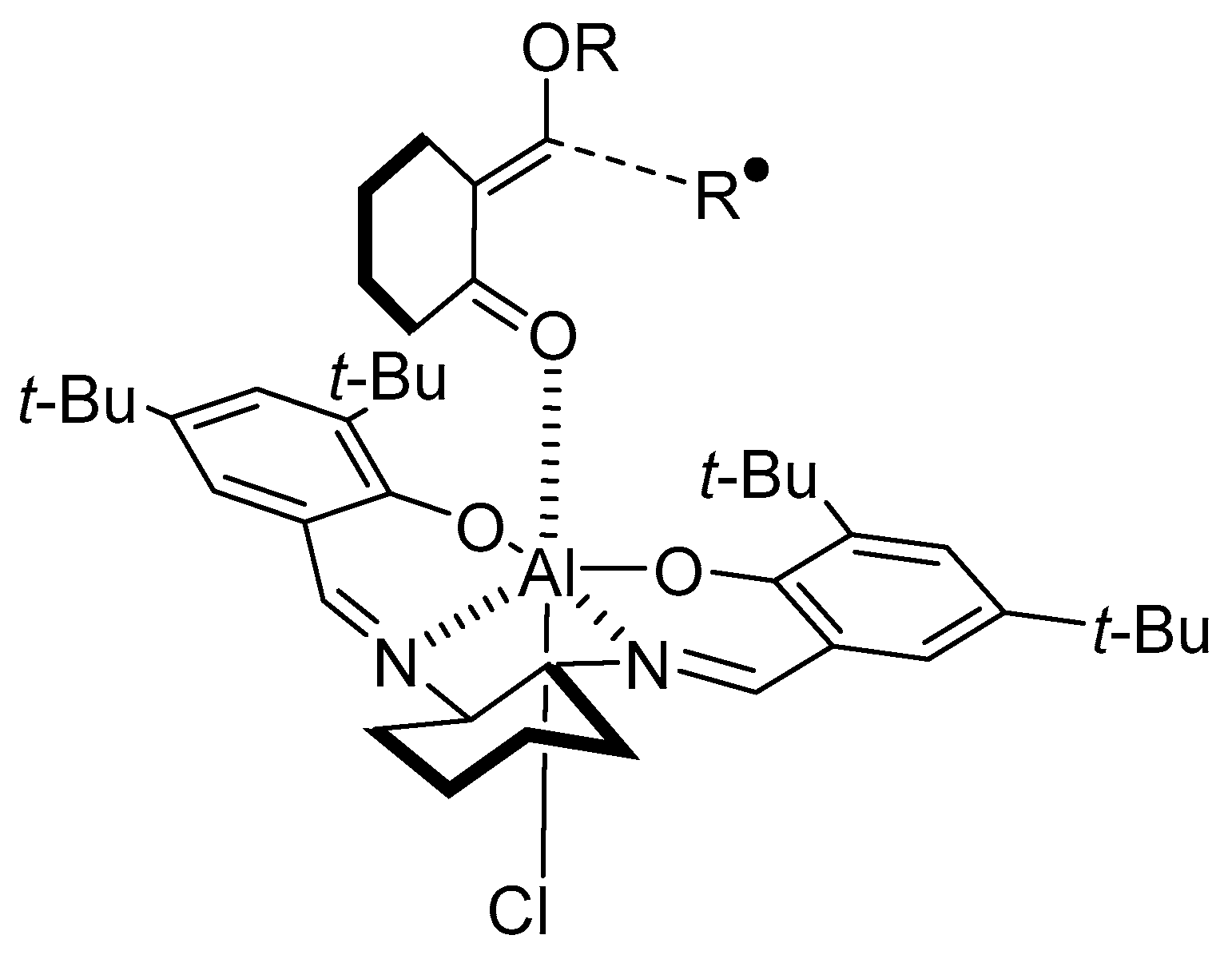
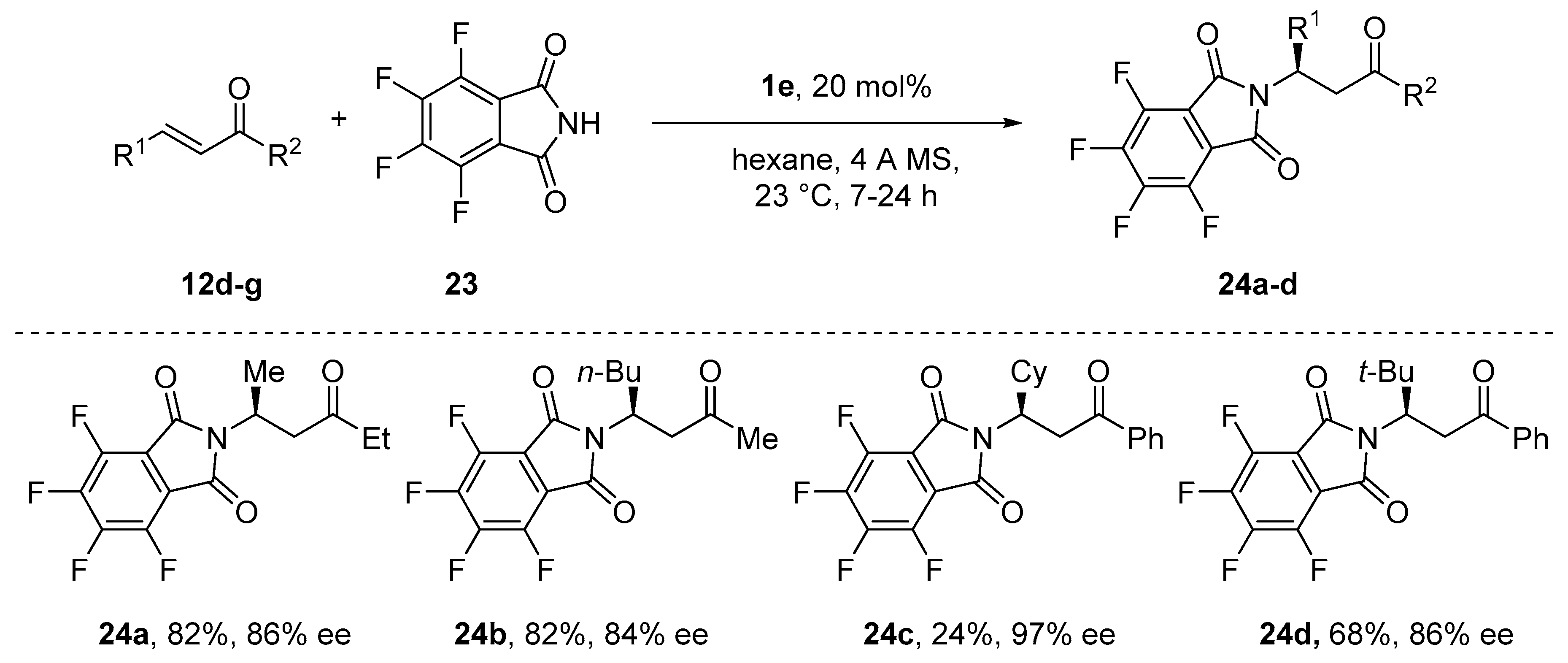
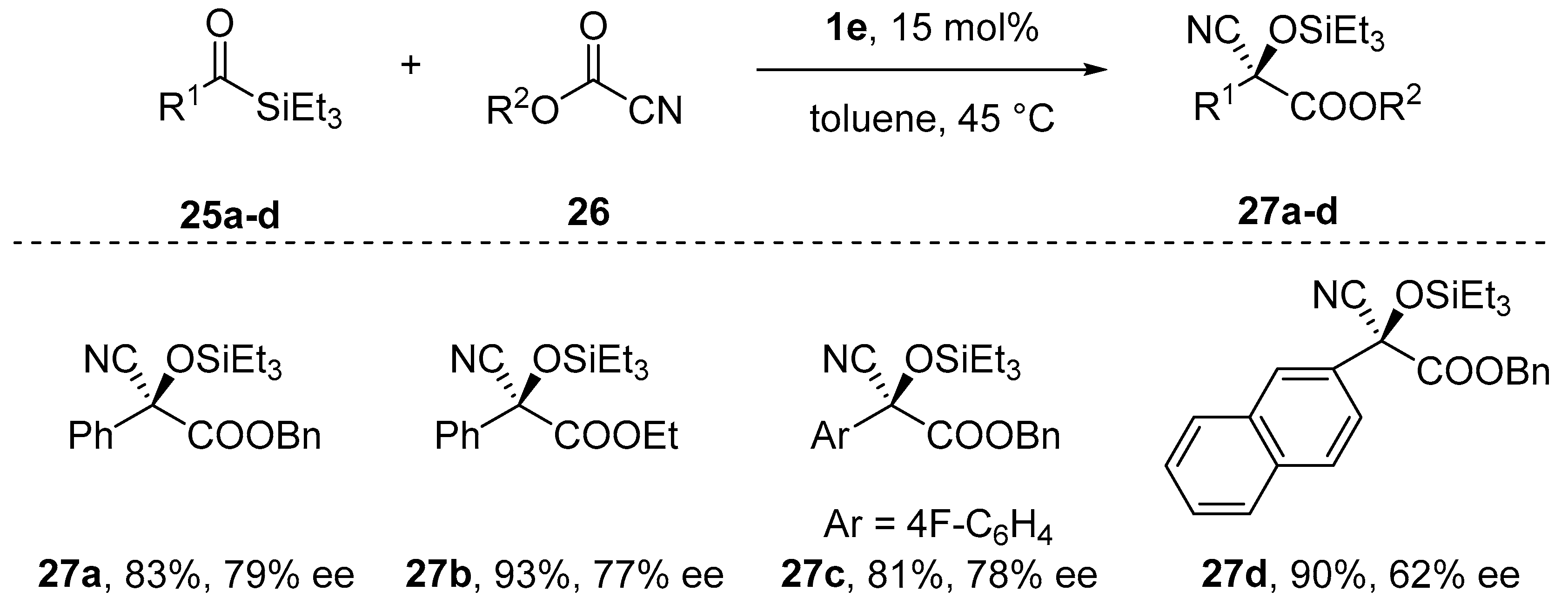
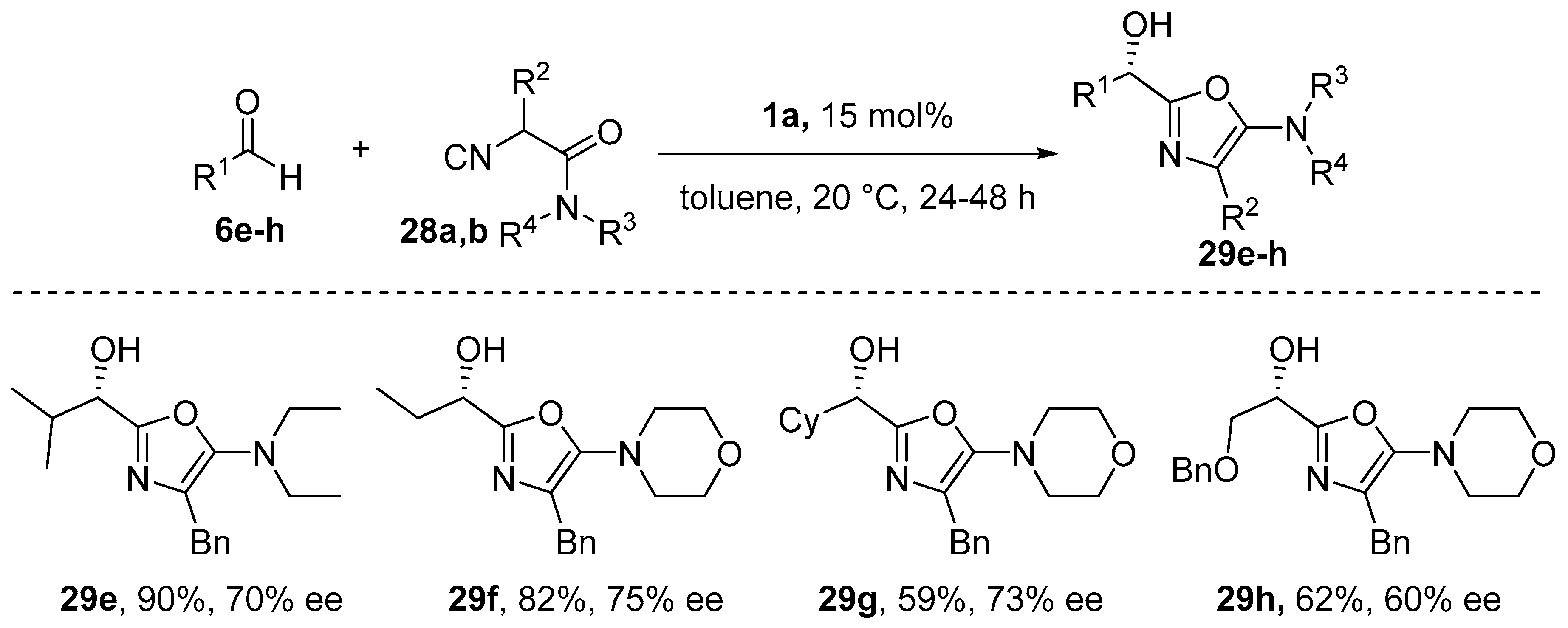
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gualandi, A.; Calogero, F.; Potenti, S.; Cozzi, P.G. Al(Salen) Metal Complexes in Stereoselective Catalysis. Molecules 2019, 24, 1716. https://doi.org/10.3390/molecules24091716
Gualandi A, Calogero F, Potenti S, Cozzi PG. Al(Salen) Metal Complexes in Stereoselective Catalysis. Molecules. 2019; 24(9):1716. https://doi.org/10.3390/molecules24091716
Chicago/Turabian StyleGualandi, Andrea, Francesco Calogero, Simone Potenti, and Pier Giorgio Cozzi. 2019. "Al(Salen) Metal Complexes in Stereoselective Catalysis" Molecules 24, no. 9: 1716. https://doi.org/10.3390/molecules24091716
APA StyleGualandi, A., Calogero, F., Potenti, S., & Cozzi, P. G. (2019). Al(Salen) Metal Complexes in Stereoselective Catalysis. Molecules, 24(9), 1716. https://doi.org/10.3390/molecules24091716