DNA Damaging Effects, Oxidative Stress Responses and Cholinesterase Activity in Blood and Brain of Wistar Rats Exposed to Δ9-Tetrahydrocannabinol

 ,
,  ,
, 
Abstract
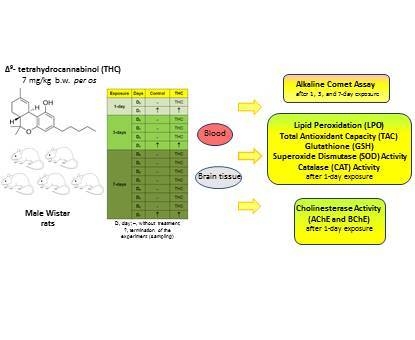
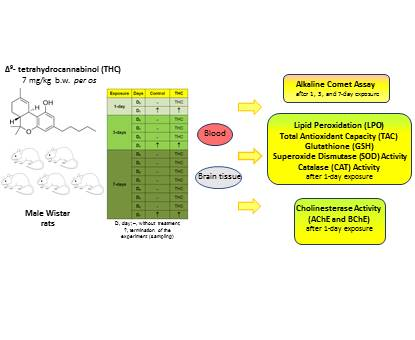
1. Introduction
2. Results
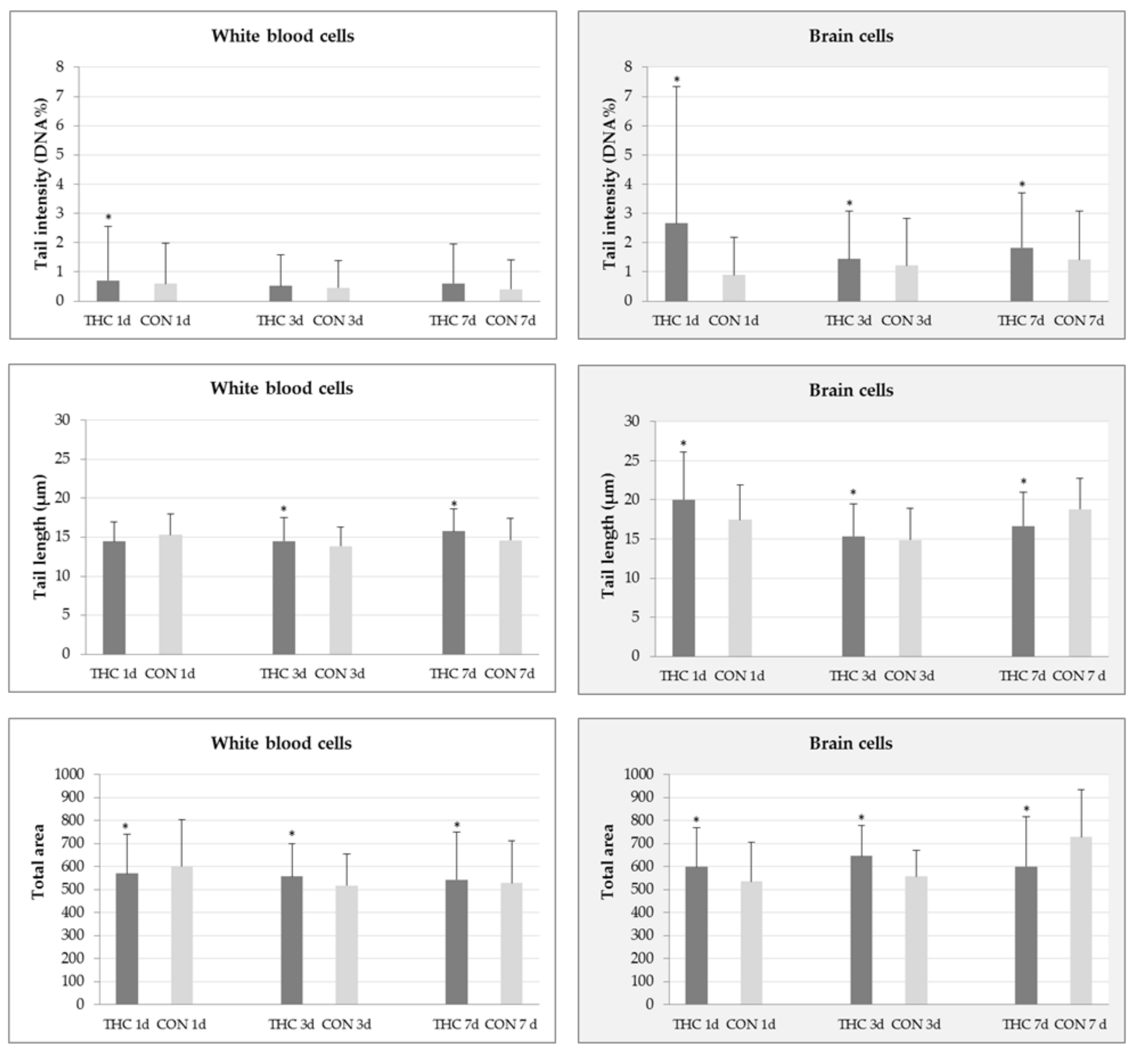
2.1. DNA Damage in White Blood and Brain Cells
2.2. Biochemical Markers of Oxidative Stress
2.3. Cholinesterase Activities
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Breeding and Housing of Animals
4.3. Experimental Design
4.4. The Alkaline Comet Assay
4.5. Biochemical Markers of Oxidative Stress
4.5.1. Thiobarbituric Reactive Substances (TBARS) Assay
4.5.2. Ferric Reducing Antioxidant Power (FRAP) Assay
4.5.3. Glutathione (GSH) Assay
4.5.4. Superoxide Dismutase (SOD) Activity
4.5.5. Catalase (CAT) Activity
4.5.6. Protein Quantification
4.6. Cholinesterase Activity Assay
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guzmán, M. Cannabis for the management of cancer symptoms: THC Version 2.0? Cannabis Cannabinoid Res. 2018, 3, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J. Metabolomics of Δ9-tetrahydrocannabinol: Implications in toxicity. Drug Metab. Rev. 2016, 48, 80–87. [Google Scholar] [CrossRef] [PubMed]
- McGilveray, I.J. Pharmacokinetics of cannabinoids. Pain Res. Manag. 2005, 10, 15A–22A. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.; Rosenkrantz, H.; Schaeppi, U.; Braude, M. Comparison of acute oral toxicity of cannabinoids in rats, dogs and monkeys. Toxicol. Appl. Pharmacol. 1973, 25, 363–372. [Google Scholar] [CrossRef]
- Rosenkrantz, H.; Heyman, I.; Braude, M. Inhalation, parenteral and oral LD50 values of delta 9-tetrahydrocannabinol in Fischer rats. Toxicol. Appl. Pharmacol. 1974, 28, 18–27. [Google Scholar] [CrossRef]
- Huestis, M.A. Human cannabinoid pharmacokinetics. Chem. Biodivers. 2007, 4, 1770–1804. [Google Scholar] [CrossRef]
- Grotenhermen, F. Clinical pharmacokinetics of cannabinoids. J. Cannabis Ther. 2003, 3, 3–51. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M.E.; Metwaly, S.; Sleem, A.A.; Morsy, F.A.; Sharaf, H.A. Cannabis sativa exacerbates hepatic injury caused by acetaminophen or carbon tetrachloride in rats. Comp. Clin. Pathol. 2013, 22, 209–218. [Google Scholar] [CrossRef]
- Musa, E.M.; EL Badwi, S.M.; Jah Elnabi, M.A.; Osman, E.A.; Dahab, M.M. Hepatoprotective and toxicity assessment of Cannabis sativa seed oil in Albino rat. Int. J. Chem. Biochem. Sci. 2012, 1, 69–76. [Google Scholar]
- Mukhtar, A.H.; Elbagir, N.M. Effect of Cannabis sativa on hematological indices in rats and men. Pak. J. Nutr. 2011, 10, 313–316. [Google Scholar] [CrossRef][Green Version]
- Okwari, O.O.; Emerole, C.G.; Dasofunjo, K.; Alagwu, E.A.; Olatunji, T.L.; Osim, E.E. Impact of repeated administration of Cannabis sativa on some biochemical parameters in albino rats. J. Pharm. Biol. Sci. 2014, 9, 51–57. [Google Scholar] [CrossRef]
- Dasofunjo, K.; Okwari, O.; Obembe, A.; Olantuji, T.; Ezugwu, H.; Osim, E. Assessment of the effect of Cannabis sativa and Nicotiana tobacum leaves on some haematological and liver function indices of albino rats. IOSR J. Pharm. Biol. Sci. 2014, 9, 33–40. [Google Scholar]
- Grotenhermen, F.; Russo, E. Cannabis and Cannabinoids: Pharmacology, Toxicology, and Therapeutic Potential; Haworth Press: Binghamton, NY, USA, 2002; ISBN 0789015072. [Google Scholar]
- Gloss, D. An overview of products and bias in research. Neurotherapeutics 2015, 12, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Molloy, L.; Pertile, J.; Iglesias, M. A review for Australian nurses: Cannabis use for anti-emesis among terminally ill patients in Australia. Aust. J. Adv. Nurs. 2017, 34, 43–47. [Google Scholar]
- Romano, L.; Hazekamp, A. Cannabis oil: Chemical evaluation of an upcoming cannabis-based medicine. Cannabinoids 2013, 1, 1–11. [Google Scholar] [CrossRef]
- McLaren, J.; Swift, W.; Dillon, P.; Allsop, S. Cannabis potency and contamination: A review of the literature. Addiction 2008, 103, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Rella, J.G. Recreational cannabis use: Pleasures and pitfalls. Cleve. Clin. J. Med. 2015, 82, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Stockburger, S. Forms of administration of cannabis and their efficacy. J. Pain Manag. 2016, 9, 381–386. [Google Scholar]
- Lucić Vrdoljak, A.; Fuchs, N.; Mikolić, A.; Žunec, S.; Brčić Karačonji, I.; Jurič, A.; Prester, L.; Micek, V.; Neuberg, M.; Čanović, S.; et al. Irinotecan and ∆9-tetrahydrocannabinol interactions in rat liver: A preliminary evaluation using biochemical and genotoxicity markers. Molecules 2018, 23, 1332. [Google Scholar] [CrossRef]
- Bridgeman, M.B.; Abazia, D.T. Medicinal cannabis: History, pharmacology, and implications for the acute care setting. Pharm. Ther. 2017, 42, 180–188. [Google Scholar] [CrossRef]
- Badowski, M.E. A review of oral cannabinoids and medical marijuana for the treatment of chemotherapy-induced nausea and vomiting: A focus on pharmacokinetic variability and pharmacodynamics. Cancer Chemother. Pharmacol. 2017, 80, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Engels, F.K.; de Jong, F.A.; Sparreboom, A.; Mathot, R.A.A.; Loos, W.J.; Kitzen, J.J.E.M.; de Bruijn, P.; Verweij, J.; Mathijssen, R.H.J. Medicinal cannabis does not influence the clinical pharmacokinetics of irinotecan and docetaxel. Oncologist 2007, 12, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Zgair, A.; Lee, J.B.; Wong, J.C.M.; Taha, D.A.; Aram, J.; di Virgilio, D.; McArthur, J.W.; Cheng, Y.K.; Hennig, I.M.; Barrett, D.A.; et al. Oral administration of cannabis with lipids leads to high levels of cannabinoids in the intestinal lymphatic system and prominent immunomodulation. Sci. Rep. 2017, 7, 14542. [Google Scholar] [CrossRef]
- Brozović, G.; Oršolić, N.; Rozgaj, R.; Knežević, F.; Horvat Knežević, A.; Maričić, M.; Krsnik, D.; Benković, V. Sevoflurane and isoflurane genotoxicity in kidney cells of mice. Arh. Hig. Rada Toksikol. 2017, 68, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Kopjar, N.; Žunec, S.; Mendaš, G.; Micek, V.; Kašuba, V.; Mikolić, A.; Tariba Lovaković, B.; Milić, M.; Pavičić, I.; Marjanović Čermak, A.M.; et al. Evaluation of chlorpyrifos toxicity through a 28-day study: Cholinesterase activity, oxidative stress responses, parent compound/metabolite levels, and primary DNA damage in blood and brain tissue of adult male Wistar rats. Chem. Biol. Interact. 2018, 279, 51–63. [Google Scholar] [CrossRef]
- Milić, M.; Žunec, S.; Micek, V.; Kašuba, V.; Mikolić, A.; Tariba Lovaković, B.; Živković Semren, T.; Pavičić, I.; Marjanović Čermak, A.M.; Pizent, A.; et al. Oxidative stress, cholinesterase activity, and DNA damage in the liver, whole blood, and plasma of Wistar rats following a 28-day exposure to glyphosate. Arch. Hig. Rada Toksikol. 2018, 69, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Ošiņa, K.; Leonova, E.; Isajevs, S.; Baumane, L.; Rostoka, E.; Sjakste, T.; Bisenieks, E.; Duburs, G.; Vigante, B.; Sjakste, N. Modifications of expression of genes and proteins involved in DNA repair and nitric oxide metabolism by carbatonides [disodium-2,6-dimethyl-1,4-dihydropyridine-3,5-bis(carbonyloxyacetate) derivatives] in intact and diabetic rats. Arch. Hig. Rada Toksikol. 2017, 68, 212–227. [Google Scholar] [CrossRef]
- Paulraj, R.; Behari, J. Single strand DNA breaks in rat brain cells exposed to microwave radiation. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2006, 596, 76–80. [Google Scholar] [CrossRef]
- Swain, U.; Subba Rao, K. Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mech. Ageing Dev. 2011, 132, 374–381. [Google Scholar] [CrossRef]
- Žunec, S.; Kopjar, N.; Želježić, D.; Kuča, K.; Musilek, K.; Lucić Vrdoljak, A. In vivo evaluation of cholinesterase activity, oxidative stress markers, cyto- and genotoxicity of K048 oxime—A promising antidote against organophosphate Poisoning. Basic Clin. Pharmacol. Toxicol. 2014, 114, 344–351. [Google Scholar] [CrossRef]
- Azqueta, A.; Collins, A.R. The essential comet assay: A comprehensive guide to measuring DNA damage and repair. Arch. Toxicol. 2013, 87, 949–968. [Google Scholar] [CrossRef]
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Langie, S.A.S.; Azqueta, A.; Collins, A.R. The comet assay: Past, present, and future. Front. Genet. 2015, 6, 1–3. [Google Scholar] [CrossRef]
- Speit, G.; Hartmann, A. The comet assay a sensitive genotoxicity test for the detection of DNA damage and repair. In Methods in Molecular Biology: DNA Repair Protocols: Mammalian Systems; Henderson, D.S., Ed.; Humana Press: New York, NY, USA, 2006; pp. 275–286. ISBN 9781588295132. [Google Scholar]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Ghani, M.A.; Barril, C.; Bedgood, D.R.; Prenzler, P.D. Measurement of antioxidant activity with the thiobarbituric acid reactive substances assay. Food Chem. 2017, 230, 195–207. [Google Scholar] [CrossRef]
- Sochor, J.; Ruttkay-Nedecky, B.; Babula, P.; Vojtech, A.; Hubalek, J.; Kizek, R. Automation of Methods for Determination of Lipid Peroxidation. In Lipid Peroxidation; Catala, A., Ed.; InTech: London, UK, 2012; pp. 131–154. ISBN 978-953-51-0716-3. [Google Scholar]
- Dominko, K.; Đikić, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arh. Hig. Rada Toksikol. 2018, 69, 1–24. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Ahmed, W.; Katz, S. Therapeutic use of cannabis in inflammatory bowel disease. Gastroenterol. Hepatol. 2016, 12, 668–679. [Google Scholar] [CrossRef]
- Fife, T.; Moawad, H.; Mosconas, C.; Shepard, K.; Hammond, N. Clinical perspectives on medical marijuana (cannabis) for neurologic disorders. Neurol. Clin. Pract. 2015, 5, 344–351. [Google Scholar] [CrossRef]
- Reece, A.S.; Hulse, G.K. Chromothripsis and epigenomics complete causality criteria for cannabis- and addiction-connected carcinogenicity, congenital toxicity and heritable genotoxicity. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2016, 789, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Lin, L.F. Genetic toxicology of abused drugs: A brief review. Mutagenesis 1998, 13, 557–565. [Google Scholar] [CrossRef]
- Kim, H.R.; Son, B.H.; Lee, S.Y.; Chung, K.H.; Oh, S.M. The role of p53 in marijuana smoke condensates-induced genotoxicity and apoptosis. Environ. Health Toxicol. 2012, 27, e2012017. [Google Scholar] [CrossRef] [PubMed]
- Al-Salmani, K.; Abbas, H.H.K.; Schulpen, S.; Karbaschi, M.; Abdalla, I.; Bowman, K.J.; So, K.K.; Evans, M.D.; Jones, G.D.D.; Godschalk, R.W.; et al. Simplified method for the collection, storage, and comet assay analysis of DNA damage in whole blood. Free Radic. Biol. Med. 2011, 51, 719–725. [Google Scholar] [CrossRef]
- Nouspikel, T.; Hanawalt, P.C. DNA repair in terminally differentiated cells. DNA Repair 2002, 1, 59–75. [Google Scholar] [CrossRef]
- McCracken, J.M.; Allen, L. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef]
- Ponath, V.; Kaina, B. Death of monocytes through oxidative burst of macrophages and neutrophils: Killing in trans. PLoS ONE 2017, 12, e0170347. [Google Scholar] [CrossRef] [PubMed]
- Suckow, M.; Weisbroth, S.; Franklin, C. The Laboratory Rat; Suckow, M., Weisbroth, S., Franklin, C., Eds.; Academic Press: Cambridge, MA, USA, 2005. [Google Scholar]
- Yusuf, I.; Fruman, D.A. Regulation of quiescence in lymphocytes. Trends Immunol. 2003, 24, 380–386. [Google Scholar] [CrossRef]
- Bauer, M.; Goldstein, M.; Christmann, M.; Becker, H.; Heylmann, D.; Kaina, B. Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 21105–21110. [Google Scholar] [CrossRef] [PubMed]
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The balancing act of neutrophils. Cell Host Microbe 2014, 15, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Mócsai, A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J. Exp. Med. 2013, 210, 1283–1299. [Google Scholar] [CrossRef]
- Chan, G.C.; Hinds, T.R.; Impey, S.; Storm, D.R. Hippocampal neurotoxicity of Δ9-tetrahydrocannabinol. J. Neurosci. 1998, 18, 5322–5332. [Google Scholar] [CrossRef]
- Campbell, F.A.; Tramèr, M.R.; Carroll, D.; Reynolds, D.J.M.; Moore, R.A. Are cannabinoids an effective and safe treatment option in the management of pain? BMJ Br. Med. J. 2001, 323, 13. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Hazekamp, A.; Perret, N.; Zurich, M.G.; Mangin, P.; Giroud, C.; Honegger, P. Delta-9-tetrahydrocannabinol accumulation, metabolism and cell-type-specific adverse effects in aggregating brain cell cultures. Toxicol. Appl. Pharmacol. 2008, 228, 8–16. [Google Scholar] [CrossRef]
- Kumaravel, T.S.; Vilhar, B.; Faux, S.P.; Jha, A.N. Comet Assay measurements: A perspective. Cell Biol. Toxicol. 2009, 25, 53–64. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Cooper, A.; Kristal, B. Multiple roles of glutathione in the central nervous system. Biol. Chem. 1997, 378, 793–802. [Google Scholar]
- Barzilai, A.; Biton, S.; Shiloh, Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair 2008, 7, 1010–1027. [Google Scholar] [CrossRef]
- Schwilke, E.W.; Schwope, D.M.; Karschner, E.L.; Lowe, R.H.; Darwin, W.D.; Kelly, D.L.; Goodwin, R.S.; Gorelick, D.A.; Huestis, M.A. Δ9-tetrahydrocannabinol (THC), 11-hydroxy-THC, and 11-nor-9-carboxy-THC plasma pharmacokinetics during and after continuous high-dose oral THC. Clin. Chem. 2009, 55, 2180–2189. [Google Scholar] [CrossRef]
- Gunasekaran, N.; Long, L.E.; Dawson, B.L.; Hansen, G.H.; Richardson, D.P.; Li, K.M.; Arnold, J.C.; McGregor, I.S. Reintoxication: The release of fat-stored Δ9-tetrahydrocannabinol (THC) into blood is enhanced by food deprivation or ACTH exposure. Br. J. Pharmacol. 2009, 158, 1330–1337. [Google Scholar] [CrossRef]
- Costa, B.; Colleoni, M. Changes in rat brain energetic metabolism after exposure to anandamide or Δ9-tetrahydrocannabinol. Eur. J. Pharmacol. 2000, 395, 1–7. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Kouyoumjian, S.; Khoshaghideh, F.; Tashkin, D.P.; Roth, M.D. Δ9-Tetrahydrocannabinol disrupts mitochondrial function and cell energetics. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L298–L306. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Hroudová, J.; Fišar, Z. Cannabinoid-induced changes in the activity of electron transport chain complexes of brain mitochondria. J. Mol. Neurosci. 2015, 56, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Wolff, V.; Schlagowski, A.-I.; Rouyer, O.; Charles, A.-L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.-S.; Zoll, J.; et al. Tetrahydrocannabinol induces brain mitochondrial respiratory chain dysfunction and increases oxidative stress: A potential mechanism involved in cannabis-related stroke. Biomed. Res. Int. 2015, 2015, 323706. [Google Scholar] [CrossRef]
- Coşkun, Z.M.; Bolkent, S. The changes of oxidative stress and Δ9-tetrahydrocannabinol accumulation in liver of type-2 diabetic rats. IUFS J. Biol. 2015, 74, 1–8. [Google Scholar]
- O’Sullivan, S.E.O.; Kendall, D.A.; Randall, M.D. Further characterization of the time-dependent vascular effects of Δ9-tetrahydrocannabinol. J. Pharmacol. Exp. Ther. 2006, 317, 428–438. [Google Scholar] [CrossRef]
- Pinto, C.E.; Moura, E.; Serrão, M.P.; Martins, M.J.; Vieira-Coelho, M. A Effect of (−)-Δ9-tetrahydrocannabinoid on the hepatic redox state of mice. Braz. J. Med. Biol. Res. 2010, 43, 325–329. [Google Scholar] [CrossRef]
- Coskun, Z.M.; Bolkent, S. Evaluation of Δ9-tetrahydrocannabinol metabolites and oxidative stress in type 2 diabetic rats. Iran. J. Basic Med. Sci. 2016, 19, 154–158. [Google Scholar]
- Kannan, R.; Kuhlenkamp, J.; Jeandidier, E.; Trinh, H.; Ookhtena, M.; Kaplowitz, N. Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. J. Clin. Investig. 1990, 85, 2009–2013. [Google Scholar] [CrossRef]
- Solowij, N.; Stephens, R.; Roffman, R.; Babor, T.; Kadden, R.; Miller, M.; Christiansen, K.; McRee, B.; Vendetti, J. Cognitive functioning of long-term heavy cannabis users seeking treatment. J. Am. Med. Assoc. 2002, 287, 1123–1131. [Google Scholar] [CrossRef]
- Court, J.M. Annotation cannabis and brain function. J. Paediatr. Child Health 1998, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Heishman, S.; Arasteh, K.; Stitzer, M. Comparative effects of alcohol and marijuana on mood, memory, and performance. Pharmacol. Biochem. Behav. 1997, 58, 93–101. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M.E.; Youness, E.R.; Khadrawy, Y.A.; Sleem, A.A. Acetylcholinesterase, butyrylcholinesterase and paraoxonase 1 activities in rats treated with cannabis, tramadol or both. Asian Pac. J. Trop. Med. 2016, 9, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Abrams, D.I.; Couey, P.; Shade, S.B.; Kelly, M.E.; Benowitz, N.L. Cannabinoid-opioid interaction in chronic pain. Clin. Pharmacol. Ther. 2011, 90, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Nava, F.; Gessa, G.L. Inhibition of hippocampal acetylcholine release after acute and repeated Δ9-tetrahydrocannabinol in rats. Brain Res. 1998, 809, 1–4. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Rubio-Casillas, A. Biphasic effects of THC in memory and cognition. Eur. J. Clin. Investig. 2018, 48, e12920. [Google Scholar] [CrossRef] [PubMed]
- Eubanks, L.M.; Rogers, C.J.; Beuscher, A.E.; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K.D. A molecular link between the active component of marijuana and Alzheimer disease patologhy. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Sharma, R.K.; Singh, N.; Sharma, B. Acetylcholinesterase from human erythrocytes membrane: A screen for evaluating the activity of some traditional plant extracts. Cell. Mol. Biol. 2012, 58, 160–169. [Google Scholar] [CrossRef]
- Ebuehi, O.A.T.; Abey, N.O. Impact of marijuana (Cannabis sativa) on some neurochemicals and cognitive function of Sprague-Dawley rats. Res. Neurosci. 2016, 5, 1–9. [Google Scholar] [CrossRef]
- Loflin, M.; Earleywine, M. A new method of cannabis ingestion: The dangers of dabs? Addict. Behav. 2014, 39, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. 1-Trans-delta9-tetrahydrocannabinol (CAS No. 1972-08-3) in F344/N Rats and B6C3F1 Mice (Gavage Studies); National Institutes of Health: Research Triangle Park, NC, USA, 1996. [Google Scholar]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef]
- Drury, J.A.; Nycyk, J.A.; Cooke, R.W.I. Comparison of urinary and plasma malondialdehyde in preterm infants. Clin. Chim. Acta 1997, 263, 177–185. [Google Scholar] [CrossRef]
- Ellman, G.L. A colorimetric method for low concentration of mercaptons. Arch. Biochem. Biophys. 1958, 74, 443–450. [Google Scholar] [CrossRef]
- Flohé, L.; Ötting, F. Superoxide dismutase assays. Methods Enzymol. 1984, 105, 93–104. [Google Scholar] [PubMed]
- Aebi, H. Oxygen radicals in biological systems. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
Sample Availability: Not available. |


{kind=link}
{kind=link}
| Sample/Parameter | FRAP (mmol/L) | TBARS (μmol/L) | GSH (μg/mL) | CAT (IU/gprotein) | SOD (IU/gprotein) |
|---|---|---|---|---|---|
| Plasma | |||||
| Control | 0.170 ± 0.060 | 4.329 ± 1.481 | 142.6 ± 65.9 | 0.018 ± 0.008 | 0.188 ± 0.055 |
| 0.153 | 4.553 | 127.1 | 0.019 | 0.204 | |
| 0.119–0.264 | 2.129–6.210 | 99.5–257.8 | 0.007–0.027 | 0.114–0.247 | |
| THC | 0.124 ± 0.022 | 3.057 ± 0.826 | 106.6 ± 18.6 | 0.011 ± 0.004 | 0.128 ± 0.030 |
| 0.129 | 2.927 | 104.1 | 0.009 | 0.131 | |
| 0.092–0.148 | 2.247–4.435 | 85.9–137.0 | 0.007–0.016 | 0.093–0.171 | |
| Brain | |||||
| Control | 0.308 ± 0.094 | 10.597 ± 4.637 | 28.027 ± 8.302 | 0.140 ± 0.009 | 3.533 ± 0.612 |
| 0.271 | 12.334 | 28.377 | 0.138 | 3.353 | |
| 0.229–0.452 | 5.349–15.670 | 14.481–36.202 | 0.131–0.155 | 2.877–4.249 | |
| THC | 0.272 ± 0.047 | 17.256 ± 1.353 * | 38.537 ± 2.293 * | 0.159 ± 0.0187 | 1.409 ± 0.330 * |
| 0.274 | 17.093 | 39.663 | 0.158 | 1.437 | |
| 0.231–0.346 | 15.899–19.418 | 34.950–40.448 | 0.142–0.190 | 1.066–1.724 | |
| Sample | Plasma | Brain | ||||
|---|---|---|---|---|---|---|
| Parameter | ChE (IU/gprotein) | AChE (IU/gprotein) | BChE (IU/gprotein) | ChE (IU/gprotein) | AChE (IU/gprotein) | BChE (IU/gprotein) |
| Control | 0.107 0.091–0.141 | 0.073 0.058–0.097 | 0.033 0.032–0.067 | 21.8 13.7–24.1 | 19.7 10.4–20.9 | 1.4 1.2–4.4 |
| THC | 0.104 0.090–0.135 | 0.073 0.056–0.102 | 0.041 0.002–0.062 | 18.1 16.0–21.1 | 16.5 13.7–19.5 | 1.6 0.5–2.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopjar, N.; Fuchs, N.; Žunec, S.; Mikolić, A.; Micek, V.; Kozina, G.; Lucić Vrdoljak, A.; Brčić Karačonji, I. DNA Damaging Effects, Oxidative Stress Responses and Cholinesterase Activity in Blood and Brain of Wistar Rats Exposed to Δ9-Tetrahydrocannabinol. Molecules 2019, 24, 1560. https://doi.org/10.3390/molecules24081560
Kopjar N, Fuchs N, Žunec S, Mikolić A, Micek V, Kozina G, Lucić Vrdoljak A, Brčić Karačonji I. DNA Damaging Effects, Oxidative Stress Responses and Cholinesterase Activity in Blood and Brain of Wistar Rats Exposed to Δ9-Tetrahydrocannabinol. Molecules. 2019; 24(8):1560. https://doi.org/10.3390/molecules24081560
Chicago/Turabian StyleKopjar, Nevenka, Nino Fuchs, Suzana Žunec, Anja Mikolić, Vedran Micek, Goran Kozina, Ana Lucić Vrdoljak, and Irena Brčić Karačonji. 2019. "DNA Damaging Effects, Oxidative Stress Responses and Cholinesterase Activity in Blood and Brain of Wistar Rats Exposed to Δ9-Tetrahydrocannabinol" Molecules 24, no. 8: 1560. https://doi.org/10.3390/molecules24081560
APA StyleKopjar, N., Fuchs, N., Žunec, S., Mikolić, A., Micek, V., Kozina, G., Lucić Vrdoljak, A., & Brčić Karačonji, I. (2019). DNA Damaging Effects, Oxidative Stress Responses and Cholinesterase Activity in Blood and Brain of Wistar Rats Exposed to Δ9-Tetrahydrocannabinol. Molecules, 24(8), 1560. https://doi.org/10.3390/molecules24081560