
Determination of Barbiturates in Biological Specimens by Flat Membrane-Based Liquid-Phase Microextraction and Liquid Chromatography-Mass Spectrometry

Abstract
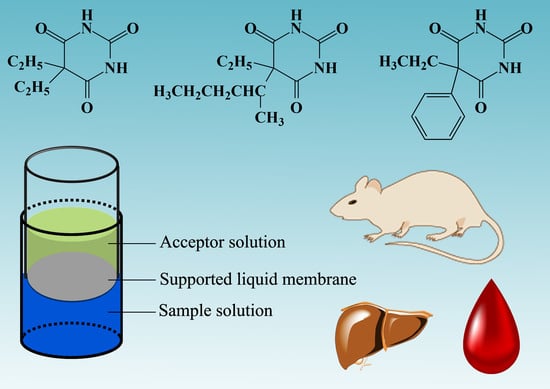
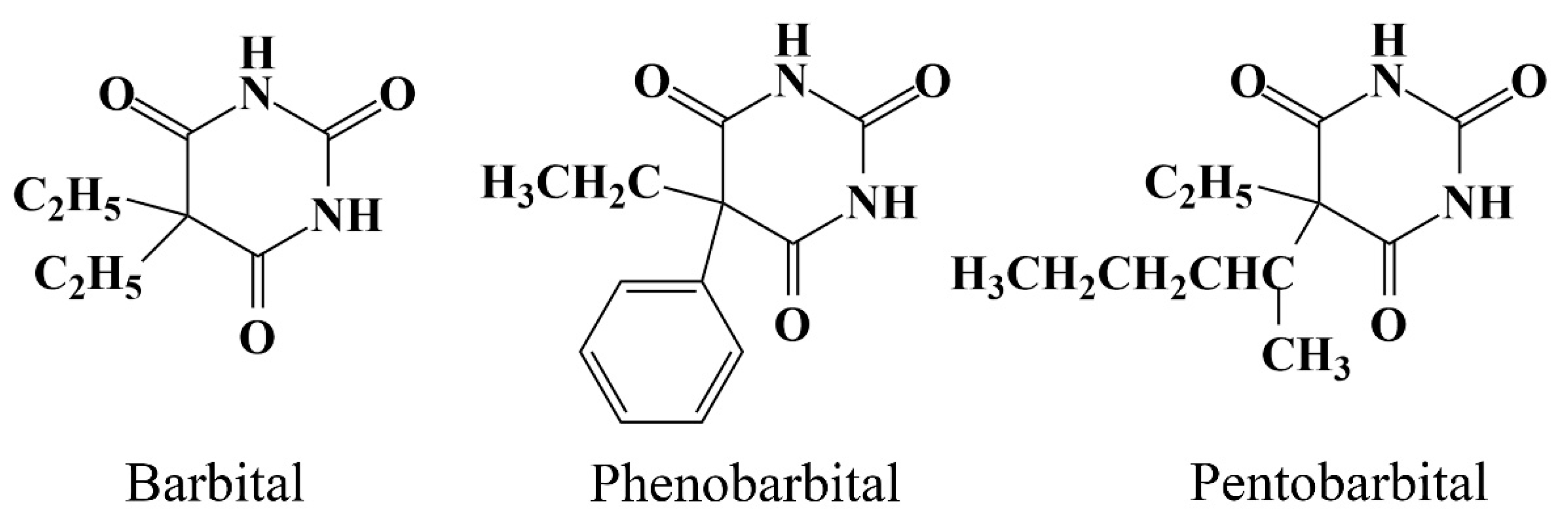
:
1. Introduction
2. Results and Discussion
2.1. Optimization of the Extraction Conditions
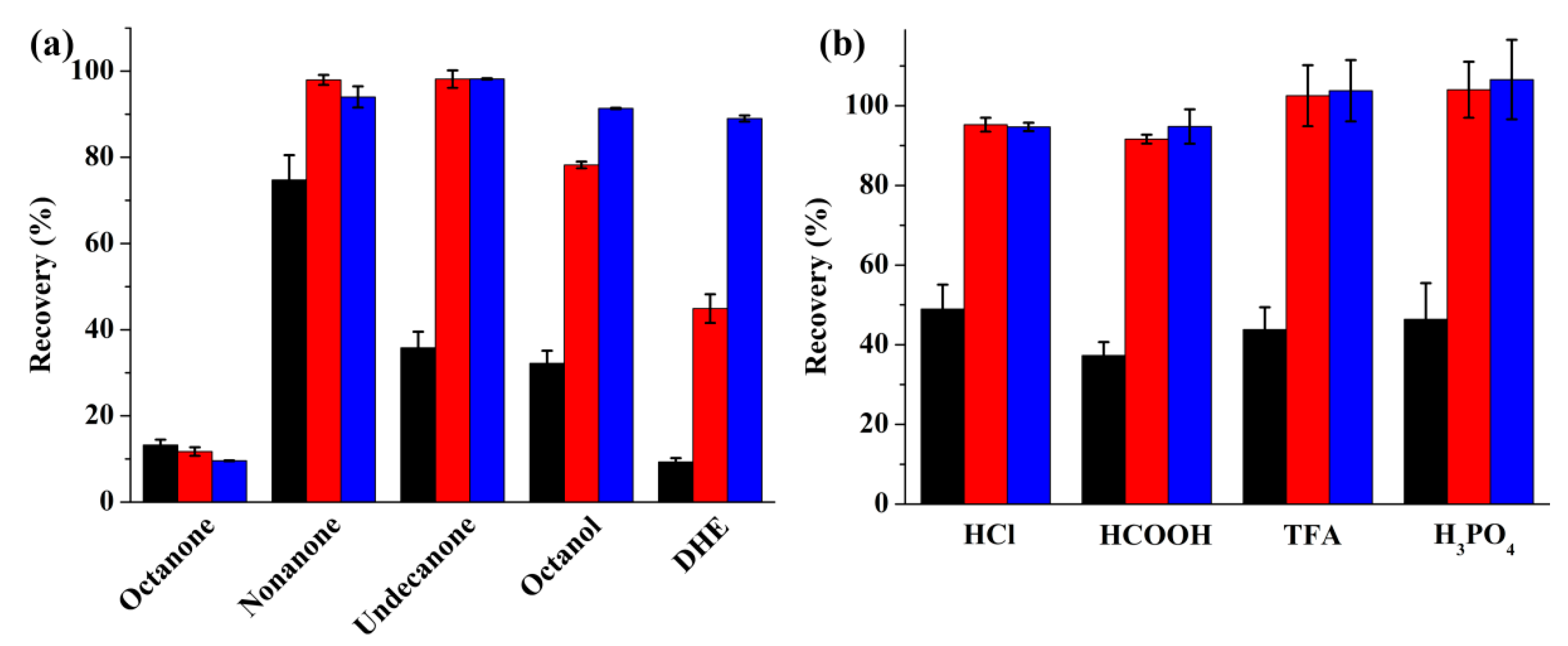
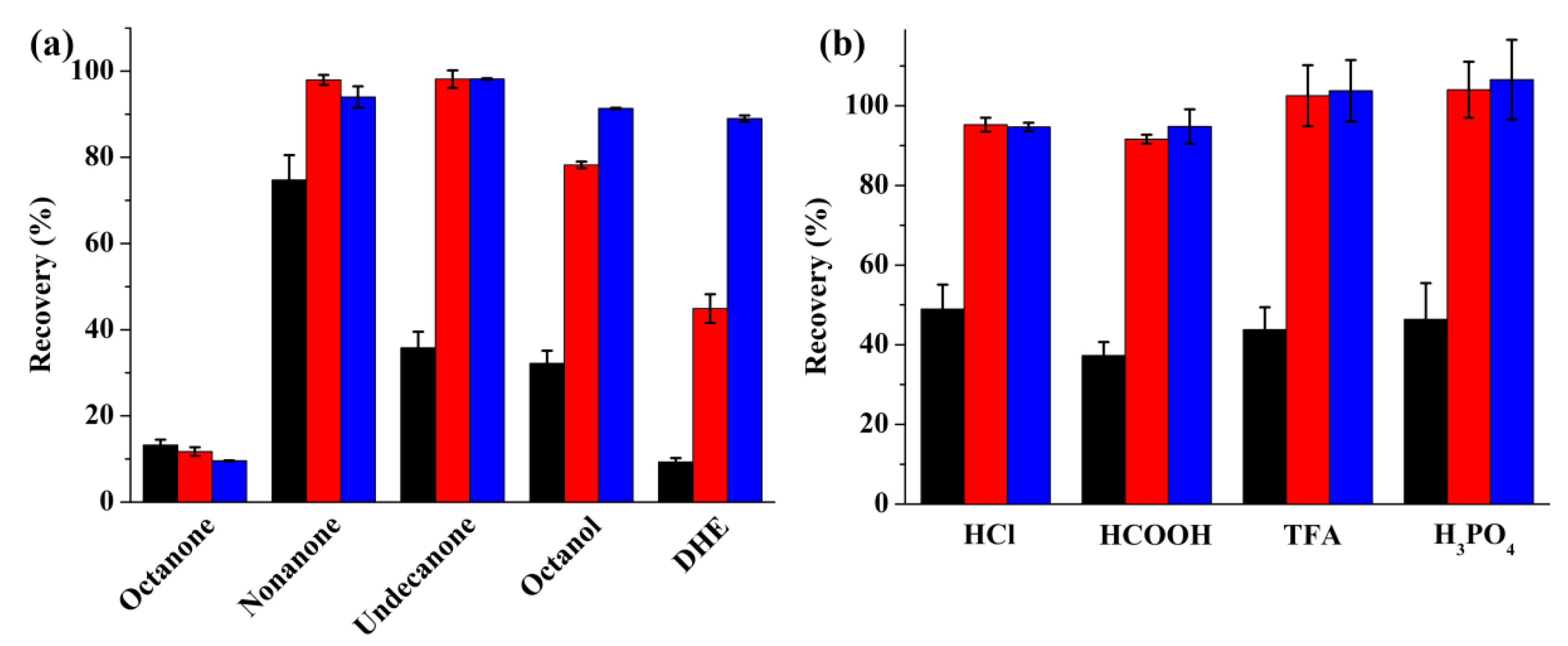
2.1.1. Selection of the Organic Extraction Solvent
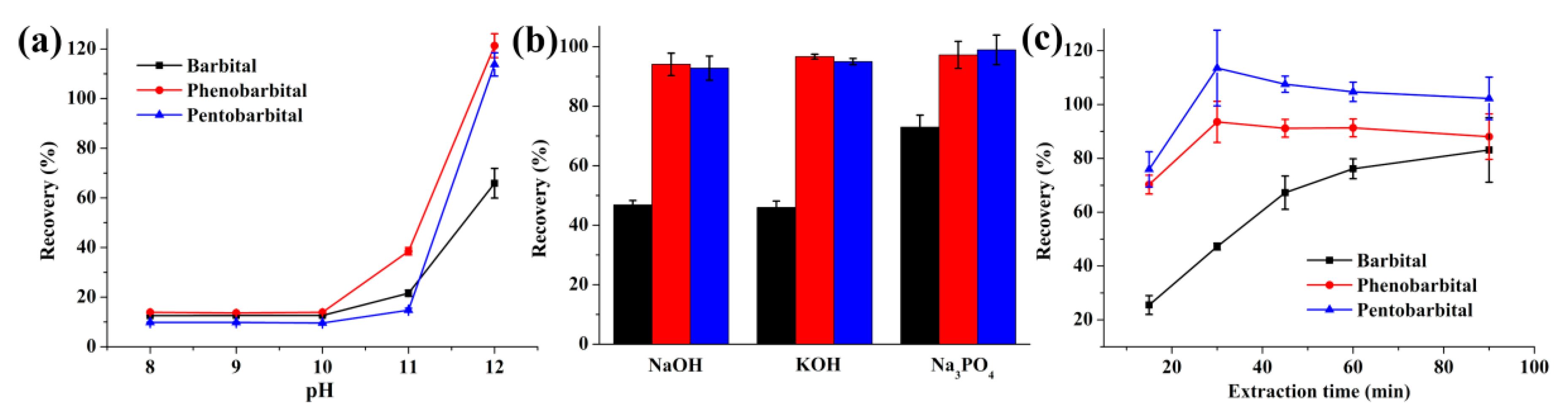
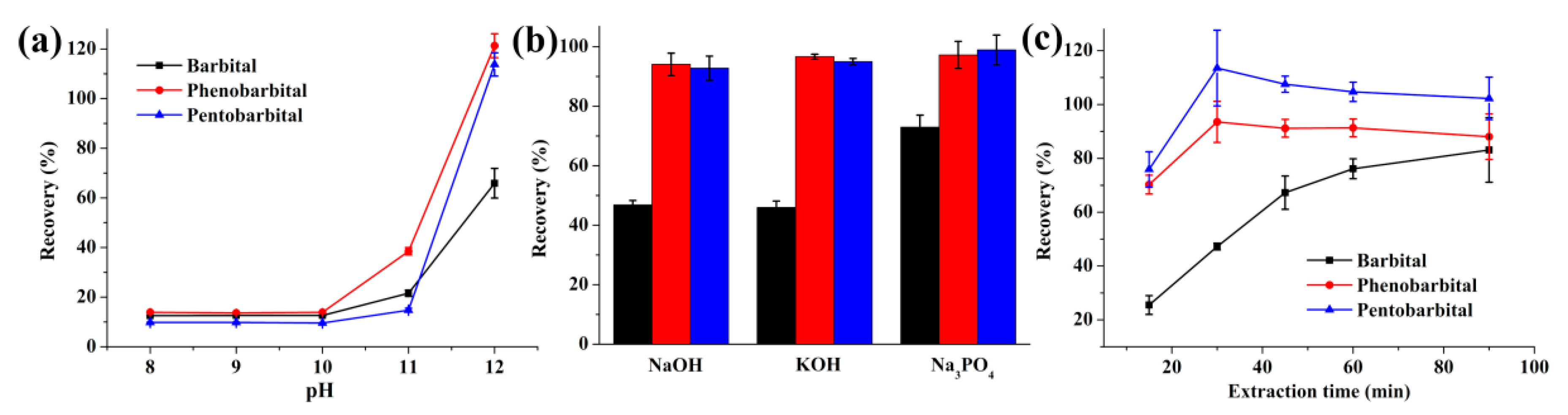
2.1.2. Optimization of the Donor Phase
2.1.3. Optimization of the Acceptor Phase
2.1.4. Effect of Stirring Rate and Extraction Time
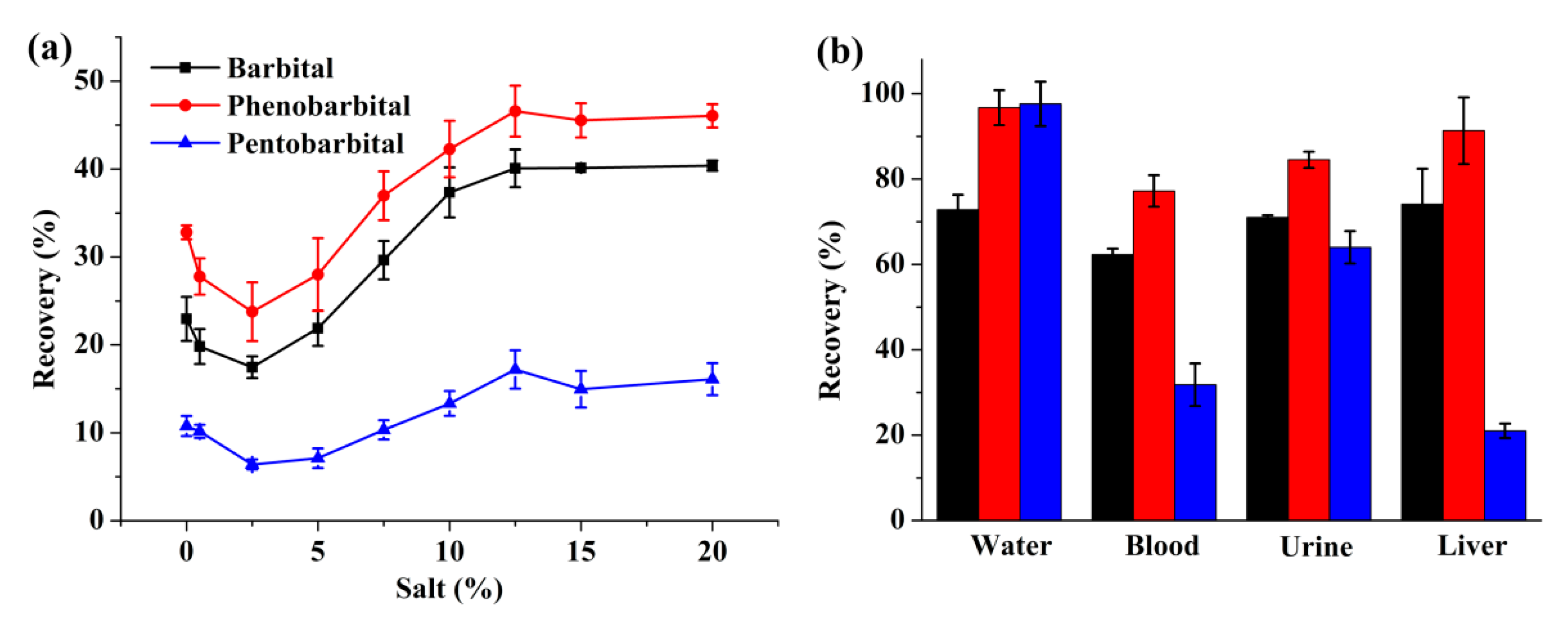
2.2. FM-LPME of Barbiturates from Biological Specimens
2.3. Method Evaluation
2.4. Application
2.5. Comparison of the Proposed Method with Other Reported Methods
3. Materials and Methods
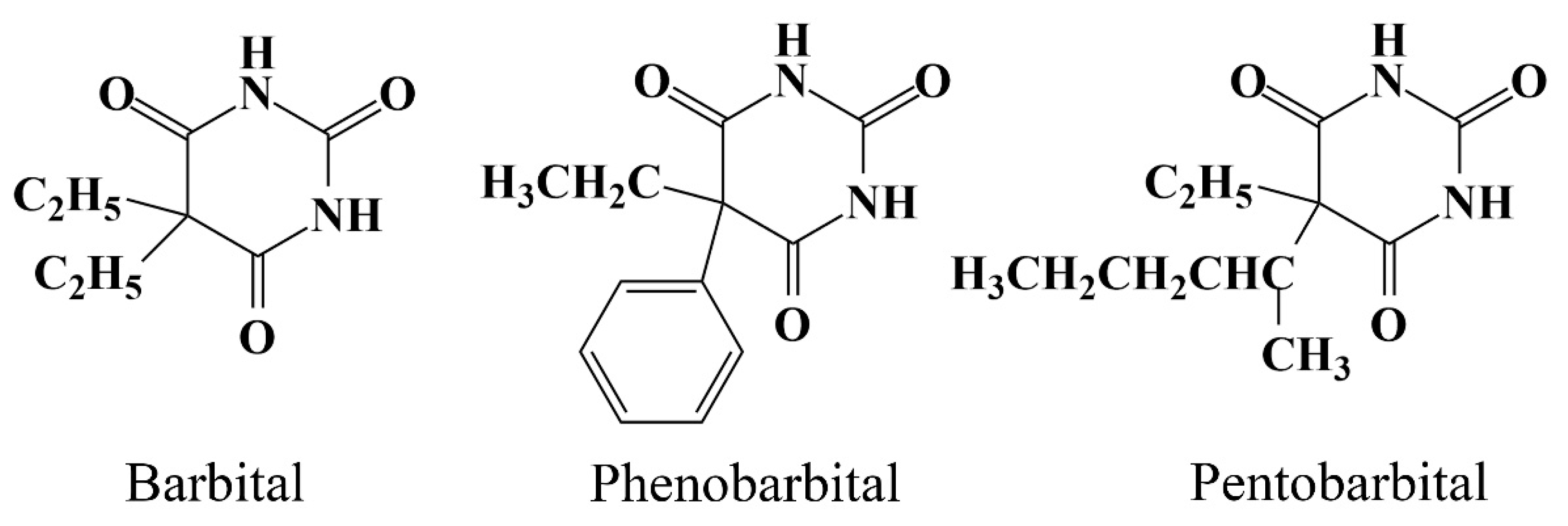
3.1. Chemicals and Materials
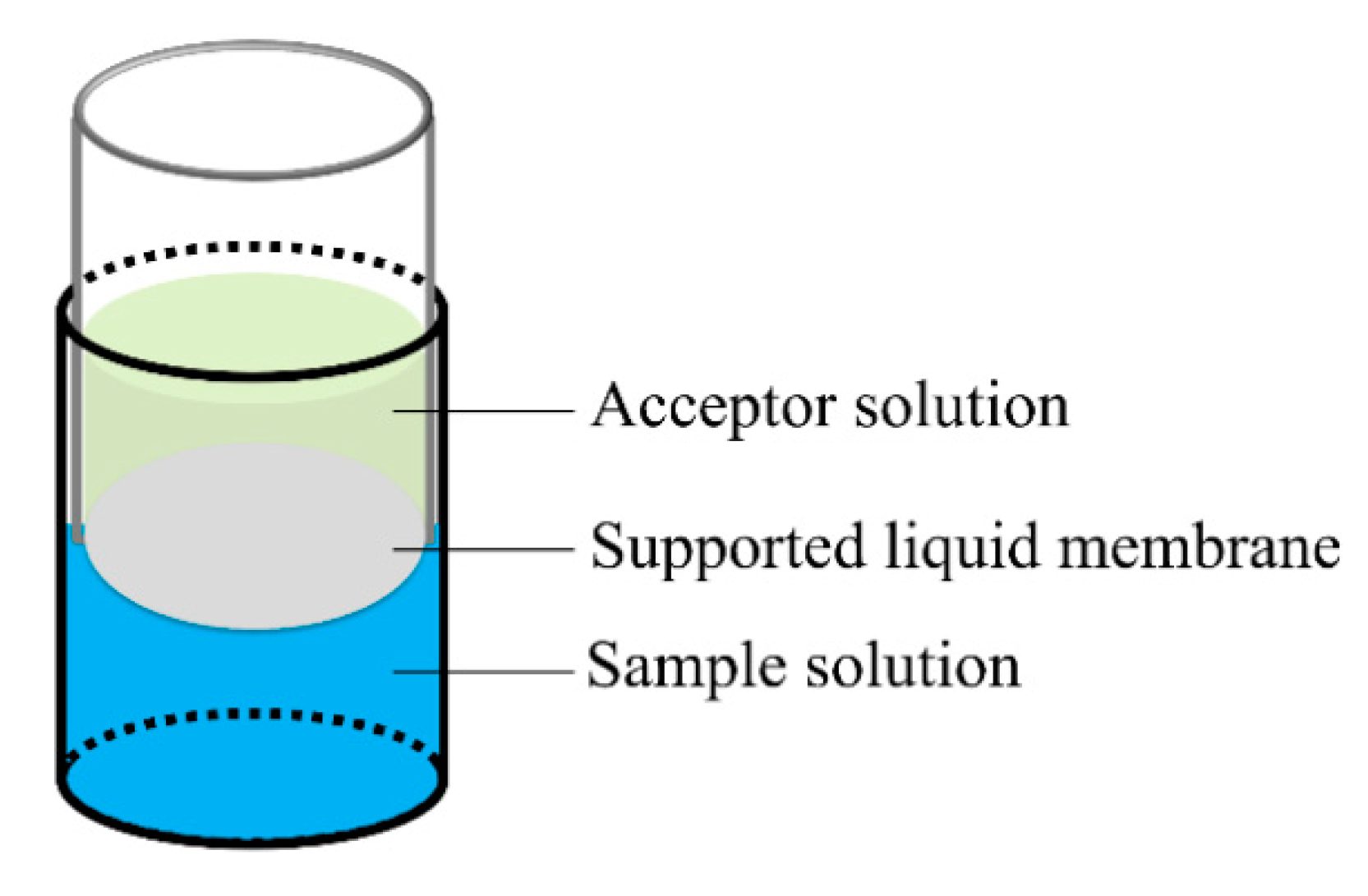
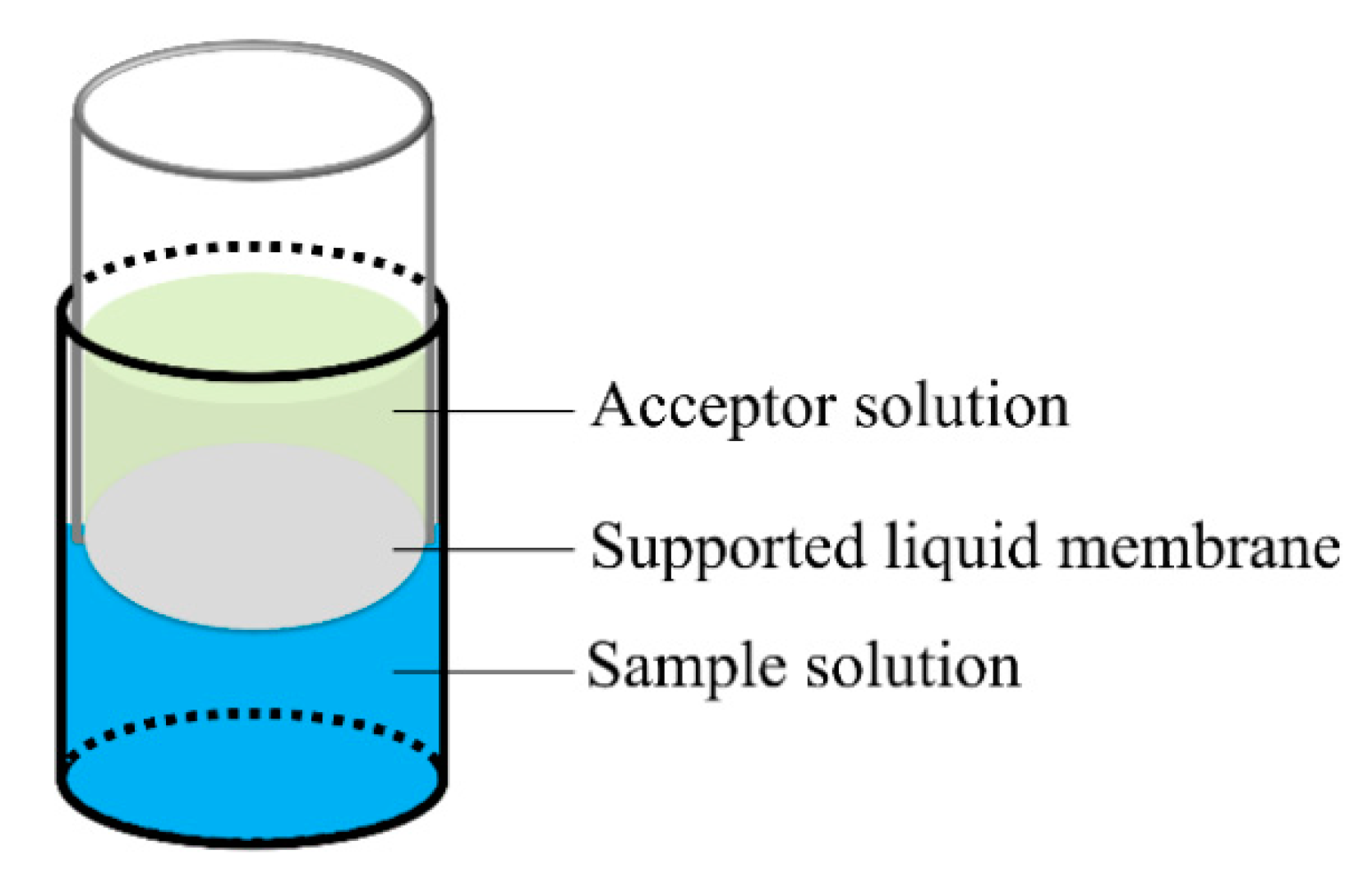
3.2. FM-LPME Setup and Extraction Procedures
3.3. Sample Preparation
3.3.1. Water Samples
3.3.2. Biological Samples
3.3.3. Method Evaluation
3.3.4. Application Experiment
3.4. HPLC-UV Analysis
3.5. LC-MS Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Swarbrick, J. Encyclopedia of Pharmaceutical Technology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Yasiry, Z.; Shorvon, S.D. How phenobarbital revolutionized epilepsy therapy: The story of phenobarbital therapy in epilepsy in the last 100 years. Epilepsia 2012, 53, 26–39. [Google Scholar] [CrossRef] [PubMed]
- López-Muñoz, F.; Ucha-Udabe, R.; Alamo, C. The history of barbiturates a century after their clinical introduction. Neuropsychiatr. Dis. Treat. 2005, 1, 329. [Google Scholar] [PubMed]
- Ito, T.; Suzuki, T.; Wellman, S.E.; Ho, K. Pharmacology of barbiturate tolerance/dependence: GABAA receptors and molecular aspects. Life Sci. 1996, 59, 169–195. [Google Scholar] [CrossRef]
- Vlasses, P.H.; Rocci, M.L., Jr.; Koffer, H.; Ferguson, R.K. Combined phenytoin and phenobarbital overdose. Drug Intell. Clin. Pharm. 1982, 16, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Fritch, D.; Blum, K.; Nonnemacher, S.; Kardos, K.; Buchhalter, A.R.; Cone, E.J. Barbiturate detection in oral fluid, plasma, and urine. Ther. Drug Monit. 2011, 33, 72–79. [Google Scholar] [CrossRef]
- Coupey, S.M. Barbiturates. Pediatr. Rev. 1997, 18, 260–264; quiz 265. [Google Scholar] [CrossRef]
- Zarei, A.R.; Gholamian, F. Development of a dispersive liquid–liquid microextraction method for spectrophotometric determination of barbituric acid in pharmaceutical formulation and biological samples. Anal. Biochem. 2011, 412, 224–228. [Google Scholar] [CrossRef]
- Li, S.; Weber, S.G. Determination of barbiturates by solid-phase microextraction and capillary electrophoresis. Anal. Chem. 1997, 69, 1217–1222. [Google Scholar] [CrossRef]
- Jiang, T.-F.; Wang, Y.-H.; Lv, Z.-H.; Yue, M.-E. Direct determination of barbiturates in urine by capillary electrophoresis using a capillary coated dynamically with polycationic polymers. Chromatographia 2007, 65, 611–615. [Google Scholar] [CrossRef]
- Botello, I.; Borrull, F.; Calull, M.; Aguilar, C.; Somsen, G.W.; de Jong, G.J. In-line solid-phase extraction–capillary electrophoresis coupled with mass spectrometry for determination of drugs of abuse in human urine. Anal. Bioanal. Chem. 2012, 403, 777–784. [Google Scholar] [CrossRef]
- Ohyama, K.; Wada, M.; Lord, G.A.; Ohba, Y.; Fujishita, O.; Nakashima, K.; Lim, C.K.; Kuroda, N. Capillary electrochromatographic analysis of barbiturates in serum. Electrophoresis 2010, 25, 594–599. [Google Scholar] [CrossRef]
- Tanaka, E.; Terada, M.; Tanno, K.; Misawa, S.; Wakasugi, C. Forensic analysis of 10 barbiturates in human biological samples using a new reversed-phase chromatographic column packed with 2-micrometre porous microspherical silica-gel. Forensic Sci. Int. 1997, 85, 73–82. [Google Scholar] [CrossRef]
- Capella-Peiró, M.E.; Gil-Agustĺ, M.; Martinavarro-Domĺnguez, A.; Esteve-Romero, J. Determination in serum of some barbiturates using micellar liquid chromatography with direct injection. Anal. Biochem. 2002, 309, 261–268. [Google Scholar] [CrossRef]
- La Marca, G.; Malvagia, S.; Filippi, L.; Luceri, F.; Moneti, G.; Guerrini, R. A new rapid micromethod for the assay of phenobarbital from dried blood spots by LC-tandem mass spectrometry. Epilepsia 2009, 50, 2658–2662. [Google Scholar] [CrossRef]
- Menck, R.A.; De Lima, D.S.; Seulin, S.C.; Leyton, V.; Pasqualucci, C.A.; Muñoz, D.R.; Osselton, M.D.; Yonamine, M. Hollow-fiber liquid-phase microextraction and gas chromatography-mass spectrometry of barbiturates in whole blood samples. J. Sep. Sci. 2012, 35, 3361–3368. [Google Scholar] [CrossRef]
- Hall, B.J.; Brodbelt, J.S. Determination of barbiturates by solid-phase microextraction (SPME) and ion trap gas chromatography–mass spectrometry. J. Chromatogr. A 1997, 777, 275–282. [Google Scholar] [CrossRef]
- Roveri, F.L.; Paranhos, B.A.P.B.; Yonamine, M. Determination of phenobarbital in hair matrix by liquid phase microextraction (LPME) and gas chromatography–mass spectrometry (GC–MS). Forensic Sci. Int. 2016, 265, 75–80. [Google Scholar] [CrossRef]
- Frison, G.; Favretto, D.; Tedeschi, L.; Ferrara, S.D. Detection of thiopental and pentobarbital in head and pubic hair in a case of drug-facilitated sexual assault. Forensic Sci. Int. 2003, 133, 171–174. [Google Scholar] [CrossRef]
- Johnson, L.L.; Garg, U. Quantitation of amobarbital, butalbital, pentobarbital, phenobarbital, and secobarbital in urine, serum, and plasma using gas chromatography-mass spectrometry (GC-MS). In Clinical Applications of Mass Spectrometry, Methods in Molecular Biology (Methods and Protocols); Garg, U., Hammett-Stabler, C., Eds.; Humana Press: New York, NY, USA, 2010; pp. 65–74. [Google Scholar]
- Deveaux, M.; Cheze, M.; Pépin, G. The role of liquid chromatography-tandem mass spectrometry (LC-MS/MS) to test blood and urine samples for the toxicological investigation of drug-facilitated crimes. Ther. Drug Monit. 2008, 30, 225–228. [Google Scholar] [CrossRef]
- Baciu, T.; Borrull, F.; Aguilar, C.; Calull, M. Recent trends in analytical methods and separation techniques for drugs of abuse in hair. Anal. Chim. Acta 2015, 856, 1–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, W.-E.; Yan, J.-Q.; Liu, M.; Zhou, Y.; Shen, X.; Ma, Y.-L.; Feng, X.-S.; Yang, J.; Li, G.-H. A review of the extraction and determination methods of thirteen essential vitamins to the human body: An update from 2010. Molecules 2018, 23, 1484. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Chen, W. A Review on the Recent Progress in Matrix Solid Phase Dispersion. Molecules 2018, 23, 2767. [Google Scholar] [CrossRef]
- Kataoka, H.; Lord, H.L.; Pawliszyn, J. Applications of solid-phase microextraction in food analysis. J. Chromatogr. A 2000, 880, 35–62. [Google Scholar] [CrossRef]
- Souza-Silva, E.A.; Jiang, R.; Rodriguez-Lafuente, A.; Gionfriddo, E.; Pawliszyn, J. A critical review of the state of the art of solid-phase microextraction of complex matrices I. Environmental analysis. TrAC Trends Anal. Chem. 2015, 71, 224–235. [Google Scholar] [CrossRef]
- Snow, N.H. Solid-phase micro-extraction of drugs from biological matrices. J. Chromatogr. A 2000, 885, 445–455. [Google Scholar] [CrossRef]
- Lee, J.; Lee, H.K.; Rasmussen, K.E.; Pedersen-Bjergaard, S. Environmental and bioanalytical applications of hollow fiber membrane liquid-phase microextraction: A review. Anal. Chim. Acta 2008, 624, 253–268. [Google Scholar] [CrossRef]
- Huang, C.; Seip, K.F.; Gjelstad, A.; Shen, X.; Pedersen-Bjergaard, S. Combination of electromembrane extraction and liquid-phase microextraction in a single step: Simultaneous group separation of acidic and basic drugs. Anal. Chem. 2015, 87, 6951–6957. [Google Scholar] [CrossRef]
- Bello-López, M.Á.; Ramos-Payán, M.; Ocaña-González, J.A.; Fernández-Torres, R.; Callejón-Mochón, M. Analytical applications of hollow fiber liquid phase microextraction (HF-LPME): A review. Anal. Lett. 2012, 45, 804–830. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, X.; Hu, S.; Bai, X. Applications of liquid-phase microextraction techniques in natural product analysis: A review. J. Chromatogr. A 2014, 1368, 1–17. [Google Scholar] [CrossRef]
- Iwai, M.; Hattori, H.; Arinobu, T.; Ishii, A.; Kumazawa, T.; Noguchi, H.; Noguchi, H.; Suzuki, O.; Seno, H. Simultaneous determination of barbiturates in human biological fluids by direct immersion solid-phase microextraction and gas chromatography–mass spectrometry. J. Chromatogr. B 2004, 806, 65–73. [Google Scholar] [CrossRef]
- Jun, X.; Jie, C.; Man, H.; Bin, H. Simultaneous quantification of amphetamines, caffeine and ketamine in urine by hollow fiber liquid phase microextraction combined with gas chromatography-flame ionization detector. Talanta 2010, 82, 969–975. [Google Scholar]
- Overstreet, D.H.; Mathe, A.A.; Nicolau, G.; Feighner, J.P.; Jimenez-Vasquez, P.A.; Hlavka, J.; Morrison, J.; Abajian, H. Liquid-phase microextraction of protein-bound drugs under non-equilibrium conditions. Analyst 2002, 127, 608–613. [Google Scholar]
- Menck, R.A.; de Oliveira, C.D.R.; de Lima, D.S.; Goes, L.E.; Leyton, V.; Pasqualucci, C.A.; Munoz, D.R.; Yonamine, M. Hollow fiber–liquid phase microextraction of barbiturates in liver samples. Forensic Toxicol. 2013, 31, 31–36. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Bioanalysis of drugs by liquid-phase microextraction coupled to separation techniques. J. Chromatogr. B 2005, 817, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hadjmohammadi, M.; Ghambari, H. Three-phase hollow fiber liquid phase microextraction of warfarin from human plasma and its determination by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2012, 61, 44–49. [Google Scholar] [CrossRef]
- Oliveira, A.F.F.; de Figueiredo, E.C.; dos Santos-Neto, Á.J. Analysis of fluoxetine and norfluoxetine in human plasma by liquid-phase microextraction and injection port derivatization GC–MS. J. Pharm. Biomed. Anal. 2013, 73, 53–58. [Google Scholar] [CrossRef]
- Shen, G.; Lee, H.K. Hollow fiber-protected liquid-phase microextraction of triazine herbicides. Anal. Chem. 2002, 74, 648–654. [Google Scholar] [CrossRef]
- Clarke, E.G.C.; Moffat, A.C.; Osselton, M.D.; Widdop, B. Clarke’s Analysis of Drugs and Poisons: In Pharmaceuticals, Body Fluids and Postmortem Material; Pharmaceutical Press: London, UK, 2004. [Google Scholar]
- Winek, C.L.; Wahba, W.W.; Winek, C.L., Jr.; Balzer, T.W. Drug and chemical blood-level data 2001. Forensic Sci. Int. 2001, 122, 107–123. [Google Scholar] [CrossRef]
- Chemical Toxicity Database. Available online: http://www.drugfuture.com/toxic/index.html (accessed on 10 March 2019).
- Zhang, X.; Lin, Z.; Li, J.; Huang, Z.; Rao, Y.; Liang, H.; Yan, J.; Zheng, F. Rapid determination of nine barbiturates in human whole blood by liquid chromatography-tandem mass spectrometry. Drug Test. Analysis 2016, 9, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Eibak, L.E.E.; Gjelstad, A.; Shen, X.; Trones, R.; Jensen, H.; Pedersen-Bjergaard, S. Development of a flat membrane based device for electromembrane extraction: A new approach for exhaustive extraction of basic drugs from human plasma. J. Chromatogr. A 2014, 1326, 7–12. [Google Scholar] [CrossRef]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrices | Analytes | Linearity (ng mL−1) | LOD (ng mL−1) | LOQ (ng mL−1) | Repeatability (%) |
|---|---|---|---|---|---|
| Blood | Barbital | 15–750 | 2.3 | 7.7 | 10 |
| Phenobarbital | 7.5–750 | 1.5 | 5.0 | 6 | |
| Pentobarbital | 15–750 | 3.1 | 10.2 | 8 | |
| Urine | Barbital | 20–500 | 3.6 | 12.0 | 11 |
| Phenobarbital | 5–500 | 1.2 | 4.0 | 5 | |
| Pentobarbital | 5–500 | 0.6 | 2.0 | 5 | |
| Liver 1 | Barbital | 50–2500 | 10.0 | 33.3 | 9 |
| Phenobarbital | 25–2500 | 5.2 | 17.3 | 11 | |
| Pentobarbital | 25–2500 | 7.4 | 24.7 | 14 |
| Analytes | Blood | Liver | |
|---|---|---|---|
| Drug Concentration 1 (µg mL−1) | Drug Concentration 1 (µg g−1) | ||
| Rat 1 | Barbital | 51.1 ± 4.9 | 17.2 ± 3.0 |
| Phenobarbital | 50.3 ± 4.1 | 18.8 ± 3.2 | |
| Pentobarbital | 36.0 ± 2.4 | 52.6 ± 8.2 | |
| Rat 2 | Barbital | 44.9 ± 3.0 | 19.6 ± 1.5 |
| Phenobarbital | 44.8 ± 6.7 | 20.0 ± 1.6 | |
| Pentobarbital | 34.2 ± 3.7 | 58.9 ± 4.4 |
| Sample | Analytes | Extraction | Detection | Linear Range (ng mL−1) | LOD (ng mL−1) | Ref |
|---|---|---|---|---|---|---|
| Urine | Phenobarbital Barbital | SPE | CE | 2–500 | 0.5–5.0 | [11] |
| Blood | Pentobarbital | SPME | GC-MS | 200–40000 | 50 | [32] |
| Serum | Barbital Phenobarbital | LLE | CE-UV | 2900–43290 | 830–1390 | [12] |
| Liver | Pentobarbital Phenobarbital | HF-LPME | GC-MS | 1000–10,000 (ng g−1) | 500 (ng g−1) | [35] |
| Blood | Pentobarbital Phenobarbital | HF-LPME | GC-MS | 1000–10,000 | 1000 | [16] |
| Blood | Barbital Phenobarbital Pentobarbital | LLE | LC-MS | 2–2000 | 0.2–0.5 | [43] |
| Blood Urine Liver | Barbital Phenobarbital Pentobarbital | FM-LPME | LC-MS | 7.5–750 1 5–500 2 25–2500 3 (ng g−1) | 1.5–3.1 1 0.6–3.6 2 5.2–10.0 3 (ng g−1) | Our work |
| Analyte | Parent (m/z) | Product (m/z) | Collision energy (eV) | Tube lens (V) | Retention Time (min) |
|---|---|---|---|---|---|
| Barbital | 183.0 | 42.4 | 80 | 52 | 1.54 |
| 140.0 | 13 | ||||
| Phenobarbital | 231.0 | 42.4 | 17 | 57 | 3.33 |
| 188.0 | 10 | ||||
| Pentobarbital | 225.0 | 42.4 | 53 | 48 | 3.78 |
| 182.0 | 18 | ||||
| IS | 294.0 | 214.0 | 24 | 74 | 4.22 |
| 249.9 | 14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, R.; Dong, Y.; Cai, X.; Huang, C. Determination of Barbiturates in Biological Specimens by Flat Membrane-Based Liquid-Phase Microextraction and Liquid Chromatography-Mass Spectrometry. Molecules 2019, 24, 1494. https://doi.org/10.3390/molecules24081494
Zhu R, Dong Y, Cai X, Huang C. Determination of Barbiturates in Biological Specimens by Flat Membrane-Based Liquid-Phase Microextraction and Liquid Chromatography-Mass Spectrometry. Molecules. 2019; 24(8):1494. https://doi.org/10.3390/molecules24081494
Chicago/Turabian StyleZhu, Ruiqin, Ying Dong, Xiangyang Cai, and Chuixiu Huang. 2019. "Determination of Barbiturates in Biological Specimens by Flat Membrane-Based Liquid-Phase Microextraction and Liquid Chromatography-Mass Spectrometry" Molecules 24, no. 8: 1494. https://doi.org/10.3390/molecules24081494
APA StyleZhu, R., Dong, Y., Cai, X., & Huang, C. (2019). Determination of Barbiturates in Biological Specimens by Flat Membrane-Based Liquid-Phase Microextraction and Liquid Chromatography-Mass Spectrometry. Molecules, 24(8), 1494. https://doi.org/10.3390/molecules24081494