
Flavonoid Glycosides from Endemic Bulgarian Astragalus aitosensis (Ivanisch.)

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
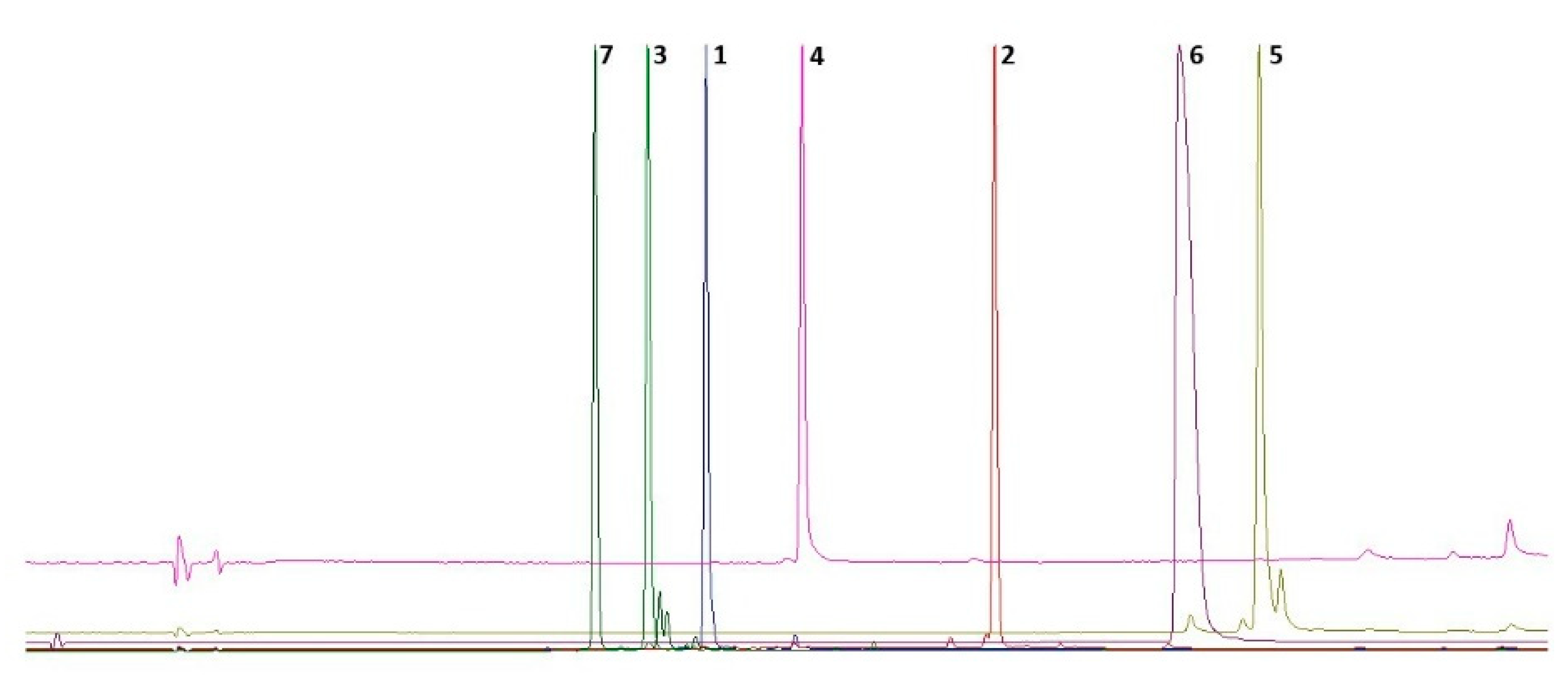
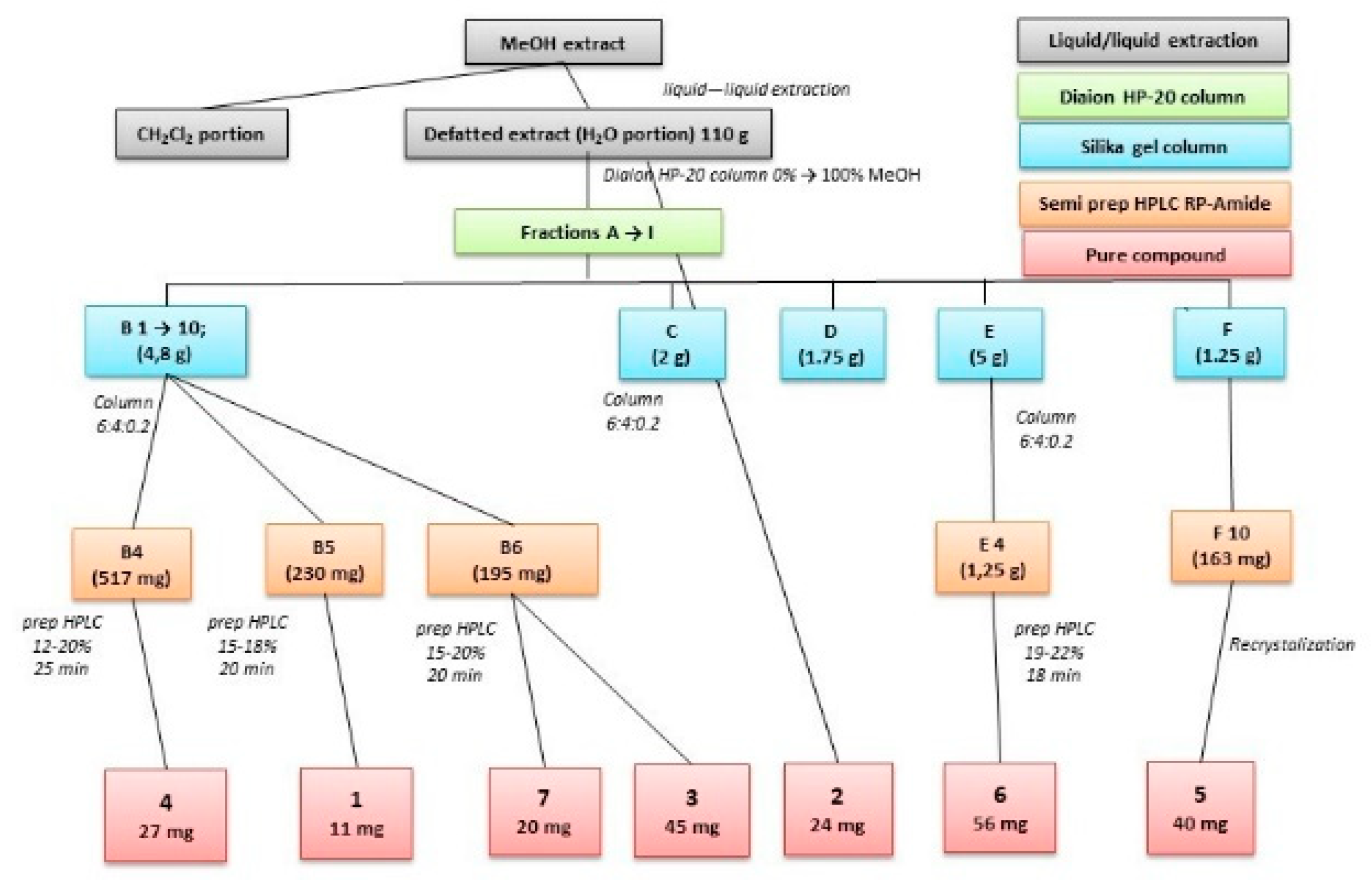
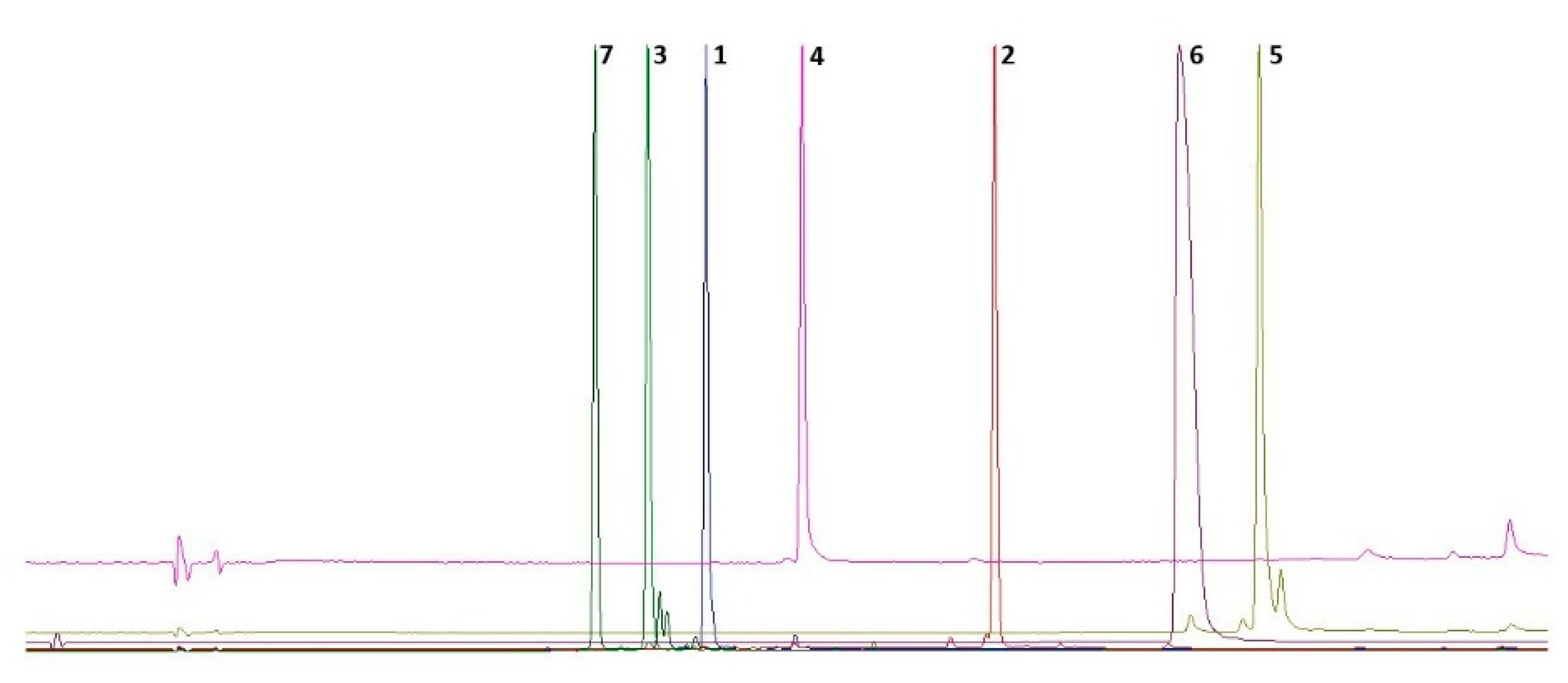
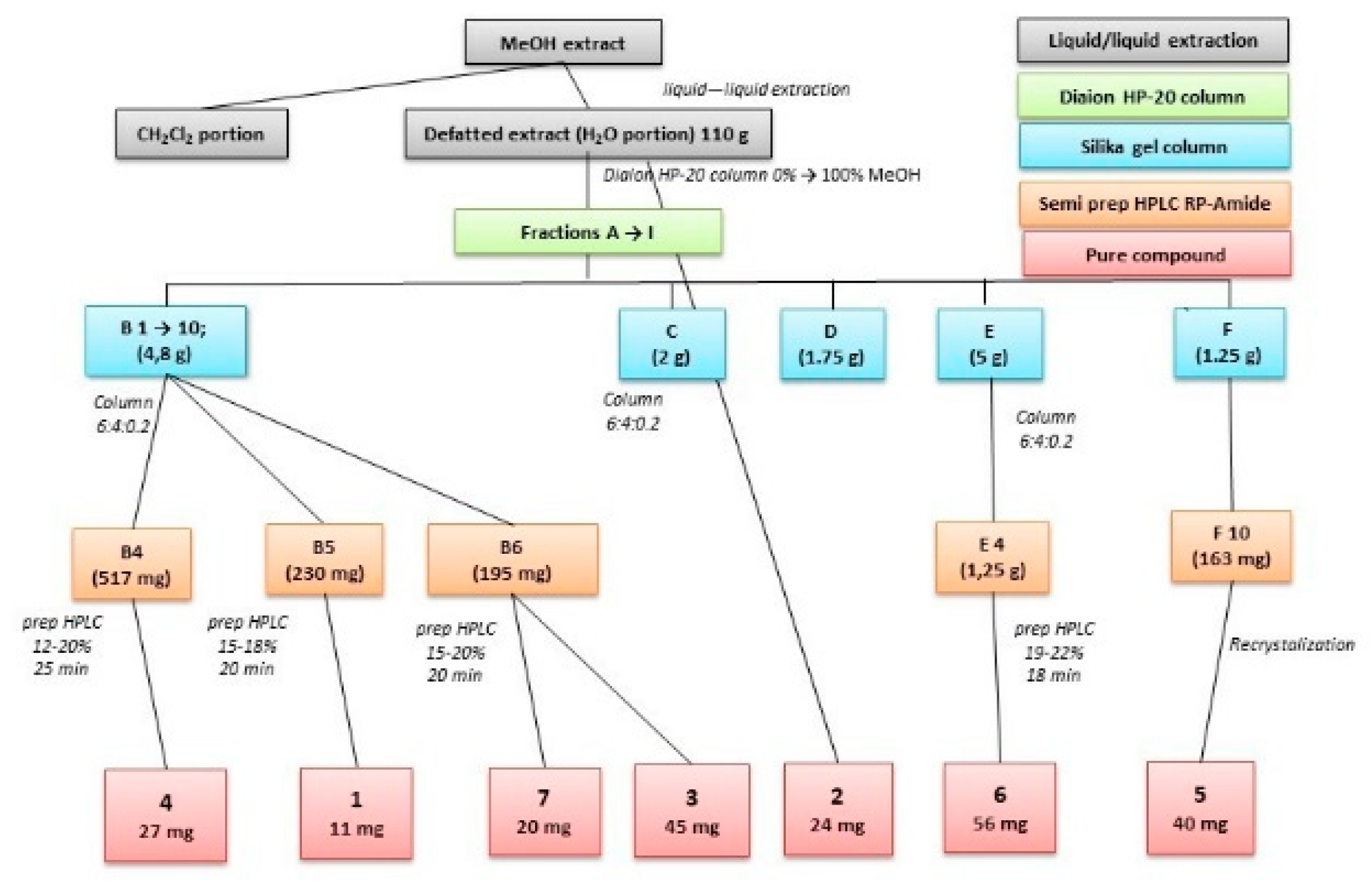
2.1. Isolation of Compounds
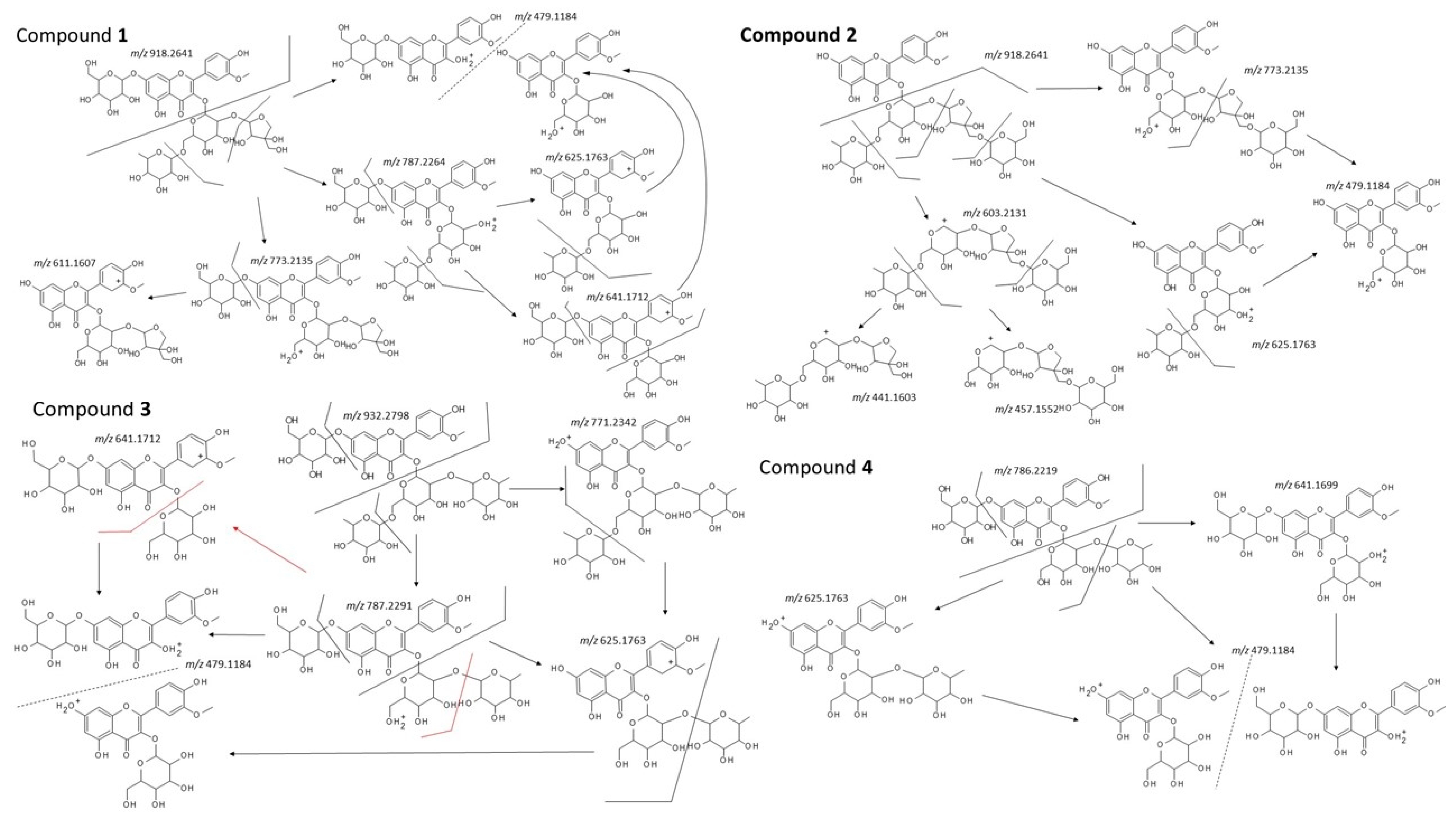
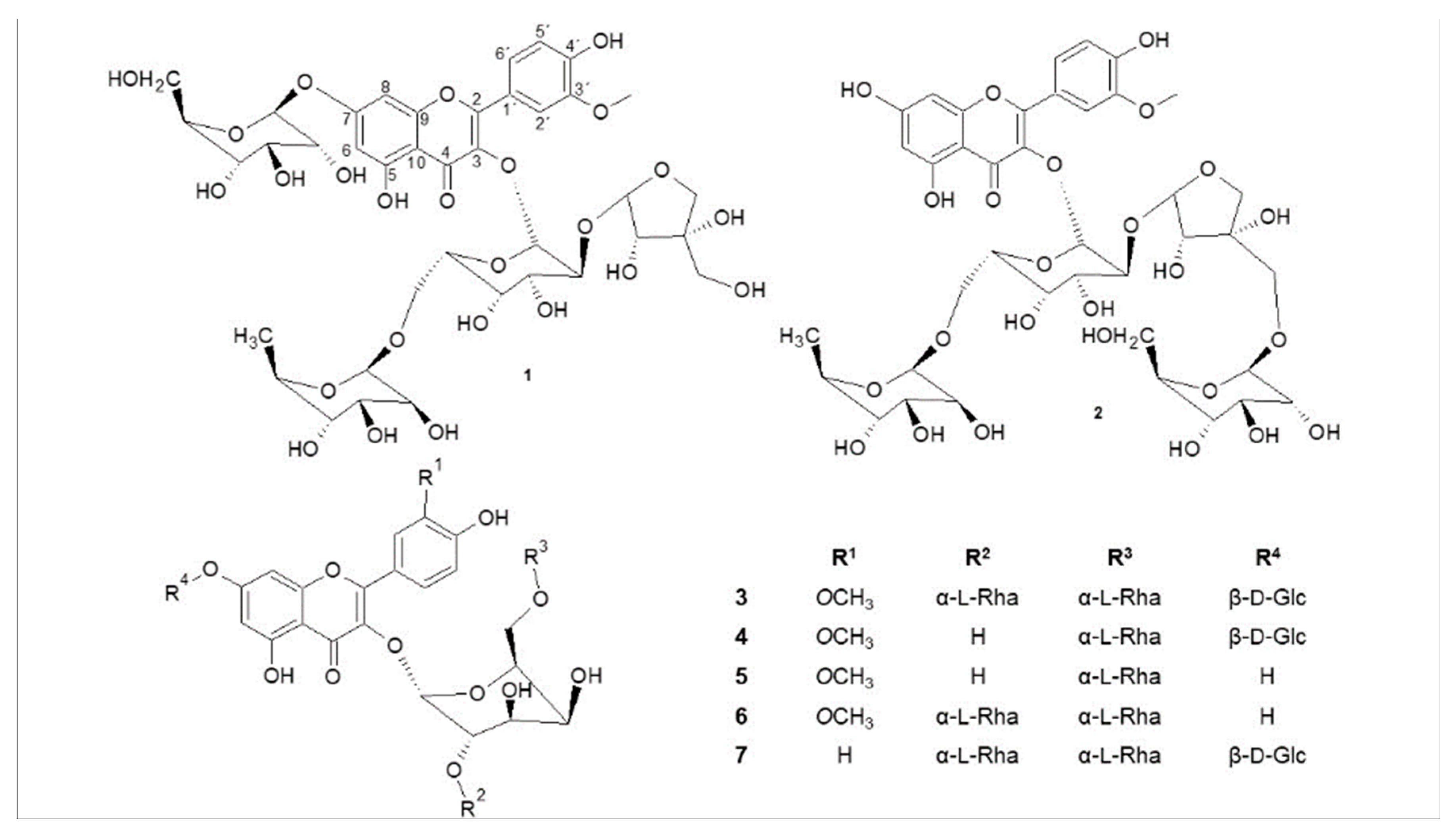
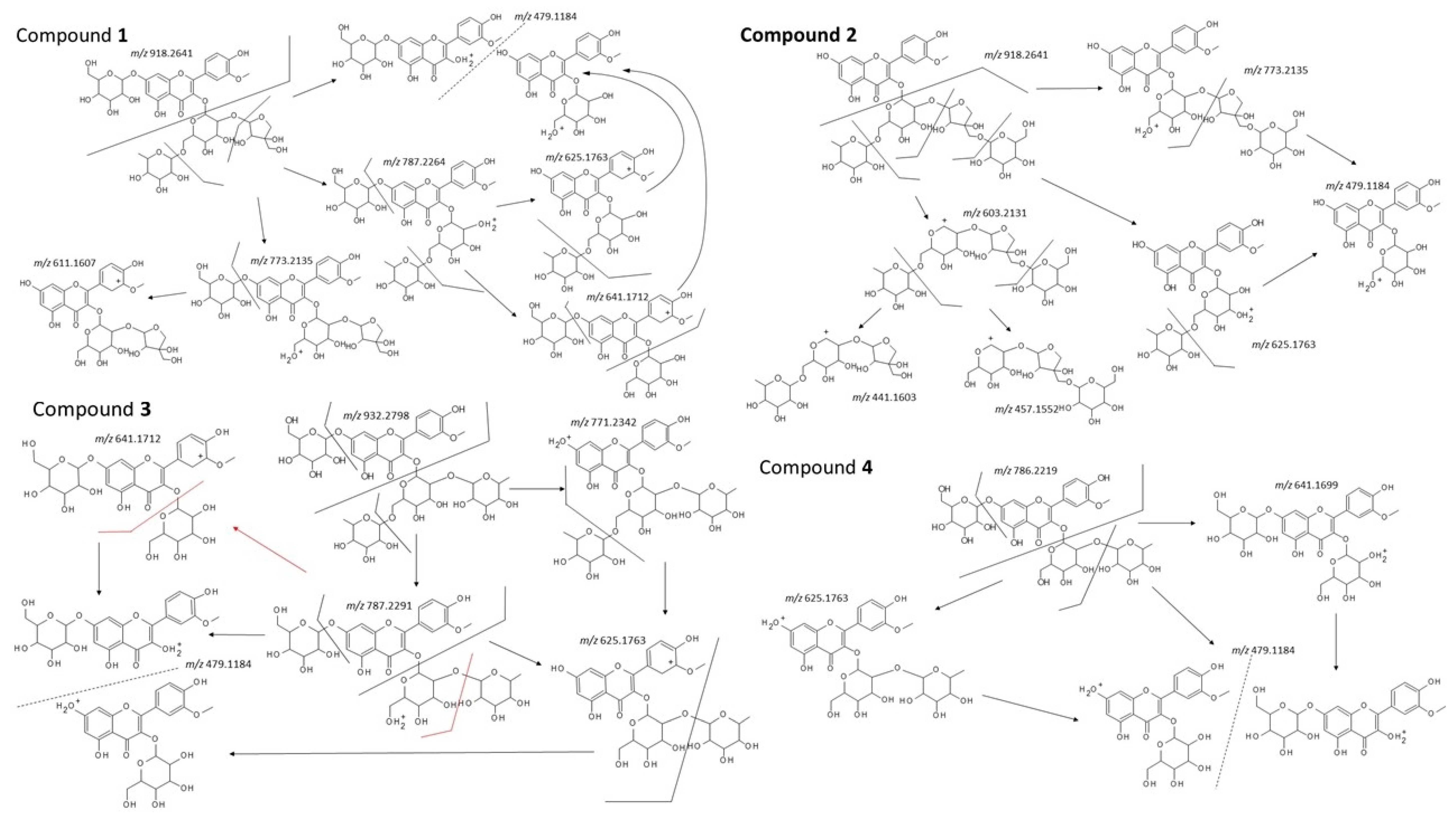
2.2. Structural Analysis
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Identification of Isolated Compounds
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pistelli, L.F. Secondary metabolites of genus Astragalus: Structure and biological activity. Stud. Nat. Prod. Chem. 2002, 27, 443–545. [Google Scholar] [CrossRef]
- Frodin, D.G. History and concepts of big plant genera. Taxon 2004, 53, 753–776. [Google Scholar] [CrossRef]
- Tutin, T.G.; Heywood, V.H.; Burges, N.A.; Moore, D.M.; Valentine, D.H.; Walters, S.M. Flora Europaea. 2; Cambridge University Press: Cambridge, UK, 1972. [Google Scholar]
- Assyov, B.; Petrova, A.; Dimitrov, D.; Vassilev, R. Conspectus of the Bulgarian vascular flora, 4rd ed.; Assyov, B., Petrova, A., Eds.; Bulgarian Biodiversity Foundation: Sofia, Bulgaria, 2012; pp. 86–89. [Google Scholar]
- Andreev, N.; Anchev, M.; Kozucharov, S.; Markova, M.; Peev, D.; Petrova, A. Field Guide of the Bulgarian Vascular Plants; Nauka i Izkustvo: Sofia, Bulgria, 1992; pp. 392–397. [Google Scholar]
- Ionkova, I. Isolation and HPLC-TLC Analysis of the Major Flavonoids from Astragalus aitosensis. Planta Med. 1990, 56, 581. [Google Scholar] [CrossRef]
- Ionkova, I. Farmacevtichno Znachimi Biologichno Aktivni Veshtestva ot Iztochnici s Optimiziran Fitohimichen Potencial (Pharmaceutically Significant Biologically Active Compounds from Sources with Optimized Phytochemical Potential). DSci Dissertation, Medical University, Sofia, Bulgaria, 2008. [Google Scholar]
- Apostolova, I. Red Data Book of the Republic of Bulgaria, Digital ed. Astracantha arnacantha. Available online: www.e-ecodb.bas.bg/rdb/en/vol1/Astarnac.html (accessed on 4 March 2019).
- Ionkova, I.; Shkondrov, A.; Krasteva, I.; Ionkov, T. Recent progress in phytochemistry, pharmacology and biotechnology of Astragalus saponins. Phytochem. Rev. 2014, 13, 343–374. [Google Scholar] [CrossRef]
- Bratkov, V.; Shkondrov, A.; Zdraveva, P.; Krasteva, I. Flavonoids from the genus Astragalus: Phytochemistry and biological activity. Pharmacogn. Rev. 2016, 10, 11–32. [Google Scholar] [PubMed]
- Krasteva, I.; Bratkov, V.; Bucar, F.; Kunert, O.; Kollroser, M.; Kondeva-Burdina, M.; Ionkova, I. Flavoalkaloids and flavonoids from Astragalus monspessulanus. J. Nat. Prod. 2015, 78, 2565–2571. [Google Scholar] [CrossRef] [PubMed]
- Buschi, C.A.; Pomilio, A.B. Isorhamnetin 3-O-robinobioside from Gomphrena martiana. J. Nat. Prod. 1982, 45, 557–559. [Google Scholar] [CrossRef]
- Yasukawa, K.E.N.; Sekine, H. Two flavonol glycosides from Lysimachia fortunei. Phytochemistry 1989, 28, 2215–2216. [Google Scholar] [CrossRef]
- Yasukawa, K.; Takido, M. A flavonol glycoside from Lysimachia mauritiana. Phytochemistry 1987, 26, 1224–1226. [Google Scholar] [CrossRef]
- Kijima, H.; Ide, T.; Otsuka, H.; Takeda, Y. Alangiflavoside, a new flavonol glycoside from the leaves of Alangium premnifolium. J. Nat. Prod. 1995, 58, 1753–1755. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Yang, L.; Zhou, D.; Zhang, J. Purification and characterization of flavonoids from the leaves of Zanthoxylum bungeanum and correlation between their structure and antioxidant activity. PLoS ONE 2014, 9, e105725/1–e105725/11. [Google Scholar] [CrossRef] [PubMed]
- Gorai, D.; Jash, S.K.; Roy, R. Flavonoids from Astragalus genus. Int. J. Pharm. Sci. Res. 2016, 7, 2732–2747. [Google Scholar]
- Krasteva, I.; Shkondrov, A.; Ionkova, I.; Zdraveva, P. Advances in phytochemistry, pharmacology and biotechnology of Bulgarian Astragalus species. Phytochem. Rev. 2016, 15, 567–590. [Google Scholar] [CrossRef]
- Li, X.; Qu, L.; Dong, Y.; Han, L.; Liu, E.; Fang, S.; Zhang, Y.; Wang, T. A review of recent research progress on the Astragalus genus. Molecules 2014, 19, 18850–18880. [Google Scholar] [CrossRef] [PubMed]
- Alaniya, M.D.; Aneli, D.N.; Patudin, A.V.; Komelin, R.V. Flavonoid glycosides of Astragalus cicer. Chem. Nat. Comp. 1983, 19, 500–501. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 157.8, C | 157.2, C | ||
| 3 | 133.6, C | 133.3, C | ||
| 4 | 178.0, C | 177.8, C | ||
| 5 | 161.3, C | 161.6, C | ||
| 6 | 99.4, CH | 6.45, d (2.02) | 98.5, CH | 6.19, d (2.00) |
| 7 | 163.0, C | 164.4, C | ||
| 8 | 94.4, CH | 6.75, d (2.02) | 93.4, CH | 6.38, d (2.00) |
| 9 | 156.5, C | 157.0, C | ||
| 10 | 106.3, C | 104.5, C | ||
| 1′ | 121.8, C | 121.9, C | ||
| 2′ | 113.3, CH | 8.05, m (1.94) | 113.7, CH | 8.04, d (1.97) |
| 3′ | 147.0, C | -OCH3 | 147.0, C | -OCH3 |
| 4′ | 149.4, C | -OH | 149.2, C | -OH |
| 5′ | 114.5, CH | 6.91, m (8.39) | 114.5, CH | 6.90, d (8.40) |
| 6′ | 122.3, CH | 7.62, m (1.92; 8.45) | 122.1, CH | 7.57, dd (1.90; 8.43) |
| -OCH3 | 55.7, CH | 4.00 s | 55.7, CH3 | 3.98, s |
| 3-O-gal | ||||
| 1 | 99.8, CH | 5.57, d (7.84) | 99.9, CH | 5.58, d (7.76) |
| 2 | 75.1, CH | 3.96, dd | 74.8, CH | 3.97, dd |
| 3 | 73.9, CH | 3.73 dd | 73.9, CH | 3.74 dd |
| 4 | 69.0, CH | 3.77 dd | 69.0, CH | 3.77 dd |
| 5 | 74.1, CH | 3.67 dt | 74.0, CH | 3.64 dt |
| 6 | 66.0, CH2 | 3.47/3.68 dd | 65.8, CH2 | 3.47/3.71 dd |
| Api (1→2) | ||||
| 1 | 109.2, CH | 5.43, d (1.55) | 109.1, CH | 5.44, d (1.73) |
| 2 | 76.6, CH | 4.01, d | 76.9, CH | 4.07, d |
| 3 | 79.5, C | -OH | 78.8, C | -OH |
| 4 | 74.2, CH2 | 3.64, d (9.58) | 74.3, CH2 | 3.66, d (n/a) |
| 3.75, d (9.59) | 4.07, d (9.70) | |||
| 5 | 65.1, CH2 | 3.64, d (11.45) | 61.3, CH2 | 3.67, d (11.95) |
| 3.75 d (11.56) | 3.85 d (11.90) | |||
| Rha (1→6) | ||||
| 1 | 100.5, CH | 4.50, d (1.56) | 100.5, CH | 4.52, d (1.53) |
| 2 | 70.7, CH | 3.51, dd | 70.7, CH | 3.57, dd |
| 3 | 70.9, CH | 3.46, dd | 70.9, CH | 3.49, dd |
| 4 | 72.4, CH | 3.25, pt | 72.4, CH | 3.26, pt |
| 5 | 68.3, CH | 3.49, dq | 68.3, CH | 3.51, dq |
| 6 | 16.5, CH3 | 1.15, d (6.17) | 16.5, CH3 | 1.16, d (6.23) |
| Glc | 7-O-Glc | Api (5→1) Glc | ||
| 1 | 100.1, CH | 5.06, d (7.53) | 103.4, CH | 4.25, d (7.54) |
| 2 | 73.3, CH | 3.49, dd | 73.6, CH | 3.17, dd |
| 3 | 77.0, CH | 3.55, dd | 76.3, CH | 3.21, dd |
| 4 | 69.9, CH | 3.38, dt | 70.4, CH | 3.27, dd |
| 5 | 76.4, CH | 3.49, dq | 76.3, CH | 3.27, dt |
| 6 | 61.1, CH2 | 3.69/3.94 dd | 72.9, CH2 | 3.68/4.15, dd |
| Compound/Position | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|
| δC, Type | δC, Type | δC, Type | δC, Type | δC, Type | |
| 2 | 157.7, C | 157.4, C | 156.8, C | 157.0, C | 158.0, C |
| 3 | 133.3, C | 133.8, C | 133.5, C | 133.0, C | 133.4, C |
| 4 | 177.9, C | 178.0, C | 177.8, C | 177.8, C | 177.1, C |
| 5 | 161.3, C | 161.3, C | 161.6, C | 161.7, C | 161.4, C |
| 6 | 99.3, CH | 99.8, CH | 99.2, CH | 98.4, CH | 99.4, CH |
| 7 | 163.0, C | 163.4, C | 164.7, C | 164.4, C | 163.0, C |
| 8 | 94.4, CH | 95.1, CH | 94.2, CH | 93.3, CH | 94.3, CH |
| 9 | 156.5, C | 156.4, C | 156.8, C | 157.0, C | 156.6, C |
| 10 | 106.3, C | 106.1, C | 104.4, C | 104.5, C | 106.2, C |
| 1′ | 121.8, C | 121.3, C | 121.5, C | 122.0, C | 121.5, C |
| 2′ | 113.3, CH | 113.9, CH | 113.9, CH | 113.3, CH | 131.0, CH |
| 3′ | 147.1, CH | 147.5, CH | 147.4, C | 147.0, C | 114.8, CH |
| 4′ | 149.3, C | 150.1, C | 149.9, C | 149.1, C | 160.1, C |
| 5′ | 114.6, CH | 115.6, CH | 114.6, CH | 114.5, CH | 114.8, CH |
| 6′ | 122.1, CH | 122.6, CH | 122.4, CH | 121.8, CH | 131.0, CH |
| OCH3 | 56.37, CH3 | 56.39, CH | 56.37, CH | 55.79, CH3 | |
| 3-O-Gal | |||||
| 1 | 99.3, CH | 102.1, CH | 102.3, CH | 99.4, CH | 99.4, CH |
| 2 | 76.4, CH | 74.1, CH | 74.0, CH | 76.4, CH | 76.1, CH |
| 3 | 74.2, CH | 71.6, CH | 71.5, CH | 74.2, CH | 74.3, CH |
| 4 | 69.2, CH | 68.4, CH | 68.4, CH | 69.1, CH | 69.4, CH |
| 5 | 74.1, CH | 73.4, CH | 73.4, CH | 73.9, CH | 74.1, CH |
| 6 | 65.9, CH2 | 65.6, CH2 | 65.6, CH2 | 65.7, CH2 | 66.0, CH2 |
| Rha (1→2) | |||||
| 1 | 101.4, CH | 101.3, CH | 101.2, CH | ||
| 2 | 71.0, CH | 71.0, CH | 71.0, CH | ||
| 3 | 71.0, CH | 71.0, CH | 70.9, CH | ||
| 4 | 72.5, CH | 72.5, CH | 72.7, CH | ||
| 5 | 68.4, CH | 68.4, CH | 68.4, CH | ||
| 6 | 16.0, CH3 | 16.0, CH3 | 16.1, CH3 | ||
| Rha (1→6) | |||||
| 1 | 100.6, CH | 100.5, CH | 100.5, CH | 100.5, CH | 100.5, CH |
| 2 | 70.7, CH | 70.9, CH | 70.9, CH | 70.7, CH | 70.7, CH |
| 3 | 70.9, CH | 71.1, CH | 71.1, CH | 70.9, CH | 70.9, CH |
| 4 | 72.4, CH | 72.2, CH | 72.3, CH | 72.6, CH | 72.5, CH |
| 5 | 68.3, CH | 68.7, CH | 68.7, CH | 68.3, CH | 68.3, CH |
| 6 | 16.6, CH3 | 18.3, CH3 | 18.3, CH3 | 16.6, CH3 | 16.6, CH3 |
| 7-O-Glc | |||||
| 1 | 100.1, CH | 100.3, CH | 100.1, CH | ||
| 2 | 73.3, CH | 73.6, CH | 73.3, CH | ||
| 3 | 77.0, CH | 76.9, CH | 77.0, CH | ||
| 4 | 69.9, CH | 70.0, CH | 69.9, CH | ||
| 5 | 76.4, CH | 77.7, CH | 76.4, CH | ||
| 6 | 61.1, CH2 | 61.2, CH2 | 61.1, CH2 |
| 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| 2 | |||||
| 3 | |||||
| 4 | |||||
| 5 | |||||
| 6 | 6.44, d (2.07) | 6.44, d (1.91) | 6.19, d (1.97) | 6.15, d (1.97) | 6.46, d (2.15) |
| 7 | |||||
| 8 | 6.75, d (2.09) | 6.77, d (1.97) | 6.42, d (1.97) | 6.37, d (1.97) | 6.75, d (2.14) |
| 9 | |||||
| 10 | |||||
| 1′ | |||||
| 2′ | 8.09, m (1.93) | 8.00, d (1.84) | 7.98, d (1.97) | 8.07, d (1.94) | 8.09, m (8.95) |
| 3′ | 6.90, m (8.90) | ||||
| 4′ | |||||
| 5′ | 6.91, m (8.44) | 6.90, d (8.39) | 6.88, d (8.44) | 6.90, d (8.42) | 6.90, m (8.90) |
| 6′ | 7.57, m (1.91; 8.42) | 7.54, dd (1.82; 8.48) | 7.49, dd (2.03; 8.39) | 7.52, dd (1.99; 8.43) | 8.09, m (8.95) |
| OCH3 | 3.83, s | 3.84, s | 3.83, s | 4.00, s | - |
| 3-O-Gal | |||||
| 1 | 5.79, d (7.84) | 5.47, d (7.73) | 5.45, d (7.67) | 5.59, d (7.83) | 5.59, d (7.74) |
| 2 | 3.97, dd | 3.60, dd | 3.57, dd | 3.96, dd | 3.95, dd |
| 3 | 3.78, dd | 3.57, dd | 3.57, dd | 3.76, dd | 3.70, dd |
| 4 | 3.80, dd | 3.62, dd | 3.63, dd | 3.81, dd | 3.75, dd |
| 5 | 3.74, dt | 3.42, dt | 3.41, dt | 3.71, dt | 3.64, dt |
| 6 | 3.54/3.73dd | 3.30/3.60 dd | 3.31/3.61 dd | 3.52/3.72 dd | 3.46/3.69 dd |
| Rha (1→2) | |||||
| 1 | 5.16, d (1.52) | 5.16, d (1.53) | 5.21, d (1.32) | ||
| 2 | 4.00, dd | 4.00, dd | 4.00, dd | ||
| 3 | 3.75, dd | 3.76, dd | 3.78, dd | ||
| 4 | 3.33, pt | 3.32, pt | 3.33, pt | ||
| 5 | 4.04, dq | 4.03, dq | 4.06, dq | ||
| 6 | 0.89, d (6.25) | 0.89, d (6.23) | 0.98, d (6.23) | ||
| Rha (1→6) | |||||
| 1 | 4.53, d (1.53) | 4.41, d (1.53) | 4.41, d (1.53) | 4.55, d (1.55) | 4.49, d (1.24) |
| 2 | 3.54, dd | 3.37, dd | 3.38, dd | 3.59, dd | 3.48, dd |
| 3 | 3.49, dd | 3.28, dd | 3.28, dd | 3.51, dd | 3.46, dd |
| 4 | 3.25, pt | 3.06, pt | 3.07, pt | 3.26, pt | 3.25, pt |
| 5 | 3.52, dq | 3.36, dq | 3.34, dq | 3.54, dq | 3.49, dq |
| 6 | 1.16, d (6.08) | 1.04, d (6.18) | 1.03, d (6.25) | 1.17, d (6.22) | 1.16, d (6.21) |
| 7-O-Glc | |||||
| 1 | 5.06, d (7.24) | 5.05, d (7.89) | 5.07, d (7.45) | ||
| 2 | 3.48, dd | 3.24, dd | 3.48, dd | ||
| 3 | 3.53, dd | 3.27, dd | 3.53, dd | ||
| 4 | 3.39, dd | 3.15, dd | 3.38, dd | ||
| 5 | 3.50, dt | 3.43, dt | 3.49, dt | ||
| 6 | 3.69/3.92, dd | 3.44/3.68, dd | 3.69/3.91, dd |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilev, H.; Ross, S.; Šmejkal, K.; Maršík, P.; Jankovská, D.; Havlík, J.; Veselý, O. Flavonoid Glycosides from Endemic Bulgarian Astragalus aitosensis (Ivanisch.). Molecules 2019, 24, 1419. https://doi.org/10.3390/molecules24071419
Vasilev H, Ross S, Šmejkal K, Maršík P, Jankovská D, Havlík J, Veselý O. Flavonoid Glycosides from Endemic Bulgarian Astragalus aitosensis (Ivanisch.). Molecules. 2019; 24(7):1419. https://doi.org/10.3390/molecules24071419
Chicago/Turabian StyleVasilev, Hristo, Samir Ross, Karel Šmejkal, Petr Maršík, Dagmar Jankovská, Jaroslav Havlík, and Ondřej Veselý. 2019. "Flavonoid Glycosides from Endemic Bulgarian Astragalus aitosensis (Ivanisch.)" Molecules 24, no. 7: 1419. https://doi.org/10.3390/molecules24071419
APA StyleVasilev, H., Ross, S., Šmejkal, K., Maršík, P., Jankovská, D., Havlík, J., & Veselý, O. (2019). Flavonoid Glycosides from Endemic Bulgarian Astragalus aitosensis (Ivanisch.). Molecules, 24(7), 1419. https://doi.org/10.3390/molecules24071419