
A Simple and Efficient Method for the Partial Synthesis of Pure (3R,3’S)-Astaxanthin from (3R,3’R,6’R)-Lutein and Lutein Esters via (3R,3’S)-Zeaxanthin and Theoretical Study of Their Formation Mechanisms


Abstract
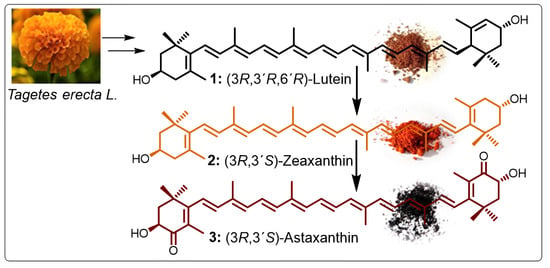
1. Introduction
2. Results and Discussion
2.1. Synthesis and Formation Mechanism of Meso-zeaxanthin
2.1.1. High Yield Preparation of Meso-zeaxanthin
2.1.2. Theoretical Mechanism of the Conversion of (3R,3’R,6’R)-Lutein to Meso-zeaxanthin
2.2. Synthesis and Formation Mechanism of Astaxanthin from Zeaxanthin
2.2.1. Synthesis of (3R,3’S)-Astaxanthin
2.2.2. Theoretical Mechanism for the Conversion of (3R,3’S)-Zeaxanthin to (3R,3’S)-Astaxanthin
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Free (3R,3’R,6’R)-Lutein (1) from Oleoresin
3.3. Synthesis of (3R,3’S)-Zeaxanthin (2)
3.4. Synthesis of (3R,3’S)-astaxanthin (3)
3.5. Computational Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Álvarez, R.; Vaz, B.; Gronemeyer, H.; de Lera, A.R. Functions, therapeutic applications, and synthesis of retinoids and carotenoids. Chem. Rev. 2014, 114, 1–125. [Google Scholar] [CrossRef] [PubMed]
- Bauerfeind, J.; Hintze, V.; Kschonsek, J.; Killenberg, M.; Böhm, V. Use of photochemiluminescence for the determination of antioxidant capacities of selected tomato products. J. Agric. Food. Chem. 2014, 62, 7452–7459. [Google Scholar] [CrossRef]
- Britton, G.; Liaaen-Jensen, S.; Pfander, H. Carotenoids: Nutrition and Health; Birkhäuser Verlag: Basel, Switzerland, 2009; pp. 45–57. [Google Scholar]
- Cazzonelli, C.I. Carotenoids in nature: Insights from plants and beyond. Funct. Plant Biol. 2011, 38, 833–847. [Google Scholar] [CrossRef]
- Britton, G.; Liaaen-Jensen, S.; Pfander, H. Carotenoids: Synthesis; Birkhäuser Verlag: Basel, Switzerland, 2009; pp. 1–6. [Google Scholar]
- Rivera, S.M. Guide for carotenoid identification in biological samples. J. Nat. Prod. 2016, 79, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Ciaparra, I.; Félix-Valenzuela, L.; Goycoolea, F.M. Astaxanthin: A review of its chemistry and applications. Crit. Rev. Food Sci. Nutr. 2006, 46, 185–196. [Google Scholar] [CrossRef]
- Visioli, F.; Artaria, C. Astaxanthin in cardiovascular health and disease: Mechanism of action, therapeutic merits, and knowledge gaps. Food Funct. 2017, 8, 39–63. [Google Scholar] [CrossRef]
- Torres-Cardona, M.D. Process for the isomerization of lutein. U.S. Patent 5,523,494, 4 June 1996. [Google Scholar]
- Bernhard, K.U.S. Process for the manufacturing of zeaxanthin from lutein. U.S. Patent 5,780,693, 14 July 1998. [Google Scholar]
- Andrews, A.G. Isomerization of ε-Carotene to β-Carotene and Lutein to Zeaxanthin. Acta Chem. Scand. 1974, 28, 137–138. [Google Scholar] [CrossRef][Green Version]
- Buchecker, R.; Eugster, C.H.; Weber, A. Absolute Konfiguration von α-Doradexanthin und von Fritschiellaxanthin, einem neuen Carotinoid aus Fritschella tuberosa IYENG. Helv. Chim. Acta 1978, 61, 1962–1968. [Google Scholar]
- Khachick, F. Process for making a (3R,3’R)-zeaxanthin precursor. U.S. Patent 6,818,798, 16 November 2004. [Google Scholar]
- Khachik, F. An efficient conversion of (3R,3’R,6’R)-lutein to (3R,3’S,6’R)-lutein (3’-epilutein) and (3R,3’R)-zeaxanthin. J. Nat. Prod. 2003, 66, 67–72. [Google Scholar] [CrossRef]
- Deli, J.; Molnar, P.; Osz, E.; Tóth, G.; Zsila, F. Epimerisation of lutein to 3’-epilutein in processed foods. Bioorg. Med. Chem. Lett. 2004, 14, 925–928. [Google Scholar] [CrossRef]
- Khachik, F.; Chang, A.-N.; Gana, A.; Mazzola, E. Partial synthesis of (3R,6’R)-α-cryptoxanthin from (3R,3’R,6’R)-lutein. J. Nat. Prod. 2007, 70, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Khachik, F. Partial synthesis of serum carotenoids and their metabolites. Acta Biochim. Polon. 2012, 59, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Health protective effects of carotenoids and their interactions with other biological antioxidants. Eur. J. Med. Chem. 2013, 70, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.V.; Rao, L.G. Carotenoids and Human Health. Pharmacol. Res. 2007, 55, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-H.; Ho, C.-T. Chemopreventive effects of natural dietary compounds on cancer development. Chem. Soc. Rev. 2008, 37, 2558–2574. [Google Scholar] [CrossRef]
- Panis, G.; Rosales, J. Commercial Astaxanthin production derived by green alga Haematococcus pluvialis: A microalgae process model and a technoeconomic assessment all through production line. Algal Res. 2016, 18, 175–190. [Google Scholar] [CrossRef]
- Liu, X.; Song, M.; Gao, Z.; Cai, X.; Dixon, W.; Chen, X.; Cao, Y.; Xiao, H. Stereoisomers of Astaxanthin inhibit Human colon cancer cell growth by inducing G2/M cell cycle arrest and apoptosis. J. Agric. Food Chem. 2016, 64, 7750–7759. [Google Scholar] [CrossRef] [PubMed]
- Nagendrapabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expression of ERK-2, NFkB and COX-2. Investig. New Drugs 2011, 29, 207–224. [Google Scholar] [CrossRef]
- Fasset, R.G.; Coombes, J.S. Astaxanthin: A potential Therapeutic Agent in Cardiovascular Disease. Mar. Drugs 2011, 9, 447–465. [Google Scholar] [CrossRef]
- Wang, J.-J.; Chen, Z.-Q.; Lu, W.-Q. Hypoglycemic effect of astaxanthin from shrimp waste in alloxan-induced diabetic mica. Med. Chem. Res. 2012, 21, 2363–2367. [Google Scholar] [CrossRef]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef]
- Preuss, H.G.; Echard, B.; Yamashita, E.; Perricone, N.V. High dose Astaxanthin lowers blood pressure and increases insulin sensitivity in rats: Are these effects interdependent? Int. J. Med. Sci. 2011, 8, 126–138. [Google Scholar] [CrossRef]
- Gross, G.J.; Lockwood, S.F. Cardioprotection and myocardial salvage by a disodium disuccinate Astaxanthin derivate (Cardax). Life Sci. 2004, 75, 215–224. [Google Scholar] [CrossRef]
- Gross, G.J.; Lockwood, S.F. Acute and chronic administration of disodium disuccinate astaxanthin (Cardax) produces marked cardioprotection in dog hearts. Mol. Cell. Biochem. 2005, 272, 221–227. [Google Scholar] [CrossRef]
- Lauver, D.A.; Lockwood, S.F.; Lucchesi, B.R. Disodium disuccinate Astaxanthin (Cardax) attenuates complement activation and reduces myocardial injury following ischemia/reperfusion. J. Pharmacol. Exp. Ther. 2005, 314, 686–692. [Google Scholar] [CrossRef]
- Widmer, V.E.; Zell, R.; Albin, E.; Crameri, Y.; Wagner, H.P.; Dinkel, J.; Schlageter, M.; Lukat, T. Technische Verfahren zur Synthese von Carotinoiden und verwandten Verbindungen aus 6-Oxo-isophoron. II. Ein neues Konzept für die synthese von (3RS,3’RS)-Astaxanthin. Helv. Chim. Acta 1981, 64, 2436–2446. [Google Scholar] [CrossRef]
- Schloemer, G.C.; Davis, J.L. Preparación de astaxantina. Patente ES2 312 433, 22 January 2003. [Google Scholar]
- Bernhard, K.; Miller, R.K.; Spruijtenburg, R. Cyclohexenone derivatives and process for making same. U.S. Patent 4,585,885, 29 April 1986. [Google Scholar]
- Chen, K.; Zhang, P.; Wang, Y.; Li, H. Metal free allylic/benzylic oxidation strategies with molecular oxygen: Recent advances and future prospects. Green Chem. 2014, 16, 2344–2374. [Google Scholar] [CrossRef]
- Ding, R.; Grant, J.L.; Metzger, R.M.; Kispert, L.D. Carotenoid cation radicals produced by the interaction of carotenoids with iodine. J. Phys. Chem. 1988, 92, 4600–4606. [Google Scholar] [CrossRef]
- Lutnaes, B.F.; Krane, J.; Liaaen-Jensen, S. On the structure of carotenoid iodine complexes. Org. Biomol. Chem. 2004, 2, 2821–2828. [Google Scholar] [CrossRef]
- Liaaen-Jensen, S. Studies on Allylic Oxidation of Compounds. Acta Chem. Scand. 1965, 19, 1166–1174. [Google Scholar] [CrossRef]
- Yuan, J.-P.; Chen, F. Isomerization of trans-astaxanthin to cis-astaxanthin in organic solvents. J. Agric. Food Chem. 1999, 47, 3656–3660. [Google Scholar] [CrossRef]
- Holtin, K.; Kuehnle, M.; Rehbein, J.; Schuler, P.; Nicholson, G.; Albert, K. Determination of Astaxanthin esters in the microalgae Haematococcus pluvialis by LC-(APCI)MS and characterization of predominant carotenoid isomers by NMR spectroscopy. Anal. Bioanal. Chem. 2009, 395, 1613–1622. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; Brittain, D.R.B.; Hodgson, J.L.; Krenske, E.H.; Lin, C.Y.; Namazian, M.; Coote, M.L. Should Contemporary Density Functional Theory Methods Be Used to Study the Thermodynamics of Radial Reactions? J. Phys. Chem. A 2007, 111, 10754–10768. [Google Scholar] [CrossRef]
- Otaka, J.; Seo, S.; Nishimura, M. Lutein, a natural carotenoid, induces α-1,3-glucan accumulation on the cell wall surface of fungal plant pathogens. Molecules 2016, 21, 980. [Google Scholar] [CrossRef]
- Englert, G.; Noack, K.; Broger, E.A.; Glinz, E.; Vecchi, M.; Zell, R. Synthesis, isolation, and full spectroscopic characterization of eleven Z-isomers of (3R,3’R)-zeaxanthin. Helv. Chim. Acta 1991, 74, 969–982. [Google Scholar] [CrossRef]
- Englert, G.; Kienzle, F.; Noack, K. 1H-NMR, 13C-NMR, UV, und CD. Daten von synthestichem (3S,3’S)-astaxanthin, seinem 15-cis-isomeren un einegen analogen verbindungen. Helv. Chim. Acta 1977, 60, 1209–1219. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and non-covalent interactions. J. Chem. Phys. 2006, 125, 1–18. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. A new definition of cavities for the computation of solvatation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Case, B.; Parson, R. The real energy free of solvation of ions in some non-aqueous and mixed solvents. Trans. Faraday Soc. 1967, 63, 1224–1239. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
Sample Availability: All compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Base | Solvent | Temperature | Time (Hours) | Yield (%) |
|---|---|---|---|---|---|
| 1 | KOH | MeOH | Reflux | 12 | 65 |
| 2 | KOH | EtOH | Reflux | 12 | 60 |
| 3 | KOH | Water | Reflux | 16 | nr |
| 4 | KOH | n-BuOH | 115 | 12 | 92 |
| 5 | KOH | PG | 115 | 12 | 62 |
| 6 | NaOH | n-BuOH | 115 | 12 | 85 |
| 7 | DBU | n-BuOH | 100 | 20 | nr |
| 8 | TEA | n-BuOH | 100 | 24 | nr |
| 9 | a K2CO3 | n-BuOH | 115 | 12 | 22 |
| 10 | DIPEA | n-BuOH | 115 | 24 | nr |
| 11 | t-BuOK | n-BuOH | 115 | 20 | - b |

| Entry | Base | Solvent | Time(Hours) | Yield (%) |
|---|---|---|---|---|
| 1 | KOH | n-BuOH | 12 | 86 |
| 2 | KOH | n-BuOH | 16 | 95 |
| 3 | KOH | PG | 16 | 72 |

| DFT Method | 6-311 + G(2d) | 6-31 + G(d) | Def2-svp | |
|---|---|---|---|---|
| 1 | M06L | 4.75 | 4.92 | 4.80 |
| 2 | cam-b3lyp | 4.87 | 4.90 | 4.90 |
| 3 | ωB97XD | 4.89 | 5.02 | 4.94 |

| Entry | I2 % mol | Solvent | Time Hours | Yield (%) |
|---|---|---|---|---|
| 1 | 20 | CH2Cl2 | 4 | 46 |
| 2 | 10 | CH2Cl2 | 4 | 52 |
| 3 | 10 | CH2Cl2 | 2.5 | 72 |
| 4 | 5 | CH2Cl2 | 2.5 | 55 |
| 5 | 2 | CH2Cl2 | 2.5 | 60 |
| 6 | 10 | CH2Cl2 | 2.5 | 80 a |
| 7 | 10 | acetone | 6 | nr b |
| 8 | 10 | water | 6 | nr b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-deLeón, E.; Jiménez-Halla, J.O.C.; Báez, J.E.; Bah, M.M. A Simple and Efficient Method for the Partial Synthesis of Pure (3R,3’S)-Astaxanthin from (3R,3’R,6’R)-Lutein and Lutein Esters via (3R,3’S)-Zeaxanthin and Theoretical Study of Their Formation Mechanisms. Molecules 2019, 24, 1386. https://doi.org/10.3390/molecules24071386
Rodríguez-deLeón E, Jiménez-Halla JOC, Báez JE, Bah MM. A Simple and Efficient Method for the Partial Synthesis of Pure (3R,3’S)-Astaxanthin from (3R,3’R,6’R)-Lutein and Lutein Esters via (3R,3’S)-Zeaxanthin and Theoretical Study of Their Formation Mechanisms. Molecules. 2019; 24(7):1386. https://doi.org/10.3390/molecules24071386
Chicago/Turabian StyleRodríguez-deLeón, Eloy, J. Oscar. C. Jiménez-Halla, José E. Báez, and M. Moustapha Bah. 2019. "A Simple and Efficient Method for the Partial Synthesis of Pure (3R,3’S)-Astaxanthin from (3R,3’R,6’R)-Lutein and Lutein Esters via (3R,3’S)-Zeaxanthin and Theoretical Study of Their Formation Mechanisms" Molecules 24, no. 7: 1386. https://doi.org/10.3390/molecules24071386
APA StyleRodríguez-deLeón, E., Jiménez-Halla, J. O. C., Báez, J. E., & Bah, M. M. (2019). A Simple and Efficient Method for the Partial Synthesis of Pure (3R,3’S)-Astaxanthin from (3R,3’R,6’R)-Lutein and Lutein Esters via (3R,3’S)-Zeaxanthin and Theoretical Study of Their Formation Mechanisms. Molecules, 24(7), 1386. https://doi.org/10.3390/molecules24071386