Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
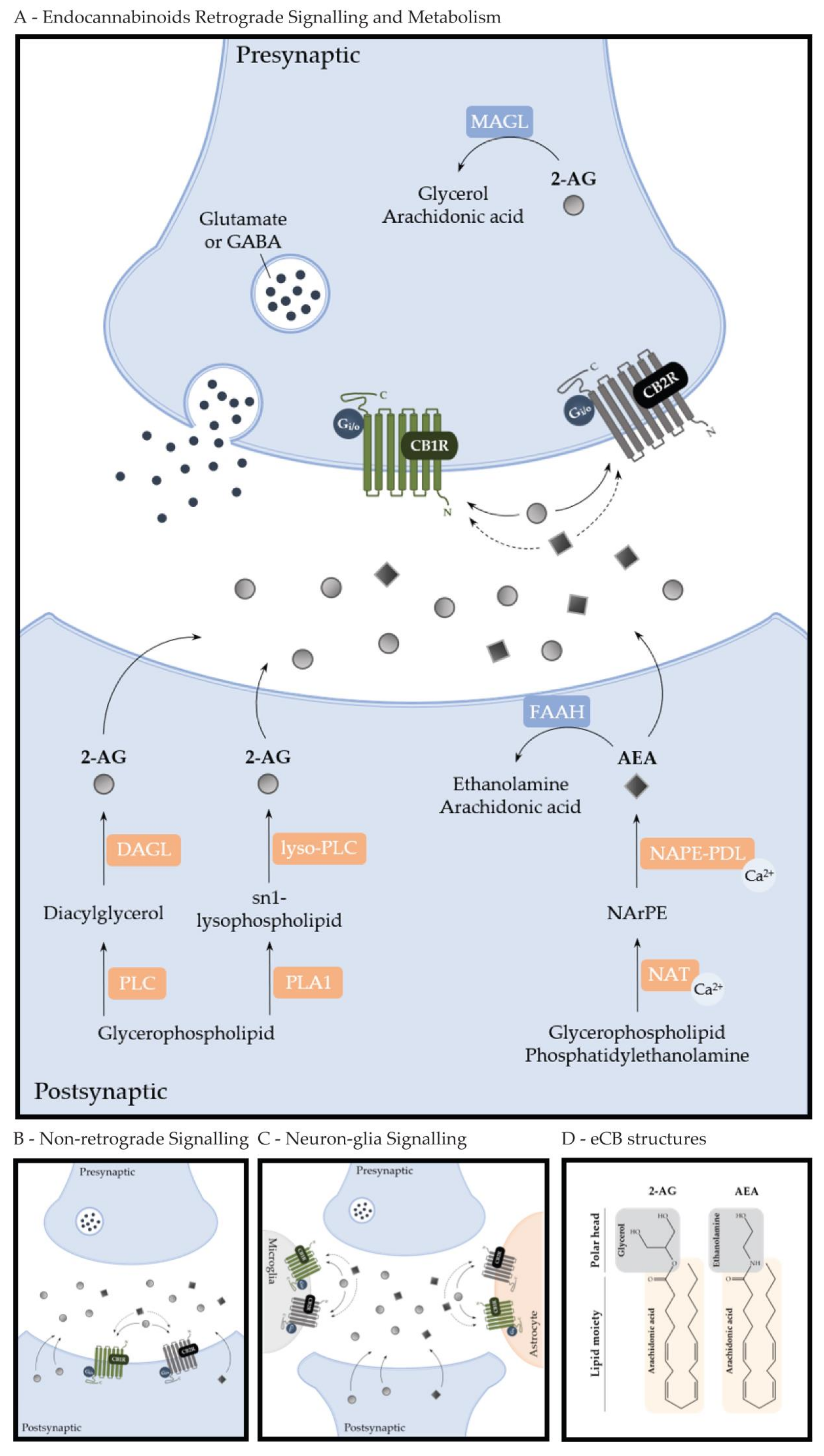
2. Endocannabinoid System and Cannabinoids
2.1. Endocannabinoid System
2.2. Cannabinoid Pharmacology and Actions
3. Neurogenesis
3.1. Neurodevelopmental Neurogenesis
3.1.1. Neurulation
3.1.2. Embryonic Neurogenesis
3.2. Adult Neurogenesis
3.2.1. Subventricular Zone
3.2.2. Subgranular Zone
3.3. Regulators of Neurogenesis
3.3.1. Morphogens and Growth Factors
3.3.2. Neurotrophins
3.3.3. Cytokines
3.3.4. Neurotransmitters
3.3.5. Extracellular Matrix
3.3.6. Cell-cell Signaling Molecules
3.3.7. Systemic Factors
4. Cannabinoid Regulation of Neurogenesis
4.1. Cannabinoid Actions in Embryonic Neurogenesis
4.2. Cannabinoid Actions in Postnatal Neurogenesis
5. Role of Cannabinoids in Neurogenesis and Pathophysiology
5.1. Cannabinoids and Neuroprotection
5.2. Cannabinoids and Brain Disorders
5.2.1. Alzheimer’s Disease
5.2.2. Parkinson’s Disease
5.2.3. Multiple Sclerosis
5.2.4. Epilepsy
5.2.5. Anxiety/Depression
6. Perspectives and Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| (N-acyl-phosphatidylethanolamine)-specific phospholipase D | NAPE-PLD |
| 2-arachidonoglycerol | 2-AG |
| 6-hydroxydopamine | 6-OHDA |
| Acetylcholinesterase | AChE |
| Adult hippocampal neurogenesis | AHN |
| Alpha-synuclein | αSyn |
| Alzheimer’s disease | AD |
| Amyloid precursor protein/presenilin 1 | APP/PS1 |
| Amyloid-β | Aβ |
| Anandamide | AEA |
| Brain-derived neurotrophic factor | BDNF |
| Cajal-Retzius cell | CRC |
| Calcium-dependent trans-acylase | NAT |
| Cannabichromene | CBC |
| Cannabidiol | CBD |
| Cannabidivarin | CBDV |
| Cannabigerol | CBG |
| Cannabinoid receptor type 1 | CB1R |
| Cannabinoid receptor type 2 | CB2R |
| Cannabinol | CBN |
| Cannabivarin | CBV |
| CB1R interacting protein 1a | CRIP1a |
| Central nervous system | CNS |
| Chronic mild stress | CMS |
| Cornu Ammonis 3 | CA3 |
| Dentate Gyrus | DG |
| Depolarization-induced suppression of excitation | DSE |
| Depolarization-induced suppression of inhibition | DSI |
| Diacylglycerol lipase | DAGL |
| Dopaminergic | DA |
| Doublecortin | DCX |
| Endocannabinoid membrane transporter | EMT |
| Endocannabinoid system | ECS |
| Endocannabinoids | eCBs |
| Epidermal growth factor | EGF |
| Experimental autoimmune encephalomyelitis | EAE |
| Fatty acid amide hydrolase | FAAH |
| Fibroblast growth factor | FGF |
| G protein-coupled receptors | GPCR |
| Glial fibrillary acidic protein | GFAP |
| Glycogen synthase kinase 3β | GSK-3β |
| Granule cell layer | GCL |
| Granule cell | GC |
| Human immunodeficiency virus infection and acquired immune deficiency syndrome | HIV/AIDS |
| Interferon-γ | IFN-γ |
| Interleukin 1 | IL-1 |
| Intermediate progenitor cells | IPC |
| Long-term depression | LTD |
| Molecular layer | ML |
| Monoacylglycerol lipase | MAGL |
| Multiple sclerosis | MS |
| N-arachidonoyl-phosphatidyl ethanolamine | NArPE |
| Nerve growth factor | NGF |
| Neural stem cells | NCS |
| Neuroepithelial progenitor cells | NEC |
| Neurotrophin-3 | NT-3 |
| Olfactory bulb | OB |
| Oligodendrocyte progenitor cells | OPC |
| Parkinson’s disease | PD |
| Peripheral nervous system | PNS |
| Peroxisome proliferator-activated receptor | PPAR |
| Phosphatidylinositol 3-kinase | PI3K |
| Phospholipase A1 | PLA1 |
| Phospholipase C | PLC |
| Polysialic acid neural cell adhesion molecule | PSA-NCAM |
| Preplate | PP |
| Protein kinase C | PKC |
| Radial glial cell | RGC |
| Reactive oxygen species | ROS |
| Rostral migratory stream | RMS |
| Short-term depression | STD |
| Sonic hedgehog | shh |
| Striatum | STR |
| Subgranular Zone | SGZ |
| Substantia nigra pars compacta | SN |
| Subventricular Zone | SVZ |
| Theiler murine encephalomyelitis virus-induced demyelinating disease | TMEV-IDD |
| Transient receptor potential vanilloid receptor type 1 | TRPV1R |
| Tumor necrosis factor α | TNF-α |
| Ventricular zone | VZ |
| β-caryophyllene | BCP |
| Δ8-tetrahydrocannabinol | Δ8-THC |
| Δ9-tetrahydrocannabinol | Δ9-THC |
| Δ9-tetrahydrocannabivarin | THCV |
References
- Solymosi, K.; Köfalvi, A. Cannabis: A treasure trove or pandora’s box? Mini-Rev. Med. Chem. 2017, 17, 1–70. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.-M.K.; Downer, E.J. Toll-like receptor signalling as a cannabinoid target in Multiple Sclerosis. Neuropharmacology 2017, 113, 618–626. [Google Scholar] [CrossRef]
- Rice, J.; Cameron, M. Cannabinoids for treatment of MS symptoms: State of the evidence. Curr. Neurol. Neurosci. Rep. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Maa, E.; Figi, P. The case for medical marijuana in epilepsy. Epilepsia 2014, 55, 783–786. [Google Scholar] [CrossRef]
- Pamplona, F.A.; Coan, A.C. Potential clinical benefits of CBD-rich Cannabis extracts over purified CBD in treatment-resistant epilepsy: Observational data meta-analysis. Front. Neurol. 2017. [Google Scholar] [CrossRef]
- Fagan, S.G.; Campbell, V.A. The influence of cannabinoids on generic traits of neurodegeneration. Br. J. Pharmacol. 2014, 171, 1347–1360. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. Brain Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Carter, G.T.; Javaher, S.P.; Nguyen, M.H.; Garret, S.; Carlini, B.H. Re-branding cannabis: The next generation of chronic pain medicine? Pain Manag. 2015, 5, 13–21. [Google Scholar] [CrossRef]
- Pascual, D.; Sánchez-Robles, E.M.; García, M.M.; Goicoechea, C. Chronic pain and cannabinoids. Great expectations or a christmas carol. Biochem. Pharmacol. 2018, 157, 33–42. [Google Scholar] [CrossRef]
- Abush, H.; Akirav, I. Short- and long-term cognitive effects of chronic cannabinoids administration in late-adolescence rats. PLoS ONE 2012, 7, e31731. [Google Scholar] [CrossRef] [PubMed]
- Borgelt, L.M.; Franson, K.L.; Nussbaum, A.M.; Wang, G.S. The pharmacologic and clinical effects of medical cannabis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2013, 33, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, S.; Engström, A.; Allebeck, P.; Rydberg, U. Cannabis and schizophrenia: A longitudinal study of swedish conscripts. Lancet 1987, 330, 1483–1486. [Google Scholar] [CrossRef]
- Hall, W.; Degenhardt, L. Adverse health effects of non-medical cannabis use. Lancet 2009, 374, 9. [Google Scholar] [CrossRef]
- Khan, M.A.; Akella, S. Cannabis-induced bipolar disorder with psychotic features: A case report. Psychiatry (Edgmont) 2009, 6, 44–48. [Google Scholar]
- Lichtman, A.H.; Martin, B.R. Cannabinoid tolerance and dependence. In Cannabinoids; Pertwee, R.G., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2005; Volume 168, pp. 691–717. ISBN 978-3-540-22565-2. [Google Scholar]
- Mouro, F.M.; Ribeiro, J.A.; Sebastião, A.M.; Dawson, N. Chronic, intermittent treatment with a cannabinoid receptor agonist impairs recognition memory and brain network functional connectivity. J. Neurochem. 2018, 147, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mouro, F.M.; Batalha, V.L.; Ferreira, D.G.; Coelho, J.E.; Baqi, Y.; Müller, C.E.; Lopes, L.V.; Ribeiro, J.A.; Sebastião, A.M. Chronic and acute adenosine A2A receptor blockade prevents long-term episodic memory disruption caused by acute cannabinoid CB1 receptor activation. Neuropharmacology 2017, 117, 316–327. [Google Scholar] [CrossRef] [PubMed]
- United Nations Office on Drugs and Crime World Drug Report 2017. In SIRIUS – Zeitschrift für Strategische Analysen; United Nations publication: Berlin/Boston, 2017; ISBN 978-92-1-148291-1.
- Adams, I.B.; Martin, B.R. Cannabis: Pharmacology and toxicology in animals and humans. Addiction 1996, 91, 1585–1614. [Google Scholar] [CrossRef] [PubMed]
- Gaoni, Y.; Mechoulam, R. Isolation, structure and partial synthesis of anactive constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Russo, E.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular targets of the phytocannabinoids: A complex picture. In Phytocannabinoids; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Vol. 103, pp. 103–131. ISBN 978-3-319-45539-6. [Google Scholar]
- Fride, E. Endocannabinoids in the central nervous system--an overview. Prostaglandins. Leukot. Essent. Fatty Acids 2002, 66, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Elphick, M.R.; Egertova, M. The neurobiology and evolution of cannabinoid signalling. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2001, 356, 381–408. [Google Scholar] [CrossRef] [PubMed]
- Salzet, M.; Breton, C.; Bisogno, T.; Di Marzo, V. Comparative biology of the endocannabinoid system: Possible role in the immune response. Eur. J. Biochem. 2000, 267, 4917–4927. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 3662–3665. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef]
- Maccarrone, M. The endocannabinoid system and its manifold central actions. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids; Lajtha, A., Tettamanti, G., Goracci, G., Eds.; Springer US: Boston, MA, USA, 2009; pp. 385–405. ISBN 978-0-387-30378-9. [Google Scholar]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Ross, R.A. Cannabinoid receptors and their ligands. Prostaglandins, Leukot. Essent. Fat. Acids 2002, 66, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Breivogel, C.S.; Childers, S.R.; Deadwyler, S.A.; Hampson, R.E.; Porrino, L.J. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology 2004, 47 (Suppl. 1), 345–358. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Regehr, W.G. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J. Neurosci. 2001, 21, 174. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Huestis, M.A.; Boyd, S.J.; Heishman, S.J.; Preston, K.L.; Bonnet, D.; Le Fur, G.; Gorelick, D.A. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology 2007, 194, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 1998, 82, 1131–1149. [Google Scholar] [CrossRef]
- Romero, J.; Garcia-Palomero, E.; Berrendero, F.; Garcia-Gil, L.; Hernandez, M.L.; Ramos, J.A.; Fernández-Ruiz, J.J. Atypical location of cannabinoid receptors in white matter areas during rat brain development. Synapse 1997, 26, 317–323. [Google Scholar] [CrossRef]
- Vendel, E.; de Lange, E.C.M. Functions of the CB1 and CB2 receptors in neuroprotection at the level of the blood–brain barrier. Neuromol. Med. 2014, 16, 620–642. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 receptor signaling in the brain: Extracting specificity from ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-R.; Pan, C.-H.; Hishimoto, A.; Li, C.-Y.; Xi, Z.-X.; Llorente-Berzal, A.; Viveros, M.-P.; Ishiguro, H.; Arinami, T.; Onaivi, E.S.; et al. Species differences in cannabinoid receptor 2 (CNR2 gene): Identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, S.; Marciano-Cabral, F.; Staab, A.; Ludwick, C.; Cabral, G. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2002, 2, 69–82. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Min, X.; Cabral, G.A.; Lokensgard, J.R.; Peterson, P.K. Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1-stimulated human astrocytes. Glia 2005, 49, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Savonenko, A.V.; Melnikova, T.; Wang, Y.; Ravert, H.; Gao, Y.; Koppel, J.; Lee, D.; Pletnikova, O.; Cho, E.; Sayyida, N.; et al. Cannabinoid CB2 receptors in a mouse model of Aβ amyloidosis: Immunohistochemical analysis and suitability as a PET biomarker of neuroinflammation. PLoS ONE 2015, 10, e0129618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Gao, M.; Liu, Q.-R.; Bi, G.-H.; Li, X.; Yang, H.-J.; Gardner, E.L.; Wu, J.; Xi, Z.-X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 5007–5015. [Google Scholar] [CrossRef]
- Van Sickle, M.D. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kim, J. Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience 2015, 311, 253–267. [Google Scholar] [CrossRef]
- Andó, R.D.; Bíró, J.; Csölle, C.; Ledent, C.; Sperlágh, B. The inhibitory action of exo- and endocannabinoids on [3H]GABA release are mediated by both CB1 and CB2 receptors in the mouse hippocampus. Neurochem. Int. 2012, 60, 145–152. [Google Scholar] [CrossRef]
- Köfalvi, A.; Lemos, C.; Martín-Moreno, A.M.; Pinheiro, B.S.; García-García, L.; Pozo, M.A.; Valério-Fernandes, Â.; Beleza, R.O.; Agostinho, P.; Rodrigues, R.J.; et al. Stimulation of brain glucose uptake by cannabinoid CB2 receptors and its therapeutic potential in Alzheimer’s disease. Neuropharmacology 2016, 110, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2015, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gu, S.; Liu, Q.-R. CNS effects of CB2 cannabinoid receptors: Beyond neuro-immuno-cannabinoid activity. J. Psychopharmacol. 2012, 26, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Moro, M.A.; Martínez-Orgado, J. Cannabinoids in neurodegenerative disorders and stroke/brain trauma: From preclinical models to clinical applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Cannabinoid receptor homo- and heterodimerization. Life Sci. 2005, 77, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.; Rice, A.S.C.; Egertová, M.; Elphick, M.R.; Winter, J.; Michael, G.J. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience 2003, 119, 803–812. [Google Scholar] [CrossRef]
- Hermann, H.; Marsicano, G.; Lutz, B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 2002, 109, 451–460. [Google Scholar] [CrossRef]
- Sjöström, P.J.; Turrigiano, G.G.; Nelson, S.B. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 2003, 39, 641–654. [Google Scholar] [CrossRef]
- Loewe, S. Studies on the pharmacology and acute toxicity of compounds with marihuana activity. J. Pharmacol. Exp. Ther. 1946, 88, 154–161. [Google Scholar]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147, 163–171. [Google Scholar] [CrossRef]
- Petitet, F.; Jeantaud, B.; Reibaud, M.; Imperato, A.; Dubroeucq, M.-C. Complex pharmacology of natural cannabivoids: Evidence for partial agonist activity of Δ9-tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci. 1998, 63, 1–6. [Google Scholar] [CrossRef]
- Strougo, A.; Zuurman, L.; Roy, C.; Pinquier, J.; van Gerven, J.; Cohen, A.; Schoemaker, R. Modelling of the concentration—Effect relationship of THC on central nervous system parameters and heart rate—Insight into its mechanisms of action and a tool for clinical research and development of cannabinoids. J. Psychopharmacol. 2008, 22, 717–726. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M.; Glass, M.; Pertwee, R.G. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: Interspecies differences. Br. J. Pharmacol. 2007, 152, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2009, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.A. 9-Tetrahydrocannbinol accounts for the antinociceptive, hypothermic, and cataleptic effects of marijuana in mice. J. Pharmacol. Exp. Ther. 2005, 314, 329–337. [Google Scholar] [CrossRef]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro: Cannabinoid antagonism by cannabidiol. Br. J. Pharmacol. 2009, 150, 613–623. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor: Negative allosteric modulation of CB1 by cannabidiol. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- McHugh, D.; Page, J.; Dunn, E.; Bradshaw, H.B. Δ9-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells: Novel CB pharmacology at GPR18. Br. J. Pharmacol. 2012, 165, 2414–2424. [Google Scholar] [CrossRef]
- McHugh, D.; Roskowski, D.; Xie, S.; Bradshaw, H.B. Δ9-THC and N-arachidonoyl glycine regulate BV-2 microglial morphology and cytokine release plasticity: Implications for signaling at GPR18. Front. Pharmacol. 2014, 4. [Google Scholar] [CrossRef]
- Gonca, E.; Darıcı, F. The effect of cannabidiol on ischemia/reperfusion-induced ventricular arrhythmias: The role of adenosine A1 receptors. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Brivanlou, A.H. Proposal of a model of mammalian neural induction. Dev. Biol. 2007, 308, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, L. Chapter 12: Development of the nervous system. In Principles of development; Oxford University Press: Oxford, UK, 2011; pp. 468–504. ISBN 0-19-954907-9. [Google Scholar]
- Wilson, L.; Maden, M. The mechanisms of dorsoventral patterning in the vertebrate neural tube. Dev. Biol. 2005, 282, 1–13. [Google Scholar] [CrossRef]
- Yamaguchi, T.P. Heads or tails: Wnts and anterior-posterior patterning. Curr. Biol. 2001, 11, 713–724. [Google Scholar] [CrossRef]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Baye, L.M.; Link, B.A. Interkinetic nuclear migration and the selection of neurogenic cell divisions during vertebrate retinogenesis. J. Neurosci. 2007, 27, 10143–10152. [Google Scholar] [CrossRef] [PubMed]
- Huttner, W.B.; Brand, M. Asymmetric division and polarity of neuroepithelial cells. Curr. Opin. Neurobiol. 1997, 7, 29–39. [Google Scholar] [CrossRef]
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013, 14, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Homem, C.C.F.; Repic, M.; Knoblich, J.A. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015, 16, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008, 31, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.L.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Budday, S.; Steinmann, P.; Kuhl, E. Physical biology of human brain development. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Mountcastle, V.B. The columnar organization of the neocortex. Brain 1997, 120, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Florio, M.; Huttner, W.B. Neural progenitors, neurogenesis and the evolution of the neocortex. Development 2014, 141, 2182–2194. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Mitra, P.P.; Wang, S.S.-H. Scalable architecture in mammalian brains. Nature 2001, 411, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Tallinen, T.; Chung, J.Y.; Rousseau, F.; Girard, N.; Lefèvre, J.; Mahadevan, L. On the growth and form of cortical convolutions. Nat. Phys. 2016, 12, 588. [Google Scholar] [CrossRef]
- Dehay, C.; Kennedy, H.; Kosik, K.S. The outer subventricular zone and primate-specific cortical complexification. Neuron 2015, 85, 683–694. [Google Scholar] [CrossRef]
- Smart, I.H.M.; Dehay, C.; Giroud, P.; Berland, M.; Kennedy, H. Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb. Cortex 2002, 12, 37–53. [Google Scholar] [CrossRef]
- Farnsworth, D.R.; Doe, C.Q. Opportunities lost and gained: Changes in progenitor competence during nervous system development. Neurogenesis 2017, 4, e1324260. [Google Scholar] [CrossRef]
- Kriegstein, A.R.; Noctor, S.C. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004, 27, 392–399. [Google Scholar] [CrossRef]
- Marín, O.; Rubenstein, J.L.R. Cell migration in the forebrain. Annu. Rev. Neurosci. 2003, 26, 441–483. [Google Scholar] [CrossRef] [PubMed]
- Cavallucci, V.; Fidaleo, M.; Pani, G. Neural stem cells and nutrients: Poised between quiescence and exhaustion. Trends Endocrinol. Metab. 2016, 27, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Paredes, M.F.; Sorrells, S.F.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Brain size and limits to adult neurogenesis. J. Comp. Neurol. 2016, 524, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Inta, D.; Lang, U.E.; Borgwardt, S.; Meyer-Lindenberg, A.; Gass, P. Adult neurogenesis in the human striatum: Possible implications for psychiatric disorders. Mol. Psychiatry 2016, 21, 446–447. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Frisén, J. Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol. 2015, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Ming, G.L.; Song, H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.-M.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Chaker, Z.; Codega, P.; Doetsch, F. A mosaic world: Puzzles revealed by adult neural stem cell heterogeneity. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 640–658. [Google Scholar] [CrossRef]
- Doetsch, F.; Caillé, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem Cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Conover, J.C.; Todd, K.L. Development and aging of a brain neural stem cell niche. Exp. Gerontol. 2017, 94, 9–13. [Google Scholar] [CrossRef]
- Yabut, O.R.; Pleasure, S.J. The crossroads of neural stem cell development and tumorigenesis. Opera Medica Physiol. 2016, 2, 181–187. [Google Scholar]
- Zhao, C.; Teng, E.M.; Summers, R.G.; Ming, G.-L.; Gage, F.H. Distinct Morphological Stages of Dentate Granule Neuron Maturation in the Adult Mouse Hippocampus. J. Neurosci. 2006, 26, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Altman, J. Autoradiographic investigation of cell proliferation in the brains of rats and cats. Anat. Rec. 1963, 145, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.A.; Nottebohm, F. Neuronal production, migration, and differentiation in a vocal control nucleus of the adult female canary brain. Proc. Natl. Acad. Sci. USA 1983, 80, 2390–2394. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; McEwen, B.S.; Tanapat, P.; Galea, L.A.; Fuchs, E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 1997, 17, 2492–2498. [Google Scholar] [CrossRef] [PubMed]
- Bunk, E.C.; Stelzer, S.; Hermann, S.; Schäfers, M.; Schlatt, S.; Schwamborn, J.C. Cellular organization of adult neurogenesis in the Common Marmoset. Aging Cell 2011, 10, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Reeves, A.J.; Fallah, M.; Tanapat, P.; Gross, C.G.; Fuchs, E. Hippocampal neurogenesis in adult Old World primates. Proc. Natl. Acad. Sci. USA 1999, 96, 5263–5267. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 2018, 22, 589–599. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Lucassen, P.J.; Toni, N.; Kempermann, G.; Frisen, J.; Gage, F.H.; Swaab, D.F. Limits to human neurogenesis—Really? Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Steiner, E.; Tata, M.; Frisén, J. A fresh look at adult neurogenesis. Nat. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Boekhoorn, K.; Joels, M.; Lucassen, P.J. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis. 2006, 24, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, C.S.; Apostolopoulou, M.; Tian, Y.; Temple, S. It takes a village: Constructing the neurogenic niche. Dev. Cell 2015, 32, 435–446. [Google Scholar] [CrossRef]
- Lledo, P.-M.; Alonso, M.; Grubb, M.S. Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 2006, 7, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, F.; García-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. 1997, 17, 5046–5061. [Google Scholar] [CrossRef] [PubMed]
- Lledo, P.-M.; Merkle, F.T.; Alvarez-Buylla, A. Origin and function of olfactory bulb interneuron diversity. Trends Neurosci. 2008, 31, 392–400. [Google Scholar] [CrossRef]
- Lledo, P.-M.; Valley, M. Adult olfactory bulb neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018945. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Bonaguidi, M.A.; Ming, G.-L.; Song, H. Adult neural stem cells in the mammalian central nervous system. Cell Res. 2009, 19, 672–682. [Google Scholar] [CrossRef]
- Chang, E.H.; Adorjan, I.; Mundim, M.V.; Sun, B.; Dizon, M.L.V.; Szele, F.G. Traumatic brain injury activation of the adult subventricular zone neurogenic niche. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Gonzalez-Perez, O. Neural stem cells in the adult human brain. Biol. Biomed. Rep. 2012, 2, 59–69. [Google Scholar] [PubMed]
- Quiñones-Hinojosa, A.; Sanai, N.; Soriano-Navarro, M.; Gonzalez-Perez, O.; Mirzadeh, Z.; Gil-Perotin, S.; Romero-Rodriguez, R.; Berger, M.S.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and cytoarchitecture of the adult human subventricular zone: A niche of neural stem cells. J. Comp. Neurol. 2006, 494, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.A.; Kam, M.; Faull, R.L.M. Neurogenesis in humans. Eur. J. Neurosci. 2011, 33, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Pencea, V.; Bingaman, K.D.; Freedman, L.J.; Luskin, M.B. Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp. Neurol. 2001, 172, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Peretto, P.; Merighi, A.; Fasolo, A.; Bonfanti, L. Glial tubes in the rostral migratory stream of the adult rat. Brain Res. Bull. 1997, 42, 9–21. [Google Scholar] [CrossRef]
- Kam, M.; Curtis, M.A.; McGlashan, S.R.; Connor, B.; Nannmark, U.; Faull, R.L.M. The cellular composition and morphological organization of the rostral migratory stream in the adult human brain. J. Chem. Neuroanat. 2009, 37, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Alkass, K.; Bernard, S.; Salehpour, M.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; Frisén, J. Neurogenesis in the striatum of the adult human brain. Cell 2014, 156, 1072–1083. [Google Scholar] [CrossRef]
- Benarroch, E.E. Adult neurogenesis in the dentate gyrus: General concepts and potential implications. Neurology 2013, 81, 1443–1452. [Google Scholar] [CrossRef]
- Piatti, V.C.; Ewell, L.A.; Leutgeb, J.K. Neurogenesis in the dentate gyrus: Carrying the message or dictating the tone. Front. Neurosci. 2013, 7. [Google Scholar] [CrossRef]
- Wu, M.V.; Sahay, A.; Duman, R.S.; Hen, R. Functional differentiation of adult-born neurons along the septotemporal axis of the dentate gyrus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018978. [Google Scholar] [CrossRef]
- Berg, D.A.; Bond, A.M.; Ming, G.; Song, H. Radial glial cells in the adult dentate gyrus: What are they and where do they come from? F1000Research 2018, 7, 277. [Google Scholar] [CrossRef] [PubMed]
- Hevner, R.F. Evolution of the mammalian dentate gyrus: Evolution of the mammalian dentate gyrus. J. Comp. Neurol. 2016, 524, 578–594. [Google Scholar] [CrossRef]
- Vivar, C.; van Praag, H. Functional circuits of new neurons in the dentate gyrus. Front. Neural Circuits 2013, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Skilling, Q.; Roach, J.; Althaus, A.L.; Murphy, G.G.; Sander, L.; Zochowski, M. Modifications in network structure and excitability may drive differential activity-dependent integration of granule cells into dentate gyrus circuits during normal and pathological adult neurogenesis. In The Rewiring Brain: A Computational Approach to Structural Plasticity in the Adult Brain; Academic Press: New York, NY, USA, 2017; pp. 409–423. ISBN 978-0-12-803872-7. [Google Scholar]
- Cahill, S.P.; Yu, R.Q.; Green, D.; Todorova, E.V.; Snyder, J.S. Early survival and delayed death of developmentally-born dentate gyrus neurons. Hippocampus 2017, 27, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Hanson, N.D.; Owens, M.J.; Nemeroff, C.B. Depression, antidepressants and neurogenesis: A critical reappraisal. Neuropsychopharmacology 2011, 36, 2589–2602. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Maletic-Savatic, M. Adult human neurogenesis: From microscopy to magnetic resonance imaging. Front. Neurosci. 2011, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.G.; Toda, T.; Gage, F.H. Adult hippocampal neurogenesis: A coming-of-age story. J. Neurosci. 2018, 38, 10401–10410. [Google Scholar] [CrossRef]
- Tiberi, L.; Vanderhaeghen, P.; van den Ameele, J. Cortical neurogenesis and morphogens: Diversity of cues, sources and functions. Curr. Opin. Cell Biol. 2012, 24, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef]
- Lee, E.-G.; Son, H. Adult hippocampal neurogenesis and related neurotrophic factors. BMB Rep. 2009, 42, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Borsini, A.; Zunszain, P.A.; Thuret, S.; Pariante, C.M. The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 2015, 38, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Platel, J.-C.; Stamboulian, S.; Nguyen, I.; Bordey, A. Neurotransmitter signaling in postnatal neurogenesis: The first leg. Brain Res. Rev. 2010, 63, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Streuli, C. Extracellular matrix remodelling and cellular differentiation. Curr. Opin. Cell Biol. 1999, 11, 634–640. [Google Scholar] [CrossRef]
- Villeda, S.A.; Wyss-Coray, T. The circulatory systemic environment as a modulator of neurogenesis and brain aging. Autoimmun. Rev. 2013, 12, 674–677. [Google Scholar] [CrossRef]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cellcycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, C.N.; Mutch, C.A.; Swaminathan, S.; Taketo, M.M.; Chenn, A. Persistent expression of stabilized β-catenin delays maturation of radial glial cells into intermediate progenitors. Dev. Biol. 2007, 309, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Hébert, J.M.; Mishina, Y.; McConnell, S.K. BMP signaling is required locally to pattern the dorsal telencephalic midline. Neuron 2002, 35, 1029–1041. [Google Scholar] [CrossRef]
- Furuta, Y.; Piston, D.W.; Hogan, B.L. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 1997, 124, 2203–2212. [Google Scholar] [PubMed]
- Poo, M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.-S.; Li, W.-P.; Yao, Z.-B.; Zhou, X.-F. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 1999, 835, 259–265. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J. Upregulation of the transport system for TNFa at the blood-brain barrier. Arch. Physiol. Biochem. 2001, 109, 350–353. [Google Scholar] [CrossRef] [PubMed]
- McAfoose, J.; Baune, B.T. Evidence for a cytokine model of cognitive function. Neurosci. Biobehav. Rev. 2009, 33, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Finch, C.E.; Cohen, H.J. Cytokines and cognition - the case for a head-to-toe inflammatory paradigm. J. Am. Geriatr. Soc. 2002, 50, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Rizk, P.; Muriel, M.P.; Duyckaerts, C.; Oertel, W.H.; Caille, I.; Hirsch, E.C. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 2004, 7, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Banasr, M.; Hery, M.; Printemps, R.; Daszuta, A. Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology 2004, 29, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Dayer, A.G.; Sauvain, M.-O.; Potter, G.; Jenny, B.; De Roo, M.; Zgraggen, E.; Demaurex, N.; Muller, D.; Kiss, J.Z. GABA regulates dendritic growth by stabilizing lamellipodia in newly generated interneurons of the olfactory bulb. J. Neurosci. 2006, 26, 12956–12966. [Google Scholar] [CrossRef] [PubMed]
- Schlett, K. Glutamate as a modulator of embryonic and adult neurogenesis. Curr. Top. Med. Chem. 2006, 6, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; Baron, V. Interactions between mitogenic stimuli, or, a thousand and one connections. Curr. Opin. Cell Biol. 1999, 11, 197–202. [Google Scholar] [CrossRef]
- Aaku-Saraste, E.; Hellwig, A.; Huttner, W.B. Loss of occludin and functional tight junctions, but not ZO-1, during neural tube closure—remodeling of the neuroepithelium prior to neurogenesis. Dev. Biol. 1996, 180, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Niwa, A.; Nishibori, M.; Hamasaki, S.; Kobori, T.; Liu, K.; Wake, H.; Mori, S.; Yoshino, T.; Takahashi, H. Voluntary exercise induces neurogenesis in the hypothalamus and ependymal lining of the third ventricle. Brain Struct. Funct. 2016, 221, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.J.; Cameron, H.A. Adult neurogenesis and mental illness. Neuropsychopharmacology 2014, 40, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Xie, L.; Mao, X.O.; Greenberg, D.A. Alzheimer’s disease drugs promote neurogenesis. Brain Res. 2006, 1085, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Berrendero, F.; Sepe, N.; Ramos, J.A.; Di Marzo, V.; Fernández-Ruiz, J.J. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse 1999, 33, 181–191. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Berrendero, F.; Hernández, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef]
- Jiang, S.; Fu, Y.; Williams, J.; Wood, J.; Pandarinathan, L.; Avraham, S.; Makriyannis, A.; Avraham, S.; Avraham, H.K. Expression and function of cannabinoid receptors CB1 and CB2 and their cognate cannabinoid ligands in murine embryonic stem cells. PLoS ONE 2007, 2, e641. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Aguado, T.; Egia, A.; Mechoulam, R.; Guzmán, M.; Galve-Roperh, I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006, 20, 2405–2407. [Google Scholar] [CrossRef] [PubMed]
- Zurolo, E.; Iyer, A.M.; Spliet, W.G.M.; Van Rijen, P.C.; Troost, D.; Gorter, J.A.; Aronica, E. CB1 and CB2 cannabinoid receptor expression during development and in epileptogenic developmental pathologies. Neuroscience 2010, 170, 28–41. [Google Scholar] [CrossRef]
- Compagnucci, C.; Di Siena, S.; Bustamante, M.B.; Di Giacomo, D.; Di Tommaso, M.; Maccarrone, M.; Grimaldi, P.; Sette, C. Type-1 (CB1) cannabinoid receptor promotes neuronal differentiation and maturation of neural stem cells. PLoS ONE 2013, 8, e54271. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Rubio-Araiz, A.; García-Ovejero, D.; Williams, R.J.; Moore, J.D.; Arévalo-Martín, A.; Gómez-Torres, O.; Molina-Holgado, E. CB2 cannabinoid receptors promote mouse neural stem cell proliferation. Eur. J. Neurosci. 2007, 25, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Steger, M.; Mitrugno, V.M.; Bartesaghi, R.; Ciani, E. CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3beta/beta-catenin signaling. J. Biol. Chem. 2010, 285, 10098–10109. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Monory, K.; Palazuelos, J.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005, 19, 1704–1706. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alonso, J.; Aguado, T.; Wu, C.-S.; Palazuelos, J.; Hofmann, C.; Garcez, P.; Guillemot, F.; Lu, H.-C.; Lutz, B.; Guzmán, M.; et al. The CB1 cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. J. Neurosci. 2012, 32, 16651–16665. [Google Scholar] [CrossRef] [PubMed]
- Tapia, M.; Dominguez, A.; Zhang, W.; Del Puerto, A.; Ciorraga, M.; Benitez, M.J.; Guaza, C.; Garrido, J.J. Cannabinoid Receptors Modulate Neuronal Morphology and AnkyrinG Density at the Axon Initial Segment. Front. Cell. Neurosci. 2017, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Dobszay, M.B.; Wang, X.; Spano, S.; Ledda, F.; Sousa, K.M.; Schulte, G.; Ernfors, P.; Mackie, K.; Paratcha, G.; et al. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 19115–19120. [Google Scholar] [CrossRef]
- Díaz-Alonso, J.; de Salas-Quiroga, A.; Paraíso-Luna, J.; García-Rincón, D.; Garcez, P.P.; Parsons, M.; Andradas, C.; Sánchez, C.; Guillemot, F.; Guzmán, M.; et al. Loss of cannabinoid CB1 receptors induces cortical migration malformations and increases seizure susceptibility. Cereb. Cortex 2017, 27, 5303–5317. [Google Scholar] [PubMed]
- Alpár, A.; Tortoriello, G.; Calvigioni, D.; Niphakis, M.J.; Milenkovic, I.; Bakker, J.; Cameron, G.A.; Hanics, J.; Morris, C.V.; Fuzik, J.; et al. Endocannabinoids modulate cortical development by configuring Slit2/Robo1 signalling. Nat. Commun. 2014, 5, 4421. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.; Chambers, D.; Hobbs, C.; Doherty, P.; Graham, A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol. Cell. Neurosci. 2008, 38, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Zhu, J.; Wager-Miller, J.; Wang, S.; O’Leary, D.; Monory, K.; Lutz, B.; Mackie, K.; Lu, H.-C. Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur. J. Neurosci. 2010, 32, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, D.E.; Martin, B.R.; Gamagaris, Z.; Miller, N.; Fico, T. Plasma concentrations of delta-9-tetrahydrocannabinol in dams and fetuses following acute or multiple prenatal dosing in rats. Life Sci. 1989, 44, 697–701. [Google Scholar] [CrossRef]
- Huizink, A.C. Prenatal cannabis exposure and infant outcomes: Overview of studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 52, 45–52. [Google Scholar] [CrossRef] [PubMed]
- El Marroun, H.; Hudziak, J.J.; Tiemeier, H.; Creemers, H.; Steegers, E.A.P.; Jaddoe, V.W.V.; Hofman, A.; Verhulst, F.C.; van den Brink, W.; Huizink, A.C. Intrauterine cannabis exposure leads to more aggressive behavior and attention problems in 18-month-old girls. Drug Alcohol Depend. 2011, 118, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Fried, P.A.; Watkinson, B. Differential effects on facets of attention in adolescents prenatally exposed to cigarettes and marihuana. Neurotoxicol. Teratol. 2001, 23, 421–430. [Google Scholar] [CrossRef]
- Fried, P.A.; Watkinson, B.; Gray, R. A follow-up study of attentional behavior in 6-year-old children exposed prenatally to marihuana, cigarettes, and alcohol. Neurotoxicol. Teratol. 1992, 14, 299–311. [Google Scholar] [CrossRef]
- Smith, A.M.; Fried, P.A.; Hogan, M.J.; Cameron, I. Effects of prenatal marijuana on visuospatial working memory: An fMRI study in young adults. Neurotoxicol. Teratol. 2006, 28, 286–295. [Google Scholar] [CrossRef]
- Jutras-Aswad, D.; DiNieri, J.A.; Harkany, T.; Hurd, Y.L. Neurobiological consequences of maternal cannabis on human fetal development and its neuropsychiatric outcome. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 395–412. [Google Scholar] [CrossRef]
- Mathews, C.A.; Scharf, J.M.; Miller, L.L.; Macdonald-Wallis, C.; Lawlor, D.A.; Ben-Shlomo, Y. Association between pre- and perinatal exposures and Tourette syndrome or chronic tic disorder in the ALSPAC cohort. Br. J. Psychiatry J. Ment. Sci. 2014, 204, 40–45. [Google Scholar] [CrossRef]
- Fried, P.A.; Watkinson, B. 36- and 48-month neurobehavioral follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol. J. Dev. Behav. Pediatr. 1990, 11, 49–58. [Google Scholar] [CrossRef]
- Fried, P.A.; Watkinson, B.; Gray, R. Differential effects on cognitive functioning in 13- to 16-year-olds prenatally exposed to cigarettes and marihuana. Neurotoxicol. Teratol. 2003, 25, 427–436. [Google Scholar] [CrossRef]
- Saez, T.M.M.; Aronne, M.P.; Caltana, L.; Brusco, A.H. Prenatal exposure to the CB1 and CB2 cannabinoid receptor agonist WIN 55,212-2 alters migration of early-born glutamatergic neurons and GABAergic interneurons in the rat cerebral cortex. J. Neurochem. 2014, 129, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Roland, A.B.; Ricobaraza, A.; Carrel, D.; Jordan, B.M.; Rico, F.; Simon, A.; Humbert-Claude, M.; Ferrier, J.; McFadden, M.H.; Scheuring, S.; et al. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. Elife 2014, 3, e03159. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, T.; Tomasini, M.C.; Tattoli, M.; Cassano, T.; Tanganelli, S.; Finetti, S.; Mazzoni, E.; Trabace, L.; Steardo, L.; Cuomo, V.; et al. Prenatal exposure to the CB1 receptor agonist WIN 55,212-2 causes learning disruption associated with impaired cortical NMDA receptor function and emotional reactivity changes in rat offspring. Cereb. Cortex 2005, 15, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Mereu, G.; Fà, M.; Ferraro, L.; Cagiano, R.; Antonelli, T.; Tattoli, M.; Ghiglieri, V.; Tanganelli, S.; Gessa, G.L.; Cuomo, V. Prenatal exposure to a cannabinoid agonist produces memory deficits linked to dysfunction in hippocampal long-term potentiation and glutamate release. Proc. Natl. Acad. Sci. USA 2003, 100, 4915–4920. [Google Scholar] [CrossRef] [PubMed]
- Amlani, A.; Hornick, M.G.; Cooper, K.; Prazad, P.; Donovan, R.; Gulati, A. Maternal cannabinoid use alters cannabinoid (CB1) and endothelin (ETB) receptor expression in the brains of dams but not their offspring. Dev. Neurosci. 2017, 39, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Morgan, D.; Jew, C.P.; Haskins, C.; Andrews, M.-J.; Leishman, E.; Spencer, C.M.; Czyzyk, T.; Bradshaw, H.; Mackie, K.; et al. Long-term consequences of perinatal fatty acid amino hydrolase inhibition. Br. J. Pharmacol. 2014, 171, 1420–1434. [Google Scholar] [CrossRef]
- de Salas-Quiroga, A.; Díaz-Alonso, J.; García-Rincón, D.; Remmers, F.; Vega, D.; Gómez-Cañas, M.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 13693–13698. [Google Scholar] [CrossRef]
- Downer, E.J.; Gowran, A.; Campbell, V.A. A comparison of the apoptotic effect of Delta(9)-tetrahydrocannabinol in the neonatal and adult rat cerebral cortex. Brain Res. 2007, 1175, 39–47. [Google Scholar] [CrossRef]
- Suárez, I.; Bodega, G.; Fernández, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Stanslowsky, N.; Jahn, K.; Venneri, A.; Naujock, M.; Haase, A.; Martin, U.; Frieling, H.; Wegner, F. Functional effects of cannabinoids during dopaminergic specification of human neural precursors derived from induced pluripotent stem cells. Addict. Biol. 2017, 22, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Aguado, T.; Palazuelos, J.; Guzmán, M. The endocannabinoid system and neurogenesis in health and disease. Neuroscientist 2007, 13, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Harkany, T.; Guzmán, M.; Galve-Roperh, I.; Berghuis, P.; Devi, L.A.; Mackie, K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci. 2007, 28, 83–92. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Chiurchiù, V.; Díaz-Alonso, J.; Bari, M.; Guzmán, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 2013, 52, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Falenta, K.; Lalli, G. Endocannabinoid signalling in neuronal migration. Int. J. Biochem. Cell Biol. 2014, 47, 104–108. [Google Scholar] [CrossRef]
- Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. Endocannabinoids via CB1 receptors act as neurogenic niche cues during cortical development. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2012, 367, 3229–3241. [Google Scholar] [CrossRef] [PubMed]
- Prenderville, J.A.; Kelly, Á.M.; Downer, E.J. The role of cannabinoids in adult neurogenesis. Br. J. Pharmacol. 2015, 172, 3950–3963. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Guzmán, M.; Mackie, K.; Doherty, P.; Harkany, T. Programming of neural cells by (endo)cannabinoids: From physiological rules to emerging therapies. Nat. Rev. Neurosci. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Xapelli, S.; Agasse, F.; Sardà-Arroyo, L.; Bernardino, L.; Santos, T.; Ribeiro, F.F.; Valero, J.; Bragança, J.; Schitine, C.; de Melo Reis, R.A.; et al. Activation of type 1 cannabinoid receptor (CB1R) promotes neurogenesis in murine subventricular zone cell cultures. PLoS ONE 2013, 8, e63529. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Titterness, A.K.; Morrish, A.C.; Carrier, E.J.; Lee, T.T.-Y.; Gil-mohapel, J.; Gorzalka, B.B.; Hillard, C.J.; Christie, B.R.; Tiffany, T.; et al. Endogenous cannabinoid signaling is required for voluntary exercise-induced enhancement of progenitor cell proliferation in the hippocampus. Hippocampus 2010, 523, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Maroso, M.; Beer, A.; Baddenhausen, S.; Ludewig, S.; Fan, W.; Vennin, C.; Loch, S.; Berninger, B.; Hofmann, C.; et al. Neural stem cell lineage-specific cannabinoid type-1 receptor regulates neurogenesis and plasticity in the adult mouse hippocampus. Cereb. cortex 2018, 28, 4454–4471. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Bick-Sander, A.; Fabel, K.; Leal-Galicia, P.; Tauber, S.; Ramirez-Rodriguez, G.; Müller, A.; Melnik, A.; Waltinger, T.P.; Ullrich, O.; et al. Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun. Signal. 2010, 8, 12. [Google Scholar] [CrossRef]
- Campos, A.C.; Ortega, Z.; Palazuelos, J.; Fogaça, M.V.; Aguiar, D.C.; Díaz-Alonso, J.; Ortega-Gutiérrez, S.; Vázquez-Villa, H.; Moreira, F.A.; Guzmán, M.; et al. The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: Involvement of the endocannabinoid system. Int. J. Neuropsychopharmacol. 2013, 16, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, A.P.; Bonato, J.M.; Milani, H.; Guimarães, F.S.; de Oliveira, R.M.W. Influence of single and repeated cannabidiol administration on emotional behavior and markers of cell proliferation and neurogenesis in non-stressed mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Pazos, M.R.; García-Arencibia, M.; Sagredo, O.; Ramos, J. a Role of CB2 receptors in neuroprotective effects of cannabinoids. Mol. Cell. Endocrinol. 2008, 286, S91–S96. [Google Scholar] [CrossRef] [PubMed]
- Downer, E.J. High hopes for CB2 receptors in neurogenesis. Br. J. Pharmacol. 2014, 171, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Ortega, Z.; Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 2012, 287, 1198–1209. [Google Scholar] [CrossRef]
- Goncalves, M.B.; Suetterlin, P.; Yip, P.; Molina-Holgado, F.; Walker, D.J.; Oudin, M.J.; Zentar, M.P.; Pollard, S.; Yáñez-Muñoz, R.J.; Williams, G.; et al. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol. Cell. Neurosci. 2008, 38, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Avraham, H.K.; Jiang, S.; Fu, Y.; Rockenstein, E.; Makriyannis, A.; Zvonok, A.; Masliah, E.; Avraham, S. The cannabinoid CB2 receptor agonist AM1241 enhances neurogenesis in GFAP/Gp120 transgenic mice displaying deficits in neurogenesis. Br. J. Pharmacol. 2014, 171, 468–479. [Google Scholar] [CrossRef]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.-Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Gomez, O.; Sanchez-Rodriguez, A.; Le, M.Q.U.; Sanchez-Caro, C.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br. J. Pharmacol. 2011, 163, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Gómez Del Pulgar, T.; De Ceballos, M.L.; Guzmán, M.; Velasco, G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef] [PubMed]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef] [PubMed]
- Gomez, O.; Sanchez-Rodriguez, M.A.; Ortega-Gutierrez, S.; Vazquez-Villa, H.; Guaza, C.; Molina-Holgado, F.; Molina-Holgado, E. A basal tone of 2-arachidonoylglycerol contributes to early oligodendrocyte progenitor proliferation by activating phosphatidylinositol 3-Kinase (PI3K)/AKT and the mammalian target of rapamycin (MTOR) pathways. J. Neuroimmune Pharmacol. 2015, 10, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Gomez, O.; Arevalo-Martin, A.; Garcia-Ovejero, D.; Ortega-Gutierrez, S.; Cisneros, J.A.; Almazan, G.; Sánchez-Rodriguez, M.A.; Molina-Holgado, F.; Molina-Holgado, E. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 2010, 58, 1913–1927. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Roig, J.; Wirths, O.; Salinas-Riester, G.; Havemann-Reinecke, U. The cannabinoid CB1/CB2 agonist WIN55212.2 promotes oligodendrocyte differentiation in vitro and neuroprotection during the cuprizone-induced central nervous system demyelination. CNS Neurosci. Ther. 2016, 22, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Martín, Á.; García-Ovejero, D.; Rubio-Araiz, A.; Gómez, O.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur. J. Neurosci. 2007, 26, 1548–1559. [Google Scholar] [CrossRef]
- Solbrig, M.V.; Fan, Y.; Hermanowicz, N.; Morgese, M.G.; Giuffrida, A. A synthetic cannabinoid agonist promotes oligodendrogliogenesis during viral encephalitis in rats. Exp. Neurol. 2010, 226, 231–241. [Google Scholar] [CrossRef]
- Sonego, M.; Gajendra, S.; Parsons, M.; Ma, Y.; Hobbs, C.; Zentar, M.P.; Williams, G.; Machesky, L.M.; Doherty, P.; Lalli, G. Fascin regulates the migration of subventricular zone-derived neuroblasts in the postnatal brain. J. Neurosci. 2013, 33, 12171–12185. [Google Scholar] [CrossRef] [PubMed]
- Shinjyo, N.; Di Marzo, V. The effect of cannabichromene on adult neural stem/progenitor cells. Neurochem. Int. 2013, 63, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Sütterlin, P.; Williams, E.J.; Chambers, D.; Saraf, K.; von Schack, D.; Reisenberg, M.; Doherty, P.; Williams, G. The molecular basis of the cooperation between EGF, FGF and eCB receptors in the regulation of neural stem cell function. Mol. Cell. Neurosci. 2013, 52, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.-J.; Walsh, F.S.; Doherty, P. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J. Cell Biol. 2003, 160, 481–486. [Google Scholar] [CrossRef] [PubMed]
- García-Ovejero, D.; Arévalo-Martín, Á.; Navarro-Galve, B.; Pinteaux, E.; Molina-Holgado, E.; Molina-Holgado, F. Neuroimmmune interactions of cannabinoids in neurogenesis: Focus on interleukin-1β (IL-1β) signalling. Biochem. Soc. Trans. 2013, 41, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.S.; Ribeiro, F.F.; Ferreira, F.; Vaz, S.H.; Sebastião, A.M.; Xapelli, S. Interaction between cannabinoid type 1 and type 2 receptors in the modulation of dubventricular zone and dentate gyrus neurogenesis. Front. Pharmacol. 2017, 8, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Rivera, P.; Bindila, L.; Pastor, A.; Pérez-Martín, M.; Pavón, F.J.; Serrano, A.; de la Torre, R.; Lutz, B.; Rodríguez de Fonseca, F.; Suárez, J. Pharmacological blockade of the fatty acid amide hydrolase (FAAH) alters neural proliferation, apoptosis and gliosis in the rat hippocampus, hypothalamus and striatum in a negative energy context. Front. Cell. Neurosci. 2015, 9, 1–17. [Google Scholar] [CrossRef]
- Curtis, M.A.; Low, V.F.; Faull, R.L.M. Neurogenesis and progenitor cells in the adult human brain: A comparison between hippocampal and subventricular progenitor proliferation. Dev. Neurobiol. 2012, 72, 990–1005. [Google Scholar] [CrossRef] [PubMed]
- Casarosa, S.; Zasso, J.; Conti, L. Systems for ex-vivo isolation and culturing of neural stem cells. In Neural Stem Cells - New Perspectives; InTech: London, UK, 2014; pp. 1–27. ISBN 978-953-51-1069-9. [Google Scholar]
- Butti, E.; Cusimano, M.; Bacigaluppi, M.; Martino, G. Neurogenic and non-neurogenic functions of endogenous neural stem cells. Front. Neurosci. 2014, 8, 92. [Google Scholar] [CrossRef]
- Martinez-Canabal, A. Reconsidering hippocampal neurogenesis in Alzheimer’s disease. Front. Neurosci. 2014, 8, 147. [Google Scholar] [CrossRef]
- Prakash, N.; Wurst, W. Development of dopaminergic neurons in the mammalian brain. Cell. Mol. Life Sci. C. 2006, 63, 187–206. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Höglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef] [PubMed]
- Nickels, K.C.; Zaccariello, M.J.; Hamiwka, L.D.; Wirrell, E.C. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat. Rev. Neurol. 2016, 12, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Sarne, Y.; Asaf, F.; Fishbein, M.; Gafni, M.; Keren, O. The dual neuroprotective– neurotoxic profile of cannabinoid drugs. Br. J. Pharmacol. 2011, 163, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J. The biomedical challenge of neurodegenerative disorders: An opportunity for cannabinoid-based therapies to improve on the poor current therapeutic outcomes. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Bano, D.; Ankarcrona, M. Beyond the critical point: An overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 2018, 663, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Khaspekov, L.G.; Verca, M.S.B.; Frumkina, L.E.; Hermann, H.; Marsicano, G.; Lutz, B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 2004, 19, 1691–1698. [Google Scholar] [CrossRef]
- Docagne, F.; Muñetón, V.; Clemente, D.; Ali, C.; Loría, F.; Correa, F.; Hernangómez, M.; Mestre, L.; Vivien, D.; Guaza, C. Excitotoxicity in a chronic model of multiple sclerosis: Neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol. Cell. Neurosci. 2007, 34, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Loría, F.; Petrosino, S.; Hernangómez, M.; Mestre, L.; Spagnolo, A.; Correa, F.; Di Marzo, V.; Docagne, F.; Guaza, C. An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol. Dis. 2010, 37, 166–176. [Google Scholar] [CrossRef]
- Downer, E.J. Cannabinoids and innate immunity: Taking a toll on neuroinflammation. ScientificWorldJournal. 2011, 11, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: Pleiotropic physiological and pathological roles through complex pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2005, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Martin-Moreno, A.M.; Reigada, D.; Ramirez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; García-García, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; de Ceballos, M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation 2012, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Marchalant, Y.; Brothers, H.M.; Norman, G.J.; Karelina, K.; DeVries, A.C.; Wenk, G.L. Cannabinoids attenuate the effects of aging upon neuroinflammation and neurogenesis. Neurobiol. Dis. 2009, 34, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.B.; Zeissler, M.-L.; Hanemann, C.O.; Zajicek, J.P. Δ9-tetrahydrocannabinol (Δ9-THC) exerts a direct neuroprotective effect in a human cell culture model of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2012, 38, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Mishima, K.; Nozako, M.; Ogata, A.; Hazekawa, M.; Liu, A.-X.; Fujioka, M.; Abe, K.; Hasebe, N.; Egashira, N.; et al. Repeated treatment with cannabidiol but not Δ9-tetrahydrocannabinol has a neuroprotective effect without the development of tolerance. Neuropharmacology 2007, 52, 1079–1087. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Sagredo, O.; Pazos, M.R.; García, C.; Pertwee, R.; Mechoulam, R.; Martínez-Orgado, J. Cannabidiol for neurodegenerative disorders: Important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol. 2013, 75, 323–333. [Google Scholar] [CrossRef]
- Khaksar, S.; Bigdeli, M.R. Intra-cerebral cannabidiol infusion-induced neuroprotection is partly associated with the TNF-α/TNFR1/NF-кB pathway in transient focal cerebral ischaemia. Brain Inj. 2017, 31, 1932–1943. [Google Scholar] [CrossRef]
- Blázquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I.; et al. The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.F.; Ribeiro, F.F.; Rodrigues, R.S.; Sebastião, A.M.; Xapelli, S. Brain-derived neurotrophic factor (BDNF) role in cannabinoid-mediated neurogenesis. Front. Cell. Neurosci. 2018, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Alexander, S.P.H.; Garle, M.J.; Gibson, C.L.; Hewitt, K.; Murphy, S.P.; Kendall, D.A.; Bennett, A.J. Cannabinoid activation of PPARα; a novel neuroprotective mechanism. Br. J. Pharmacol. 2007, 152, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martínez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT(1A) and CB2 receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Tolón, M.R.; Fernández-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef]
- Aguado, T.; Romero, E.; Monory, K.; Palazuelos, J.; Sendtner, M.; Marsicano, G.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. The CB1 cannabinoid receptor mediates excitotoxicity-induced neural progenitor proliferation and neurogenesis. J. Biol. Chem. 2007, 282, 23892–23898. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.D.; Zuluaga-Ramirez, V.; Gajghate, S.; Winfield, M.; Sriram, U.; Rom, S.; Persidsky, Y. Activation of GPR55 induces neuroprotection of hippocampal neurogenesis and immune responses of neural stem cells following chronic, systemic inflammation. Brain Behav. Immun. 2019, 76, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M. Neurons on cannabinoids: Dead or alive? Br. J. Pharmacol. 2003, 140, 439–440. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Zabar, Y. Dementia: Mild Cognitive Impairment, Alzheimer’s Disease, Lewy Body Dementia, Frontotemporal Lobar Dementia, Vascular Dementia. In Netter’s Neurology; Jones, H.R., Srinivasan, J., et al., Eds.; Elsevier Saunders: St. Louis, Missouri, USA, 2012; pp. 219–243. ISBN 9781437702736. [Google Scholar]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Reports 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Kumar, K.; Kumar, A.; Keegan, R.M.; Deshmukh, R. Recent advances in the neurobiology and neuropharmacology of Alzheimer’s disease. Biomed. Pharmacother. 2018, 98, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; van der Marck, M.; van den Elsen, G.; Olde Rikkert, M. Cannabinoids in late-onset Alzheimer’s disease. Clin. Pharmacol. Ther. 2015, 97, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Huntley, J.D.; Howard, R.J. Working memory in early Alzheimer’s disease: A neuropsychological review. Int. J. Geriatr. Psychiatry 2010, 25, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Weier, M.; Hall, W. The use of cannabinoids in treating dementia. Curr. Neurol. Neurosci. Rep. 2017, 17, 56. [Google Scholar] [CrossRef] [PubMed]
- Volicer, L.; Stelly, M.; Morris, J.; McLaughlin, J.; Volicer, B.J. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 1997, 12, 913–919. [Google Scholar] [CrossRef]
- Woodward, M.R.; Harper, D.G.; Stolyar, A.; Forester, B.P.; Ellison, J.M. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am. J. Geriatr. Psychiatry 2014, 22, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Walther, S.; Mahlberg, R.; Eichmann, U.; Kunz, D. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006, 185, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Mahlberg, R.; Walther, S. Actigraphy in agitated patients with dementia. Z. Gerontol. Geriatr. 2007, 40, 178–184. [Google Scholar] [CrossRef]
- Pascual, A.C.; Gaveglio, V.L.; Giusto, N.M.; Pasquaré, S.J. 2-Arachidonoylglycerol metabolism is differently modulated by oligomeric and fibrillar conformations of amyloid beta in synaptic terminals. Neuroscience 2017, 362, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Manuel, I.; de San Román, E.G.; Giralt, M.T.; Ferrer, I.; Rodríguez-Puertas, R. Type-1 cannabinoid receptor activity during Alzheimer’s disease progression. J. Alzheimer’s Dis. 2014, 42, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, B.G.; Blázquez, C.; Gómez del Pulgar, T.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- van der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; De Filippis, D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and β-amyloid-induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. C. 2006, 63, 1410–1424. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134, 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Zheng, M.; Zhang, T.; He, G. Hippocampal neurogenesis in the APP/PS1/nestin-GFP triple transgenic mouse model of Alzheimer’s disease. Neuroscience 2016, 314, 64–74. [Google Scholar] [CrossRef]
- Hamilton, A.; Holscher, C. The effect of ageing on neurogenesis and oxidative stress in the APPswe/PS1deltaE9 mouse model of Alzheimer’s disease. Brain Res. 2012, 1449, 83–93. [Google Scholar] [CrossRef]
- Biscaro, B.; Lindvall, O.; Tesco, G.; Ekdahl, C.T.; Nitsch, R.M. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer’s disease. Neurodegener. Dis. 2012, 9, 187–198. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Jones, V.C.; Tabuchi, M.; Allan, S.M.; Knight, E.M.; LaFerla, F.M.; Oddo, S.; Verkhratsky, A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef] [PubMed]
- Valero, J.; Mastrella, G.; Neiva, I.; Sánchez, S.; Malva, J.O. Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front. Neurosci. 2014, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Liu, J.; Ruan, Z.; Tian, S.; Ma, Y.; Zhu, J.; Li, G. Intrahippocampal injection of Aβ1-42 inhibits neurogenesis and down-regulates IFN-γ and NF-κB expression in hippocampus of adult mouse brain. Amyloid 2013, 20, 13–20. [Google Scholar] [CrossRef]
- Crews, L.; Adame, A.; Patrick, C.; DeLaney, A.; Pham, E.; Rockenstein, E.; Hansen, L.; Masliah, E. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J. Neurosci. 2010, 30, 12252–12262. [Google Scholar] [CrossRef]
- Sun, B.; Halabisky, B.; Zhou, Y.; Palop, J.J.; Yu, G.; Mucke, L.; Gan, L. Imbalance between GABAergic and glutamatergic transmission impairs adult neurogenesis in an animal model of Alzheimer’s disease. Cell Stem Cell 2009, 5, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Nakajima, A.; Choi, S.H.; Xiong, X.; Sisodia, S.S.; Tang, Y.-P. Adult neurogenesis is functionally associated with AD-like neurodegeneration. Neurobiol. Dis. 2008, 29, 316–326. [Google Scholar] [CrossRef]
- Eubanks, L.M.; Rogers, C.J.; Beuscher, A.E.; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K.D.; Janda, K.D. A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J.; Chen, C. Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neuroscience 2011, 178, 159–168. [Google Scholar] [CrossRef]
- Harvey, B.S.; Ohlsson, K.S.; Mååg, J.L.V.; Musgrave, I.F.; Smid, S.D. Contrasting protective effects of cannabinoids against oxidative stress and amyloid-β evoked neurotoxicity in vitro. Neurotoxicology 2012, 33, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Tolón, R.M.; Núñez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martínez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; Di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phyther. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- d’Abramo, C.; Massone, S.; Zingg, J.-M.; Pizzuti, A.; Marambaud, P.; Dalla Piccola, B.; Azzi, A.; Marinari, U.M.; Pronzato, M.A.; Ricciarelli, R. Role of peroxisome proliferator-activated receptor γ in amyloid precursor protein processing and amyloid β-mediated cell death. Biochem. J. 2005, 391, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Camacho, I.E.; Serneels, L.; Spittaels, K.; Merchiers, P.; Dominguez, D.; De Strooper, B. Peroxisome proliferator-activated receptor induces a clearance mechanism for the amyloid-peptide. J. Neurosci. 2004, 24, 10908–10917. [Google Scholar] [CrossRef] [PubMed]
- Janefjord, E.; Mååg, J.L.V.; Harvey, B.S.; Smid, S.D. Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell. Mol. Neurobiol. 2014, 34, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Sánchez-Pla, A.; Vegas-Lozano, E.; Maldonado, R.; Ferrer, I. Cannabis-Based Medicine Reduces Multiple Pathological Processes in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2014, 43, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef]
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 2012, 63, 653–666. [Google Scholar] [CrossRef]
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB1 agonist ACEA protects neurons and reduces the cognitive impairment of AβPP/PS1 Mice. J. Alzheimer’s Dis. 2012, 30, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hocevar, M.; Foss, J.F.; Bie, B.; Naguib, M. Activation of CB2 receptor system restores cognitive capacity and hippocampal Sox2 expression in a transgenic mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2017, 811, 12–20. [Google Scholar] [CrossRef]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 Cannabinoid Receptor Agonist Ameliorates Alzheimer-Like Phenotype in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L.; Steardo, L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Andrés-Benito, P.; Ferrer, I. Delineating the efficacy of a cannabis-based medicine at advanced stages of dementia in a murine model. J. Alzheimer’s Dis. 2016, 54, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Spiro, A.S.; Jenner, A.M.; Garner, B.; Karl, T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J. Alzheimer’s Dis. 2014, 42, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Low, J.K.; Logge, W.; Garner, B.; Karl, T. Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1∆E9 mice. Psychopharmacology (Berl). 2014, 231, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, J.; Fan, N.; Teng, Z.-Q.; Wu, Y.; Yang, H.; Tang, Y.-P.; Sun, H.; Song, Y.; Chen, C. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell 2013, 155, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.-P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.-S.; Li, L.; Sun, X.-J.; Zhao, Y.-W.; Zhang, J.; Geng, Z.; Fu, J.-L.; Ren, Q.-G. Glycogen synthase kinase-3 regulates production of amyloid-β peptides and tau phosphorylation in diabetic rat brain. ScientificWorldJournal. 2014, 2014, 878123. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Fuster-Matanzo, A.; Teixeira, C.M.; Jurado-Arjona, J.; Ulloa, F.; deFelipe, J.; Rábano, A.; Hernández, F.; Soriano, E.; Ávila, J. GSK-3β overexpression causes reversible alterations on postsynaptic densities and dendritic morphology of hippocampal granule neurons in vivo. Mol. Psychiatry 2013, 18, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.M.; Pallas-Bazarra, N.; Bolós, M.; Terreros-Roncal, J.; Ávila, J.; Llorens-Martín, M. Untold new beginnings: Adult hippocampal neurogenesis and Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 64, S497–S505. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Luna-Medina, R.; Alonso-Gil, S.; Sanz-SanCristobal, M.; Palomo, V.; Gil, C.; Santos, A.; Martinez, A.; Perez-Castillo, A. Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem. Neurosci. 2012, 3, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The marijuana component cannabidiol inhibits β-amyloid-induced tau protein hyperphosphorylation through Wnt/β-catenin pathway rescue in PC12 cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Hollands, C.; Bartolotti, N.; Lazarov, O. Alzheimer’s disease and hippocampal adult neurogenesis: Exploring shared mechanisms. Front. Neurosci. 2016, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the adult hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812. [Google Scholar] [CrossRef] [PubMed]
- Tanveer, R.; Gowran, A.; Noonan, J.; Keating, S.E.; Bowie, A.G.; Campbell, V.A. The endocannabinoid, anandamide, augments Notch-1 signaling in cultured cortical neurons exposed to amyloid-β and in the cortex of aged rats. J. Biol. Chem. 2012, 287, 34709–34721. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Rubio-Casillas, A. Biphasic effects of THC in memory and cognition. Eur. J. Clin. Invest. 2018, 48, e12920. [Google Scholar] [CrossRef]
- Svízenská, I.; Dubový, P.; Sulcová, A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures - a short review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Liu, C.S.; Chau, S.A.; Ruthirakuhan, M.; Lanctôt, K.L.; Herrmann, N. Cannabinoids for the treatment of agitation and aggression in Alzheimer’s disease. CNS Drugs 2015, 29, 615–623. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.-Y.M.-Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Morris, M.; Sanchez, P.E.; Verret, L.; Beagle, A.J.; Guo, W.; Dubal, D.; Ranasinghe, K.G.; Koyama, A.; Ho, K.; Yu, G.-Q.; et al. Network dysfunction in α -synuclein transgenic mice and human Lewy body dementia. Ann. Clin. Transl. Neurol. 2015, 2, 1012–1028. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat Rev Neurol 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Spira, P.J.; Sharpe, D.M.; Halliday, G.; Cavanagh, J.; Nicholson, G. a Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann. Neurol. 2001, 49, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; Wunderle, K.B. Parkinson’s disease: Evidence for environmental risk factors. Mov. Disord. 2013, 28, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Katzenschlager, R.; Lim, S.-Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 2018, 33, 1248–1266. [Google Scholar] [CrossRef] [PubMed]
- Venderová, K.; Růžička, E.; Voříšek, V.; Višňovský, P. Survey on cannabis use in Parkinson’s disease: Subjective improvement of motor symptoms. Mov. Disord. 2004, 19, 1102–1106. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.; Johnson, M.; Melvin, L.; de Costa, B.; Rice, K. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000, 14, 1432–1438. [Google Scholar]
- Di Marzo, V.; Berrendero, F.; Bisogno, T.; González, S.; Cavaliere, P.; Romero, J.; Cebeira, M.; Ramos, J.A.; Fernández-Ruiz, J.J. Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of Δ9-tetrahydrocannabinol-tolerant rats. J. Neurochem. 2002, 74, 1627–1635. [Google Scholar] [CrossRef]
- Brotchie, J. CB1 cannabinoid receptor signalling in Parkinson’s disease. Curr. Opin. Pharmacol. 2003, 3, 54–61. [Google Scholar] [CrossRef]
- van der Stelt, M.; Di Marzo, V. The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: Implications for neurological and psychiatric disorders. Eur. J. Pharmacol. 2003, 480, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Aceves, J.J.; Rueda-Orozco, P.E.; Hernandez-Martinez, R.; Galarraga, E.; Bargas, J. Bidirectional plasticity in striatonigral synapses: A switch to balance direct and indirect basal ganglia pathways. Learn. Mem. 2011, 18, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E. Endocannabinoids in basal ganglia circuits: Implications for Parkinson disease. Neurology 2007, 69, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Parsons, L.H.; Kerr, T.M.; de Fonseca, F.R.; Navarro, M.; Piomelli, D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 1999, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.-C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; Di Filippo, M. Direct and indirect pathways of basal ganglia: A critical reappraisal. Nat. Neurosci. 2014, 17, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- More, S.V.; Choi, D.-K. Promising cannabinoid-based therapies for Parkinson’s disease: Motor symptoms to neuroprotection. Mol. Neurodegener. 2015, 10. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Molina-Holgado, F.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson’s disease. Neurobiol. Dis. 2005, 19, 96–107. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid type 2 (CB2) receptors activation protects against oxidative stress and neuroinflammation associated dopaminergic neurodegeneration in rotenone model of Parkinson’s disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [PubMed]
- Borta, A.; Höglinger, G.U. Dopamine and adult neurogenesis. J. Neurochem. 2007, 100, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Marxreiter, F.; Regensburger, M.; Winkler, J. Adult neurogenesis in Parkinson’s disease. Cell. Mol. Life Sci. 2013, 70, 459–473. [Google Scholar] [CrossRef]
- Peng, J.; Andersen, J.K. Mutant α-synuclein and aging reduce neurogenesis in the acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease: Parkinson’s disease and neurogenesis. Aging Cell 2011, 10, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Marxreiter, F.; Ettle, B.; May, V.E.L.; Esmer, H.; Patrick, C.; Kragh, C.L.; Klucken, J.; Winner, B.; Riess, O.; Winkler, J.; et al. Glial A30P alpha-synuclein pathology segregates neurogenesis from anxiety-related behavior in conditional transgenic mice. Neurobiol. Dis. 2013, 59, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Marxreiter, F.; Nuber, S.; Kandasamy, M.; Klucken, J.; Aigner, R.; Burgmayer, R.; Couillard-Despres, S.; Riess, O.; Winkler, J.; Winner, B. Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur. J. Neurosci. 2009, 29, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Mishra, A.; Srivastava, N.; Shukla, R.; Shukla, S. Acetyl-l-Carnitine via upregulating dopamine D1 receptor and attenuating microglial activation prevents neuronal loss and improves memory functions in parkinsonian rats. Mol. Neurobiol. 2018, 55, 583–602. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Singh, S.; Tiwari, V.; Shukla, S. Dopamine D1 receptor activation improves adult hippocampal neurogenesis and exerts anxiolytic and antidepressant-like effect via activation of Wnt/β-catenin pathways in rat model of Parkinson’s disease. Neurochem. Int. 2019, 122, 170–186. [Google Scholar] [CrossRef]
- Lim, J.; Bang, Y.; Choi, H.J. Abnormal hippocampal neurogenesis in Parkinson’s disease: Relevance to a new therapeutic target for depression with Parkinson’s disease. Arch. Pharm. Res. 2018, 41, 943–954. [Google Scholar] [CrossRef]
- Van Kampen, J.M.; Eckman, C.B. Dopamine D3 receptor agonist delivery to a model of Parkinson’s disease restores the nigrostriatal pathway and improves locomotor behavior. J. Neurosci. 2006, 26, 7272–7280. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Desplats, P.; Hagl, C.; Klucken, J.; Aigner, R.; Ploetz, S.; Laemke, J.; Karl, A.; Aigner, L.; Masliah, E.; et al. Dopamine receptor activation promotes adult neurogenesis in an acute Parkinson model. Exp. Neurol. 2009, 219, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S.; Shim, J.-W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Petit, G.H.; Olsson, T.T.; Brundin, P. The future of cell therapies and brain repair: Parkinson’s disease leads the way. Neuropathol. Appl. Neurobiol. 2014, 40, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Kirkeby, A.; Björklund, A.; Thompson, L.; Brundin, P. Are stem cell-based therapies for Parkinson’s disease ready for the clinic in 2016? J. Parkinsons. Dis. 2016, 6, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human trials of stem cell-derived dopamine neurons for Parkinson’s disease: Dawn of a new era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Garcia, C.; Fernandez-Ruiz, J. Cannabinoids and Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2009, 8, 432–439. [Google Scholar] [CrossRef]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple-sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Roberts, A.B.; Sporn, M.B. Handbook of experimental pharmacology - Endocannabinoids; Pertwee, R.G., Ed.; Springer: Switzerland, 2015; ISBN 978-3-319-20825-1. [Google Scholar]
- Gado, F.; Digiacomo, M.; Macchia, M.; Bertini, S.; Manera, C. Traditional uses of cannabinoids and new perspectives in the treatment of Multiple Sclerosis. Medicines 2018, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Cosroe, P.; Musty, R.; Rein, J.; Tillery, W.; Pertwee, R. The perceived effects of smoked cannabis on patients with Multiple Sclerosis. Eur. Neurol. 1997, 38, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Pryce, G.; Ludovic Croxford, J.; Brown, P.; Pertwee, R.G.; Huffman, J.W.; Layward, L. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 2000, 404, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Velayudhan, L.; Diepen, E.; Marudkar, M.; Hands, O.; Suribhatla, S.; Prettyman, R.; Murray, J.; Baillon, S.; Bhattacharyya, S. Therapeutic potential of cannabinoids in neurodegenerative disorders: A selective review. Curr. Pharm. Des. 2014, 20, 2218–2230. [Google Scholar] [CrossRef]
- Chiurchiù, V.; van der Stelt, M.; Centonze, D.; Maccarrone, M. The endocannabinoid system and its therapeutic exploitation in multiple sclerosis: Clues for other neuroinflammatory diseases. Prog. Neurobiol. 2018, 160, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Ware, M.A.; Yazer, E.; Murray, T.J.; Lynch, M.E. Patterns of cannabis use among patients with multiple sclerosis. Neurology 2004, 62, 2098–2100. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, J.P.; Apostu, V.I. Role of cannabinoids in Multiple Sclerosis. CNS Drugs 2011, 25, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Martín, Á.; Vela, M.; Molina-holgado, E.; Guaza, C. Therapeutic action of cannabinoids in a murine model of Multiple Sclerosis. J. Neurosci. 2003, 23, 2511–2516. [Google Scholar] [CrossRef]
- Cabranes, A.; Venderova, K.; De Lago, E.; Fezza, F.; Sánchez, A.; Mestre, L.; Valenti, M.; García-Merino, A.; Ramos, J.A.; Di Marzo, V.; et al. Decreased endocannabinoid levels in the brain and beneficial effects of agents activating cannabinoid and/or vanilloid receptors in a rat model of multiple sclerosis. Neurobiol. Dis. 2005, 20, 207–217. [Google Scholar] [CrossRef]
- Centonze, D.; Bari, M.; Rossi, S.; Prosperetti, C.; Furlan, R.; Fezza, F.; De Chiara, V.; Battistini, L.; Bernardi, G.; Bernardini, S.; et al. The endocannabinoid system is dysregulated in Multiple Sclerosis and in experimental autoimmune encephalomyelitis. Brain 2007, 130, 2543–2553. [Google Scholar] [CrossRef]
- Witting, A.; Chen, L.; Cudaback, E.; Straiker, A.; Walter, L.; Rickman, B.; Moller, T.; Brosnan, C.; Stella, N. Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection. Proc. Natl. Acad. Sci. USA 2006, 103, 6362–6367. [Google Scholar] [CrossRef]
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 2006, 49, 67–79. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 1–14. [Google Scholar] [CrossRef]
- Jean-Gilles, L.; Feng, S.; Tench, C.R.; Chapman, V.; Kendall, D.A.; Barrett, D.A.; Constantinescu, C.S. Plasma endocannabinoid levels in multiple sclerosis. J. Neurol. Sci. 2009, 287, 212–215. [Google Scholar] [CrossRef]
- Pryce, G.; Ahmed, Z.; Hankey, D.J.R.; Jackson, S.J.; Croxford, J.L.; Pocock, J.M.; Ledent, C.; Petzold, A.; Thompson, A.J.; Giovannoni, G.; et al. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 2003, 126, 2191–2202. [Google Scholar] [CrossRef]
- Zhang, M.; Martin, B.R.; Adler, M.W.; Razdan, R.J.; Kong, W.; Ganea, D.; Tuma, R.F. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J. Neuroimmune Pharmacol. 2009, 4, 249–259. [Google Scholar] [CrossRef]
- Maresz, K.; Pryce, G.; Ponomarev, E.D.; Marsicano, G.; Croxford, J.L.; Shriver, L.P.; Ledent, C.; Cheng, X.; Carrier, E.J.; Mann, M.K.; et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat. Med. 2007, 13, 492–497. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Cencioni, M.T.; Bisicchia, E.; De Bardi, M.; Gasperini, C.; Borsellino, G.; Centonze, D.; Battistini, L.; Maccarrone, M. Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann. Neurol. 2013, 73, 626–636. [Google Scholar] [CrossRef]
- Feliu, A.; del Río, B.I.; Carrillo-Salinas, F.; Hernández-Torres, G.; Mestre, L.; Puente, N.; Ortega-Gutiérrez, S.; López-Rodríguez, M.; Grandes, P.; Mecha, M.; et al. 2-arachidonoylglycerol reduces proteoglycans and enhances remyelination in a progressive model of demyelination. J. Neurosci. 2017, 37, 2900–2916. [Google Scholar] [CrossRef]
- Benito, C.; Romero, J.P.; Tolon, R.M.; Clemente, D.; Docagne, F.; Hillard, C.J.; Guaza, C.; Romero, J. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human Multiple Sclerosis. J. Neurosci. 2007, 27, 2396–2402. [Google Scholar] [CrossRef]
- Ortega-Gutiérrez, S.; Molina-Holgado, E.; Arévalo-Martín, Á.; Correa, F.; Viso, A.; López-Rodríguez, M.L.; Di Marzo, V.; Guaza, C. Activation of the endocannabinoid system as therapeutic approach in a murine model of multiple sclerosis. FASEB J. 2005, 19, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.M.; Singh, N.; Nagarkatti, M.; Nagarkatti, P.S. Cannabidiol attenuates experimental autoimmune encephalomyelitis model of multiple sclerosis through induction of myeloid-derived suppressor cells. Front. Immunol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kozela, E.; Juknat, A.; Kaushansky, N.; Rimmerman, N.; Ben-Nun, A.; Vogel, Z. Cannabinoids decrease the Th17 inflammatory autoimmune phenotype. J. Neuroimmune Pharmacol. 2013, 8, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Geller, E.B.; Eppihimer, M.J.; Eisenstein, T.K.; Adler, M.W.; Tuma, R.F. Win 55212-2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult. Scler. 2004, 10, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Li, H.; Tuma, R.F.; Ganea, D. Selective CB2 receptor activation ameliorates EAE by reducing Th17 differentiation and immune cell accumulation in the CNS. Cell. Immunol. 2014, 287, 1–17. [Google Scholar] [CrossRef]
- Collin, C.; Ehler, E.; Waberzinek, G.; Alsindi, Z.; Davies, P.; Powell, K.; Notcutt, W.; O’Leary, C.; Ratcliffe, S.; Nováková, I.; Zapletalova, O.; et al. A double-blind, randomized, placebo-controlled, parallel-group study of Sativex, in subjects with symptoms of spasticity due to multiple sclerosis. Neurol. Res. 2010, 32, 451–459. [Google Scholar]
- Torres-Moreno, M.C.; Papaseit, E.; Torrens, M.; Farré, M. Assessment of efficacy and tolerability of medicinal cannabinoids in patients With Multiple Sclerosis. JAMA Netw. Open 2018, 1, e183485. [Google Scholar] [CrossRef]
- Serpell, M.G.; Notcutt, W.; Collin, C. Sativex long-term use: An open-label trial in patients with spasticity due to Multiple Sclerosis. J. Neurol. 2013, 260, 285–295. [Google Scholar] [CrossRef]
- Wade, D.T.; Makela, P.; Robson, P.; House, H.; Bateman, C. Do cannabis-based medicinal extracts have general or specific effects on symptoms in multiple sclerosis? A double-blind, randomized, placebo-controlled study on 160 patients. Mult. Scler. 2004, 10, 434–441. [Google Scholar] [CrossRef]
- Giacoppo, S.; Bramanti, P.; Mazzon, E. Sativex in the management of multiple sclerosis-related spasticity: An overview of the last decade of clinical evaluation. Mult. Scler. Relat. Disord. 2017, 17, 22–31. [Google Scholar] [CrossRef]
- Turri, M.; Teatini, F.; Donato, F.; Zanette, G.; Tugnoli, V.; Deotto, L.; Bonetti, B.; Squintani, G. Pain modulation after oromucosal cannabinoid spray (SATIVEX®) in patients with Multiple Sclerosis: A study with quantitative sensory testing and laser-evoked potentials. Medicines 2018, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Rog, D.J.; Nurmikko, T.J.; Young, C.A. Oromucosal Δ9-tetrahydrocannabinol/cannabidiol for neuropathic pain associated with multiple sclerosis: An uncontrolled, open-label, 2-year extension trial. Clin. Ther. 2007, 29, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Rog, D.J.; Nurmikko, T.J.; Friede, T.; Young, C.A. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology 2005, 65, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Flachenecker, P.; Henze, T.; Zettl, U.K. Nabiximols (THC/CBD Oromucosal Spray, Sativex®) in clinical practice - results of a multicenter, non-interventional study (MOVE 2) in patients with multiple sclerosis spasticity. Eur. Neurol. 2014, 71, 271–279. [Google Scholar] [CrossRef]
- Lorente Fernández, L.; Monte Boquet, E.; Pérez-Miralles, F.; Gil Gómez, I.; Escutia Roig, M.; Boscá Blasco, I.; Poveda Andrés, J.L.; Casanova-Estruch, B. Clinical experiences with cannabinoids in spasticity management in multiple sclerosis. Neurología 2014, 29, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Bettolo, C.M.; Onesti, E.; Frasca, V.; Iacovelli, E.; Gilio, F.; Giacomelli, E.; Gabriele, M.; Aragona, M.; Tomassini, V.; et al. Cannabinoid-induced effects on the nociceptive system: A neurophysiological study in patients with secondary progressive multiple sclerosis. Eur. J. Pain 2009, 13, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, J.; Ball, S.; Wright, D.; Vickery, J.; Nunn, A.; Miller, D.; Cano, M.G.; McManus, D.; Mallik, S.; Hobart, J. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): A randomised, placebo-controlled trial. Lancet Neurol. 2013, 12, 857–865. [Google Scholar] [CrossRef]
- Notcutt, W.G. Clinical use of cannabinoids for symptom control in Multiple Sclerosis. Neurotherapeutics 2015, 12, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Picard-Riera, N.; Decker, L.; Delarasse, C.; Goude, K.; Nait-Oumesmar, B.; Liblau, R.; Pham-Dinh, D.; Baron-Van Evercooren, A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13211–13216. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riéra, N.; Kerninon, C.; Baron-Van Evercooren, A. The role of SVZ-derived neural precursors in demyelinating diseases: From animal models to multiple sclerosis. J. Neurol. Sci. 2008, 265, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M.; Reynolds, R. Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Exp. Neurol. 1999, 160, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The endocannabinoid system and oligodendrocytes in health and disease. Front. Neurosci. 2018, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia 2017, 116, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.E.; McLane, L.E.; Bercury, K.K.; Macklin, W.B.; Wood, T.L. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J. Neurosci. 2014, 34, 4453–4465. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef]
- Goebbels, S.; Oltrogge, J.H.; Kemper, R.; Heilmann, I.; Bormuth, I.; Wolfer, S.; Wichert, S.P.; Mobius, W.; Liu, X.; Lappe-Siefke, C.; et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J. Neurosci. 2010, 30, 8953–8964. [Google Scholar] [CrossRef]
- Ebner, S.; Dunbar, M.; McKinnon, R.D. Distinct roles for PI3K in proliferation and survival of oligodendrocyte progenitor cells. J. Neurosci. Res. 2000, 62, 336–345. [Google Scholar] [CrossRef]
- Vemuri, G.S.; McMorris, F. a Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development 1996, 122, 2529–2537. [Google Scholar]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.-J.; Liu, L.; Xu, H.-Q.; Shi, Y.-W.; Yi, Y.-H.; He, N.; Liao, W.-P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef]
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.M.; Kron, M.M. Neurogenesis and Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Bonansco, C.; Fuenzalida, M. Plasticity of hippocampal excitatory-inhibitory balance: Missing the synaptic control in the epileptic brain. Neural Plast. 2016, 2016, 8607038. [Google Scholar] [CrossRef]
- Hofmann, M.E.; Frazier, C.J. Marijuana, endocannabinoids, and epilepsy: Potential and challenges for improved therapeutic intervention. Exp. Neurol. 2013, 244, 43–50. [Google Scholar] [CrossRef]
- De Caro, C.; Leo, A.; Citraro, R.; De Sarro, C.; Russo, R.; Calignano, A.; Russo, E. The potential role of cannabinoids in epilepsy treatment. Expert Rev. Neurother. 2017, 17, 1069–1079. [Google Scholar] [CrossRef]
- Bhaskaran, M.D.; Smith, B.N. Cannabinoid-mediated inhibition of recurrent excitatory circuitry in the dentate gyrus in a mouse model of temporal lobe epilepsy. PLoS ONE 2010, 5, e10683. [Google Scholar] [CrossRef]
- Maglóczky, Z.; Tóth, K.; Karlócai, R.; Nagy, S.; Eross, L.; Czirják, S.; Vajda, J.; Rásonyi, G.; Kelemen, A.; Juhos, V.; et al. Dynamic changes of CB1-receptor expression in hippocampi of epileptic mice and humans. Epilepsia 2010, 51 (Suppl. 3), 115–120. [Google Scholar] [CrossRef]
- Romigi, A.; Bari, M.; Placidi, F.; Marciani, M.G.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, C.; Zannino, S.; Corte, F.; et al. Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef]
- Ludányi, A.; Eross, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef]
- O’Connell, B.K.; Gloss, D.; Devinsky, O. Cannabinoids in treatment-resistant epilepsy: A review. Epilepsy Behav. 2017, 70, 341–348. [Google Scholar] [CrossRef]
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Russo, E.; Elia, M. Cannabidiol and epilepsy: Rationale and therapeutic potential. Pharmacol. Res. 2016, 107, 85–92. [Google Scholar] [CrossRef]
- Wallace, M.J.; Blair, R.E.; Falenski, K.W.; Martin, B.R.; DeLorenzo, R.J. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J. Pharmacol. Exp. Ther. 2003, 307, 129–137. [Google Scholar] [CrossRef]
- Kozan, R.; Ayyildiz, M.; Agar, E. The effects of intracerebroventricular AM-251, a CB1-receptor antagonist, and ACEA, a CB1-receptor agonist, on penicillin-induced epileptiform activity in rats. Epilepsia 2009, 50, 1760–1767. [Google Scholar] [CrossRef]
- Friedman, D.; Devinsky, O. Cannabinoids in the treatment of Epilepsy. N. Engl. J. Med. 2015, 373, 1048–1058. [Google Scholar] [CrossRef]
- von Rüden, E.L.; Bogdanovic, R.M.; Wotjak, C.T.; Potschka, H. Inhibition of monoacylglycerol lipase mediates a cannabinoid 1-receptor dependent delay of kindling progression in mice. Neurobiol. Dis. 2015, 77, 238–245. [Google Scholar] [CrossRef]
- Wendt, H.; Soerensen, J.; Wotjak, C.T.; Potschka, H. Targeting the endocannabinoid system in the amygdala kindling model of temporal lobe epilepsy in mice. Epilepsia 2011, 52, e62–e65. [Google Scholar] [CrossRef]
- von Rüden, E.L.; Jafari, M.; Bogdanovic, R.M.; Wotjak, C.T.; Potschka, H. Analysis in conditional cannabinoid 1 receptor-knockout mice reveals neuronal subpopulation-specific effects on epileptogenesis in the kindling paradigm. Neurobiol. Dis. 2015, 73, 334–347. [Google Scholar] [CrossRef]
- Di Maio, R.; Cannon, J.R.; Greenamyre, J.T. Post-status epilepticus treatment with the cannabinoid agonist WIN 55,212-2 prevents chronic epileptic hippocampal damage in rats. Neurobiol. Dis. 2015, 73, 356–365. [Google Scholar] [CrossRef]
- Luszczki, J.J.; Czuczwar, P.; Cioczek-Czuczwar, A.; Czuczwar, S.J. Arachidonyl-2′-chloroethylamide, a highly selective cannabinoid CB1 receptor agonist, enhances the anticonvulsant action of valproate in the mouse maximal electroshock-induced seizure model. Eur. J. Pharmacol. 2006, 547, 65–74. [Google Scholar] [CrossRef]
- Luszczki, J.J.; Czuczwar, P.; Cioczek-Czuczwar, A.; Dudra-Jastrzebska, M.; Andres-Mach, M.; Czuczwar, S.J. Effect of arachidonyl-2′-chloroethylamide, a selective cannabinoid CB1 receptor agonist, on the protective action of the various antiepileptic drugs in the mouse maximal electroshock-induced seizure model. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2010, 34, 18–25. [Google Scholar] [CrossRef]
- Luszczki, J.J.; Andres-Mach, M.; Barcicka-Klosowska, B.; Florek-Luszczki, M.; Haratym-Maj, A.; Czuczwar, S.J. Effects of WIN 55,212-2 mesylate (a synthetic cannabinoid) on the protective action of clonazepam, ethosuximide, phenobarbital and valproate against pentylenetetrazole-induced clonic seizures in mice. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2011, 35, 1870–1876. [Google Scholar] [CrossRef]
- Jessberger, S.; Römer, B.; Babu, H.; Kempermann, G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp. Neurol. 2005, 196, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.M.; Yu, T.W.; Leibowitz, R.T.; Geschwind, D.H.; Sloviter, R.S.; Lowenstein, D.H. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 1997, 17, 3727–3738. [Google Scholar] [CrossRef]
- Gray, W.P.; Sundstrom, L.E. Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res. 1998, 790, 52–59. [Google Scholar] [CrossRef]
- Hattiangady, B.; Rao, M.; Shetty, A. Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol. Dis. 2004, 17, 473–490. [Google Scholar] [CrossRef]
- Zhong, Q.; Ren, B.-X.; Tang, F.-R. Neurogenesis in the hippocampus of patients with temporal lobe epilepsy. Curr. Neurol. Neurosci. Rep. 2016, 16, 20. [Google Scholar] [CrossRef]
- Mathern, G.W.; Leiphart, J.L.; De Vera, A.; Adelson, P.D.; Seki, T.; Neder, L.; Leite, J.P. Seizures decrease postnatal neurogenesis and granule cell development in the human fascia dentata. Epilepsia 2002, 43 (Suppl. 5), 68–73. [Google Scholar] [CrossRef]
- Scharfman, H.; Goodman, J.; Macleod, A.; Phani, S.; Antonelli, C.; Croll, S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. 2005, 192, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.M.; Elliott, R.C.; Pleasure, S.J.; Barbaro, N.M.; Lowenstein, D.H. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann. Neurol. 2006, 59, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Houser, C.R. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990, 535, 195–204. [Google Scholar] [CrossRef]
- Ribak, C.E.; Tran, P.H.; Spigelman, I.; Okazaki, M.M.; Nadler, J. V Status epilepticus-induced hilar basal dendrites on rodent granule cells contribute to recurrent excitatory circuitry. J. Comp. Neurol. 2000, 428, 240–253. [Google Scholar] [CrossRef]
- Yan, X.X.; Spigelman, I.; Tran, P.H.; Ribak, C.E. Atypical features of rat dentate granule cells: Recurrent basal dendrites and apical axons. Anat. Embryol. (Berl). 2001, 203, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Dashtipour, K.; Yan, X.-X.; Dinh, T.T.; Okazaki, M.M.; Nadler, J.V.; Ribak, C.E. Quantitative and morphological analysis of dentate granule cells with recurrent basal dendrites from normal and epileptic rats. Hippocampus 2002, 12, 235–244. [Google Scholar] [CrossRef]
- Walter, C.; Murphy, B.L.; Pun, R.Y.K.; Spieles-Engemann, A.L.; Danzer, S.C. Pilocarpine-induced seizures cause selective time-dependent changes to adult-generated hippocampal dentate granule cells. J. Neurosci. 2007, 27, 7541–7552. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mach, M.; Zagaja, M.; Haratym-Maj, A.; Rola, R.; Maj, M.; Haratym, J.; Dudra-Jastrzębska, M.; Łuszczki, J. A long-term treatment with arachidonyl-2′-chloroethylamide combined with valproate increases neurogenesis in a mouse pilocarpine model of Epilepsy. Int. J. Mol. Sci. 2017, 18, 900. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mach, M.; Haratym-Maj, A.; Zagaja, M.; Rola, R.; Maj, M.; Chrościńska-Krawczyk, M.; Luszczki, J.J. ACEA (a highly selective cannabinoid CB1 receptor agonist) stimulates hippocampal neurogenesis in mice treated with antiepileptic drugs. Brain Res. 2015, 1624, 86–94. [Google Scholar] [CrossRef]
- Kambli, L.; Bhatt, L.K.; Oza, M.; Prabhavalkar, K. Novel therapeutic targets for epilepsy intervention. Seizure 2017, 51, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Danzer, S.C. Adult neurogenesis: Opening the gates of Troy from the inside. Epilepsy Curr. 2015, 15, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cui, X.-L.; Wang, Y.; Li, X.-W.; Yang, F.; Wei, D.; Jiang, W. Aspirin attenuates spontaneous recurrent seizures and inhibits hippocampal neuronal loss, mossy fiber sprouting and aberrant neurogenesis following pilocarpine-induced status epilepticus in rats. Brain Res. 2012, 1469, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-H.; Chu, K.; Lee, S.-T.; Kim, J.; Sinn, D.-I.; Kim, J.-M.; Park, D.-K.; Lee, J.-J.; Kim, S.U.; Kim, M.; et al. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2006, 23, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-O.; Lybrand, Z.R.; Ito, N.; Brulet, R.; Tafacory, F.; Zhang, L.; Good, L.; Ure, K.; Kernie, S.G.; Birnbaum, S.G.; et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 2015, 6, 6606. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Sahay, A.; Hen, R. Adult hippocampal neurogenesis in depression. Nat. Neurosci. 2007, 10, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Drab, A.; Haratym-Maj, A.; Zagaja, M.; Łuszczki, J.; Andres-Mach, M. Endocannabinoid system and cannabinoids in neurogenesis – new opportunities for neurological treatment? Reports from experimental studies. J. Pre-Clinical Clin. Res. 2017, 11, 76–80. [Google Scholar] [CrossRef]
- Morena, M.; Patel, S.; Bains, J.S.; Hill, M.N. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 2016, 41, 80–102. [Google Scholar] [CrossRef]
- Rotheneichner, P.; Lange, S.; O’Sullivan, A.; Marschallinger, J.; Zaunmair, P.; Geretsegger, C.; Aigner, L.; Couillard-Despres, S. Hippocampal neurogenesis and antidepressive therapy: Shocking relations. Neural Plast. 2014, 2014, 723915. [Google Scholar] [CrossRef]
- Fogaça, M.V.; Galve-Roperh, I.; Guimarães, F.S.; Campos, A.C. Cannabinoids, neurogenesis and antidepressant drugs: Is there a link? Curr. Neuropharmacol. 2013, 11, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Hillard, C.J.; Bambico, F.R.; Patel, S.; Gorzalka, B.B.; Gobbi, G. The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol. Sci. 2009, 30, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Gorzalka, B.B.; Hill, M.N. Putative role of endocannabinoid signaling in the etiology of depression and actions of antidepressants. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2011, 35, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Miller, G.E.; Ho, W.-S.; Gorzalka, B.B.; Hillard, C.J. Serum endocannabinoid content is altered in females with depressive disorders: A preliminary report. Pharmacopsychiatry 2008, 41, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Miller, G.E.; Carrier, E.J.; Gorzalka, B.B.; Hillard, C.J. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology 2009, 34, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; McLaughlin, R.J.; Bingham, B.; Shrestha, L.; Lee, T.T.Y.; Gray, J.M.; Hillard, C.J.; Gorzalka, B.B.; Viau, V. Endogenous cannabinoid signaling is essential for stress adaptation. Proc. Natl. Acad. Sci. USA 2010, 107, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Reich, C.G.; Taylor, M.E.; McCarthy, M.M. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav. Brain Res. 2009, 203, 264–269. [Google Scholar] [CrossRef]
- Hill, M.N.; Carrier, E.J.; McLaughlin, R.J.; Morrish, A.C.; Meier, S.E.; Hillard, C.J.; Gorzalka, B.B. Regional alterations in the endocannabinoid system in an animal model of depression: Effects of concurrent antidepressant treatment. J. Neurochem. 2008, 106, 2322–2336. [Google Scholar] [CrossRef]
- Lazary, J.; Eszlari, N.; Juhasz, G.; Bagdy, G. Genetically reduced FAAH activity may be a risk for the development of anxiety and depression in persons with repetitive childhood trauma. Eur. Neuropsychopharmacol. 2016, 26, 1020–1028. [Google Scholar] [CrossRef]
- Monteleone, P.; Bifulco, M.; Maina, G.; Tortorella, A.; Gazzerro, P.; Proto, M.C.; Di Filippo, C.; Monteleone, F.; Canestrelli, B.; Buonerba, G. Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression. Pharmacol. Res. 2010, 61, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Roelke, C.T.; Rademacher, D.J.; Hillard, C.J. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling: Limbic endocannabinoid signalling and stress. Eur. J. Neurosci. 2005, 21, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, D.J.; Meier, S.E.; Shi, L.; Vanessa Ho, W.-S.; Jarrahian, A.; Hillard, C.J. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology 2008, 54, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hillard, C.J. Adaptations in endocannabinoid signaling in response to repeated homotypic stress: A novel mechanism for stress habituation. Eur. J. Neurosci. 2008, 27, 2821–2829. [Google Scholar] [CrossRef] [PubMed]
- Lutz, B.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705–718. [Google Scholar] [CrossRef]
- Rey, A.A.; Purrio, M.; Viveros, M.-P.; Lutz, B. Biphasic effects of cannabinoids in anxiety responses: CB1 and GABAB receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology 2012, 37, 2624–2634. [Google Scholar] [CrossRef]
- Viveros, M.P.; Marco, E.; File, S. Endocannabinoid system and stress and anxiety responses. Pharmacol. Biochem. Behav. 2005, 81, 331–342. [Google Scholar] [CrossRef]
- Häring, M.; Grieb, M.; Monory, K.; Lutz, B.; Moreira, F.A. Cannabinoid CB1 receptor in the modulation of stress coping behavior in mice: The role of serotonin and different forebrain neuronal subpopulations. Neuropharmacology 2013, 65, 83–89. [Google Scholar] [CrossRef] [PubMed]
- El-Alfy, A.T.; Ivey, K.; Robinson, K.; Ahmed, S.; Radwan, M.; Slade, D.; Khan, I.; ElSohly, M.; Ross, S. Antidepressant-like effect of Δ9-tetrahydrocannabinol and other cannabinoids isolated from Cannabis sativa L. Pharmacol. Biochem. Behav. 2010, 95, 434–442. [Google Scholar] [CrossRef]
- Hill, M.N.; Gorzalka, B.B. Pharmacological enhancement of cannabinoid CB1 receptor activity elicits an antidepressant-like response in the rat forced swim test. Eur. Neuropsychopharmacol. 2005, 15, 593–599. [Google Scholar] [CrossRef]
- Rutkowska, M.; Jachimczuk, O. Antidepressant-like properties of ACEA (arachidonyl-2-chloroethylamide), the selective agonist of CB1 receptors. Acta Pol. Pharm. 2004, 61, 165–167. [Google Scholar] [PubMed]
- ElBatsh, M.M.; Moklas, M.A.A.; Marsden, C.A.; Kendall, D.A. Antidepressant-like effects of Δ9-tetrahydrocannabinol and rimonabant in the olfactory bulbectomised rat model of depression. Pharmacol. Biochem. Behav. 2012, 102, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, D.J.; Hillard, C.J. Interactions between endocannabinoids and stress-induced decreased sensitivity to natural reward. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 633–641. [Google Scholar] [CrossRef]
- Segev, A.; Rubin, A.S.; Abush, H.; Richter-Levin, G.; Akirav, I. Cannabinoid receptor activation prevents the effects of chronic mild stress on emotional learning and LTP in a rat model of depression. Neuropsychopharmacology 2014, 39, 919–933. [Google Scholar] [CrossRef]
- Rubino, T.; Parolaro, D. The impact of exposure to cannabinoids in adolescence: Insights from animal models. Biol. Psychiatry 2016, 79, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Parolaro, D. Sexually dimorphic effects of cannabinoid compounds on emotion and cognition. Front. Behav. Neurosci. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ran, S.; Roerecke, M.; Le Foll, B.; George, T.P.; McKenzie, K.; Rehm, J. The association between cannabis use and depression: A systematic review and meta-analysis of longitudinal studies. Psychol. Med. 2014, 44, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, L.; Hall, W.; Lynskey, M. Exploring the association between cannabis use and depression. Addiction 2003, 98, 1493–1504. [Google Scholar] [CrossRef]
- Degenhardt, L.; Hall, W.; Lynskey, M. The relationship between cannabis use, depression and anxiety among Australian adults: Findings from the National Survey of Mental Health and Well-Being. Soc. Psychiatry Psychiatr. Epidemiol. 2001, 36, 219–227. [Google Scholar] [CrossRef]
- Hall, W.; Degenhardt, L. The adverse health effects of chronic cannabis use. Drug Test. Anal. 2014, 6, 39–45. [Google Scholar] [CrossRef]
- Degenhardt, L.; Coffey, C.; Romaniuk, H.; Swift, W.; Carlin, J.B.; Hall, W.D.; Patton, G.C. The persistence of the association between adolescent cannabis use and common mental disorders into young adulthood. Addiction 2013, 108, 124–133. [Google Scholar] [CrossRef]
- Patton, G.C.; Coffey, C.; Carlin, J.B.; Degenhardt, L.; Lynskey, M.; Hall, W. Cannabis use and mental health in young people: Cohort study. Bmj 2002, 325, 1195–1198. [Google Scholar] [CrossRef]
- Hayatbakhsh, M.R.; Najman, J.M.; Jamrozik, K.; Mamun, A.A.; Alati, R.; Bor, W. Cannabis and anxiety and depression in young adults: A large prospective study. J. Am. Acad. Child Adolesc. Psychiatry 2007, 46, 408–417. [Google Scholar] [CrossRef]
- Higuera-Matas, A.; Ucha, M.; Ambrosio, E. Long-term consequences of perinatal and adolescent cannabinoid exposure on neural and psychological processes. Neurosci. Biobehav. Rev. 2015, 55, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Abboussi, O.; Tazi, A.; Paizanis, E.; El Ganouni, S. Chronic exposure to WIN55,212-2 affects more potently spatial learning and memory in adolescents than in adult rats via a negative action on dorsal hippocampal neurogenesis. Pharmacol. Biochem. Behav. 2014, 120, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Cuccurazzu, B.; Zamberletti, E.; Nazzaro, C.; Prini, P.; Trusel, M.; Grilli, M.; Parolaro, D.; Tonini, R.; Rubino, T. Adult cellular neuroadaptations induced by adolescent THC exposure in female rats are rescued by enhancing anandamide signaling. Int. J. Neuropsychopharmacol. 2018, 21, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.T.-Y.Y.; Wainwright, S.R.; Hill, M.N.; Galea, L.A.M.M.; Gorzalka, B.B. Sex, drugs, and adult neurogenesis: Sex-dependent effects of escalating adolescent cannabinoid exposure on adult hippocampal neurogenesis, stress reactivity, and amphetamine sensitization. Hippocampus 2014, 24, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Morrish, A.C.; Hill, M.N.; Riebe, C.J.N.; Gorzalka, B.B. Protracted cannabinoid administration elicits antidepressant behavioral responses in rats: Role of gender and noradrenergic transmission. Physiol. Behav. 2009, 98, 118–124. [Google Scholar] [CrossRef]
- Bambico, F.R.; Katz, N.; Debonnel, G.; Gobbi, G. Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J. Neurosci. 2007, 27, 11700–11711. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, Y.; Xiao, L.; Van Cleemput, J.; Ji, S.-P.; Bai, G.; Zhang, X. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J. Clin. Invest. 2005, 115, 3104–3116. [Google Scholar] [CrossRef]
- Viveros, M.-P.; Marco, E.M. Age-dependent effects of cannabinoids on neurophysiological, emotional, and motivational states. In Cannabinoid Modulation of Emotion, Memory, and Motivation; Campolongo, P., Fattore, L., Eds.; Springer: New York, NY, USA, 2015; pp. 245–281. ISBN 978-1-4939-2293-2. [Google Scholar]
- Griebel, G.; Stemmelin, J.; Scatton, B. Effects of the cannabinoid CB1 receptor antagonist rimonabant in models of emotional reactivity in rodents. Biol. Psychiatry 2005, 57, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Hernández, E.; Muñoz, R.M.; del Arco, I.; Villanúa, M.A.; Carrera, M.R.; Rodríguez de Fonseca, F. Acute administration of the CB1 cannabinoid receptor antagonist SR 141716A induces anxiety-like responses in the rat. Neuroreport 1997, 8, 491–496. [Google Scholar] [CrossRef]
- Patel, S. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: Further evidence for an anxiolytic role for endogenous cannabinoid signaling. J. Pharmacol. Exp. Ther. 2006, 318, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.; Bakos, N.; Szirmay, M.; Ledent, C.; Freund, T.F. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur. J. Neurosci. 2002, 16, 1395–1398. [Google Scholar] [CrossRef]
- Beyer, C.E.; Dwyer, J.M.; Piesla, M.J.; Platt, B.J.; Shen, R.; Rahman, Z.; Chan, K.; Manners, M.T.; Samad, T.A.; Kennedy, J.D.; et al. Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol. Dis. 2010, 39, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Boekholdt, S.M.; Peters, R.J. Rimonabant: Obituary for a wonder drug. Lancet 2010, 376, 489–490. [Google Scholar] [CrossRef]
- Juhasz, G.; Chase, D.; Pegg, E.; Downey, D.; Toth, Z.G.; Stones, K.; Platt, H.; Mekli, K.; Payton, A.; Elliott, R. CNR1 gene is associated with high neuroticism and low agreeableness and interacts with recent negative life events to predict current depressive symptoms. Neuropsychopharmacology 2009, 34, 2019–2027. [Google Scholar] [CrossRef]
- Domschke, K.; Dannlowski, U.; Ohrmann, P.; Lawford, B.; Bauer, J.; Kugel, H.; Heindel, W.; Young, R.; Morris, P.; Arolt, V.; et al. Cannabinoid receptor 1 (CNR1) gene: Impact on antidepressant treatment response and emotion processing in Major Depression. Eur. Neuropsychopharmacol. 2008, 18, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Patel, S.; Carrier, E.J.; Rademacher, D.J.; Ormerod, B.K.; Hillard, C.J.; Gorzalka, B.B. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology 2005, 30, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology (Berl). 2002, 159, 379–387. [Google Scholar] [CrossRef]
- Verty, A.N.A.; Stefanidis, A.; McAinch, A.J.; Hryciw, D.H.; Oldfield, B. Anti-obesity effect of the CB2 receptor agonist JWH-015 in diet-induced obese mice. PLoS ONE 2015, 10, e0140592. [Google Scholar] [CrossRef] [PubMed]
- Bahi, A.; Al Mansouri, S.; Al Memari, E.; Al Ameri, M.; Nurulain, S.M.; Ojha, S. β-Caryophyllene, a CB2 receptor agonist produces multiple behavioral changes relevant to anxiety and depression in mice. Physiol. Behav. 2014, 135, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.-P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.-R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 receptors: Immunohistochemical localization in rat brain. Brain Res. 2006, 1071, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, K.J.; Tafesse, L.; Lee, G.; Harrison, J.E.; Boulet, J.M.; Gottshall, S.L.; Mark, L.; Pearson, M.S.; Miller, W.; Shan, S.; et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology 2005, 48, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Sejal, P.; Meozzi, P.A.; Myers, L.; Tagliaferro, P.; Hope, B.; Leonard, C.M.; Uhl, G.R.; Brusco, A.; et al. Methods to study the behavioral effects and expression of CB2 cannabinoid receptor and its gene transcripts in the chronic mild stress model of depression. Methods Mol. Med. 2006, 123, 291–298. [Google Scholar] [PubMed]
- García-Gutiérrez, M.S.; García-Bueno, B.; Zoppi, S.; Leza, J.C.; Manzanares, J. Chronic blockade of cannabinoid CB2 receptors induces anxiolytic-like actions associated with alterations in GABA(A) receptors. Br. J. Pharmacol. 2012, 165, 951–964. [Google Scholar] [CrossRef]
- Kruk-Slomka, M.; Michalak, A.; Biala, G. Antidepressant-like effects of the cannabinoid receptor ligands in the forced swimming test in mice: Mechanism of action and possible interactions with cholinergic system. Behav. Brain Res. 2015, 284, 24–36. [Google Scholar] [CrossRef]
- García-Gutiérrez, M.S.; Pérez-Ortiz, J.M.; Gutiérrez-Adán, A.; Manzanares, J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB2 receptors. Br. J. Pharmacol. 2010, 160, 1773–1784. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann. N. Y. Acad. Sci. 2008, 1139, 434–449. [Google Scholar] [CrossRef]
- Carrasquer, A.; Nebane, N.M.; Williams, W.M.; Song, Z.-H. Functional consequences of nonsynonymous single nucleotide polymorphisms in the CB2 cannabinoid receptor. Pharmacogenet Genomics 2010, 20, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Alvaro, A.; Aracil-Fernández, A.; García-Gutiérrez, M.S.; Navarrete, F.; Manzanares, J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology 2011, 36, 1489–1504. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: From mice to human subjects. PLoS ONE 2008, 3, e1640. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Elevating endocannabinoid levels: Pharmacological strategies and potential therapeutic applications. Proc. Nutr. Soc. 2014, 73, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, N.; Duan, T.; Kesner, P.; Blaskovits, F.; Liu, J.; Lu, Y.; Tong, L.; Gao, F.; Harris, C.; et al. Monoacylglycerol lipase inhibitors produce pro-or antidepressant responses via hippocampal CA1 GABAergic synapses. Mol. Psychiatry 2017, 22, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.A.; Kaiser, N.; Monory, K.; Lutz, B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology 2008, 54, 141–150. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X. FAAH inhibition produces antidepressant-like efforts of mice to acute stress via synaptic long-term depression. Behav. Brain Res. 2017, 324, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Mangieri, R.A.; Fu, J.; Kim, J.H.; Arguello, O.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry 2007, 62, 1103–1110. [Google Scholar] [CrossRef]
- Fowler, C. Monoacylglycerol lipase - a target for drug development? Br. J. Pharmacol. 2012, 166, 1568–1585. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, W.; Zhong, P.; Liu, S.J.; Long, J.Z.; Zhao, L.; Gao, H.; Cravatt, B.F.; Liu, Q.; Wang, H.; et al. Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus 2015, 25, 16–26. [Google Scholar] [CrossRef]
- Zhong, P.; Wang, W.; Pan, B.; Liu, X.; Zhang, Z.; Long, J.Z.; Zhang, H.; Cravatt, B.F.; Liu, Q. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 2014, 39, 1763–1776. [Google Scholar] [PubMed]
- Busquets-Garcia, A.; Puighermanal, E.; Pastor, A.; de la Torre, R.; Maldonado, R.; Ozaita, A. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol. Psychiatry 2011, 70, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Crippa, J.A.; Guimarães, F.S.; Campos, A.C.; Zuardi, A.W. Translational investigation of the therapeutic potential of cannabidiol (CBD): Toward a new age. Front. Immunol. 2018, 9, 2009. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Fogaça, M.V.; Sonego, A.B.; Guimarães, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Blessing, E.M.; Steenkamp, M.M.; Manzanares, J.; Marmar, C.R. Cannabidiol as a potential treatment for anxiety disorders. Neurotherapeutics 2015, 12, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Linge, R.; Jiménez-Sánchez, L.; Campa, L.; Pilar-Cuéllar, F.; Vidal, R.; Pazos, A.; Adell, A.; Díaz, Á. Cannabidiol induces rapid-acting antidepressant-like effects and enhances cortical 5-HT/glutamate neurotransmission: Role of 5-HT1A receptors. Neuropharmacology 2016, 103, 16–26. [Google Scholar] [CrossRef]
- Resstel, L.B.M.; Joca, S.R.L.; Moreira, F.A.; Corrêa, F.M.A.; Guimarães, F.S. Effects of cannabidiol and diazepam on behavioral and cardiovascular responses induced by contextual conditioned fear in rats. Behav. Brain Res. 2006, 172, 294–298. [Google Scholar] [CrossRef]
- Guimarães, F.S.; Chiaretti, T.M.; Graeff, F.G.; Zuardi, A.W. Antianxiety effect of cannabidiol in the elevated plus-maze. Psychopharmacology 1990, 100, 558–559. [Google Scholar] [CrossRef]
- Zanelati, T.; Biojone, C.; Moreira, F.; Guimarães, F.; Joca, S. Antidepressant-like effects of cannabidiol in mice: Possible involvement of 5-HT1A receptors: Cannabidiol induces antidepressant-like effect. Br. J. Pharmacol. 2010, 159, 122–128. [Google Scholar] [CrossRef]
- Sales, A.J.; Crestani, C.C.; Guimarães, F.S.; Joca, S.R.L. Antidepressant-like effect induced by Cannabidiol is dependent on brain serotonin levels. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2018, 86, 255–261. [Google Scholar]
- Sales, A.J.; Fogaça, M.V.; Sartim, A.G.; Pereira, V.S.; Wegener, G.; Guimarães, F.S.; Joca, S.R.L. Cannabidiol induces rapid and sustained antidepressant-like effects through increased BDNF signaling and synaptogenesis in the prefrontal cortex. Mol. Neurobiol. 2019, 56, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Zuardi, A.W.; Rodrigues, N.P.; Silva, A.L.; Bernardo, S.A.; Hallak, J.E.C.; Guimarães, F.S.; Crippa, J.A.S. Inverted U-shaped dose-response curve of the anxiolytic effect of cannabidiol during public speaking in real life. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: Potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, G.; Lupi, M.; Sarchione, F.; Matarazzo, I.; Santacroce, R.; Petruccelli, F.; Martinotti, G.; Di Giannantonio, M. The endocannabinoid system: A putative role in neurodegenerative diseases. Int. J. High Risk Behav. Addict. 2013, 2, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Parker, L. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.W.; Oliveira, C.L.; Guimarães, F.S.; Campos, A.C. Cannabinoid signalling in embryonic and adult neurogenesis: Possible implications for psychiatric and neurological disorders. Acta Neuropsychiatr. 2018, 1–16. [Google Scholar] [CrossRef]
- Park, J.-Y.; Wu, L.-T. Prevalence, reasons, perceived effects, and correlates of medical marijuana use: A review. Drug Alcohol Depend. 2017, 177, 1–13. [Google Scholar] [PubMed]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, R.S.; Lourenço, D.M.; Paulo, S.L.; Mateus, J.M.; Ferreira, M.F.; Mouro, F.M.; Moreira, J.B.; Ribeiro, F.F.; Sebastião, A.M.; Xapelli, S. Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology. Molecules 2019, 24, 1350. https://doi.org/10.3390/molecules24071350
Rodrigues RS, Lourenço DM, Paulo SL, Mateus JM, Ferreira MF, Mouro FM, Moreira JB, Ribeiro FF, Sebastião AM, Xapelli S. Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology. Molecules. 2019; 24(7):1350. https://doi.org/10.3390/molecules24071350
Chicago/Turabian StyleRodrigues, Rui S., Diogo M. Lourenço, Sara L. Paulo, Joana M. Mateus, Miguel F. Ferreira, Francisco M. Mouro, João B. Moreira, Filipa F. Ribeiro, Ana M. Sebastião, and Sara Xapelli. 2019. "Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology" Molecules 24, no. 7: 1350. https://doi.org/10.3390/molecules24071350
APA StyleRodrigues, R. S., Lourenço, D. M., Paulo, S. L., Mateus, J. M., Ferreira, M. F., Mouro, F. M., Moreira, J. B., Ribeiro, F. F., Sebastião, A. M., & Xapelli, S. (2019). Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology. Molecules, 24(7), 1350. https://doi.org/10.3390/molecules24071350