
Hippocampal Stratum Oriens Somatostatin-Positive Cells Undergo CB1-Dependent Long-Term Potentiation and Express Endocannabinoid Biosynthetic Enzymes



Abstract
1. Introduction
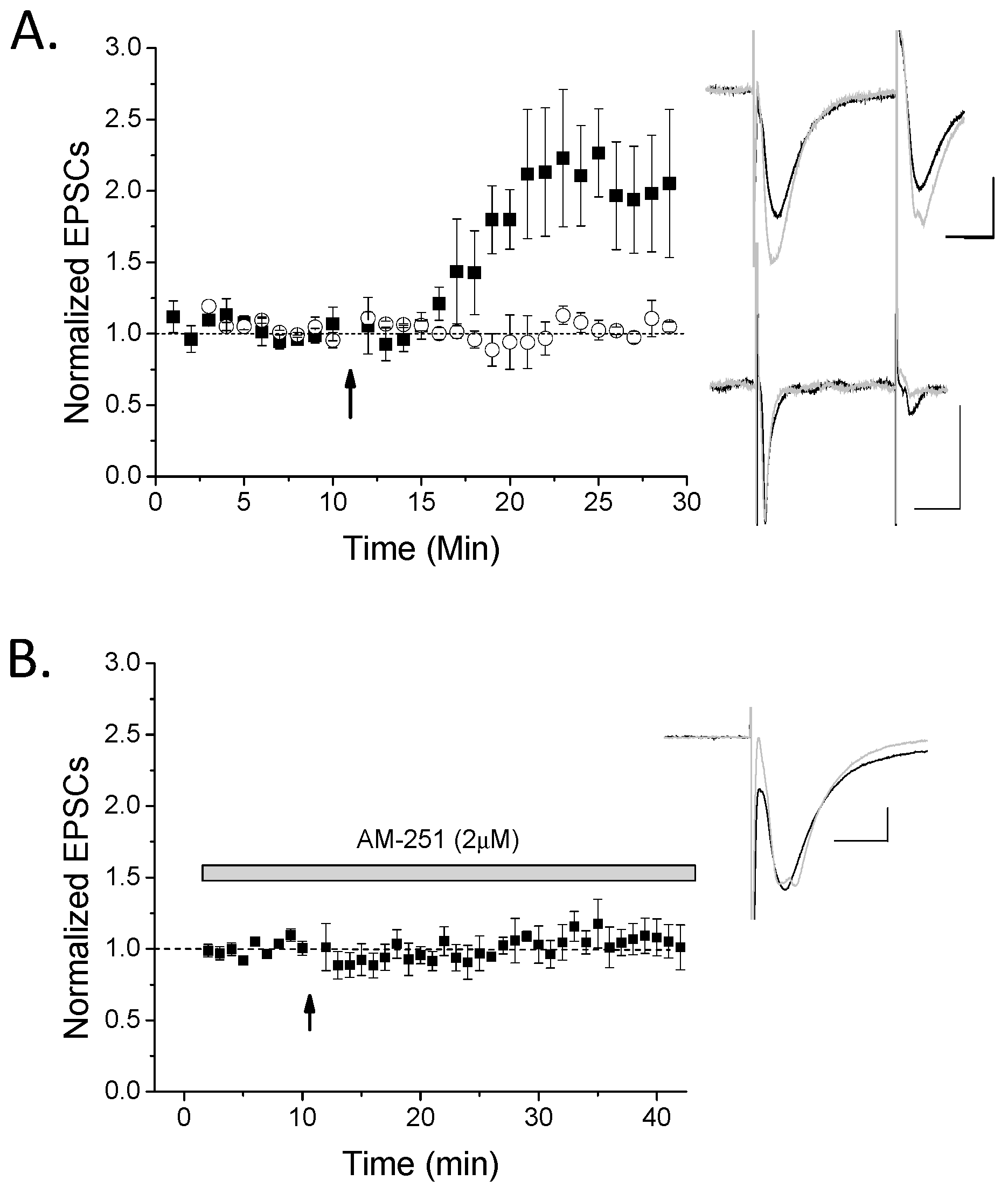
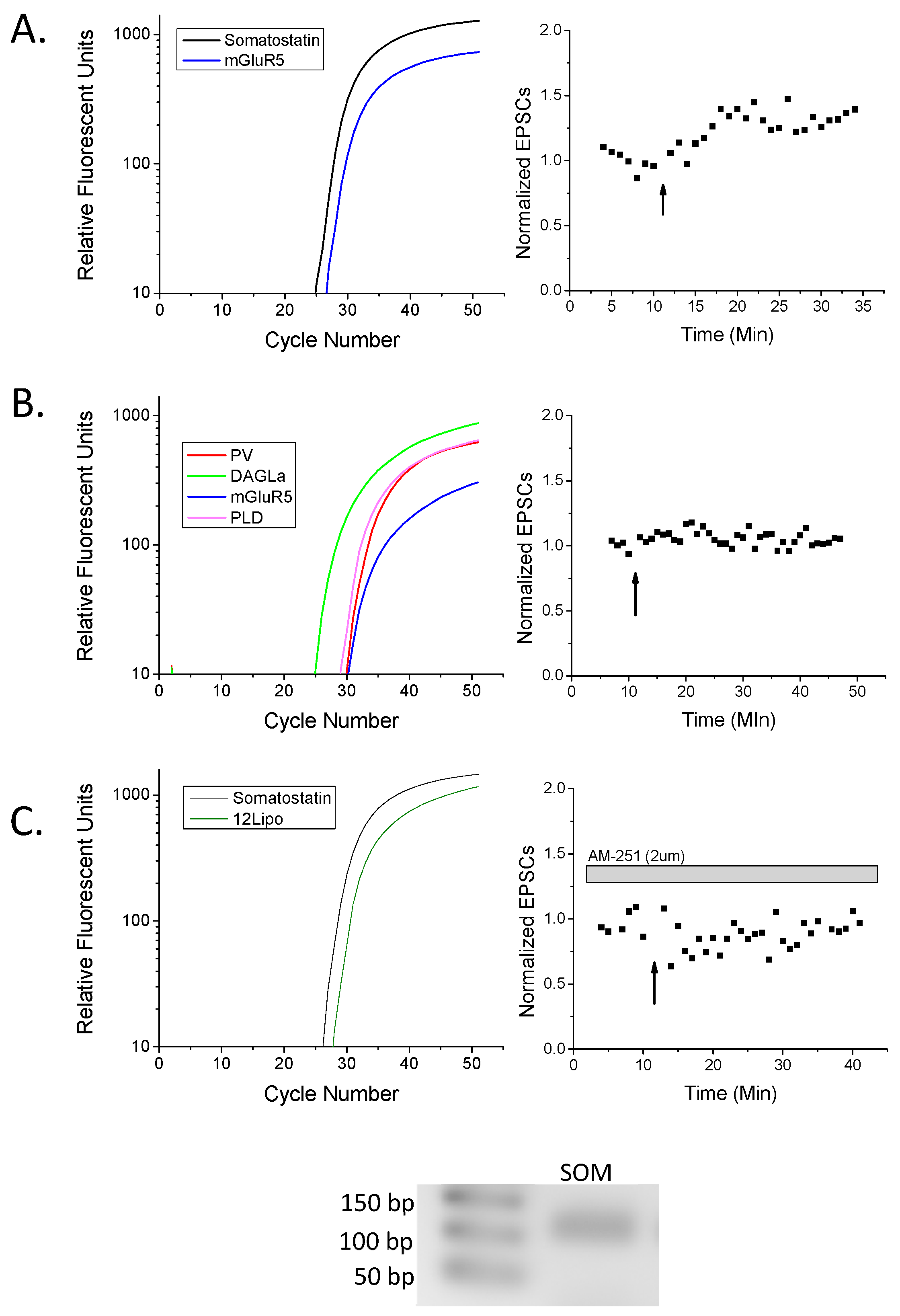
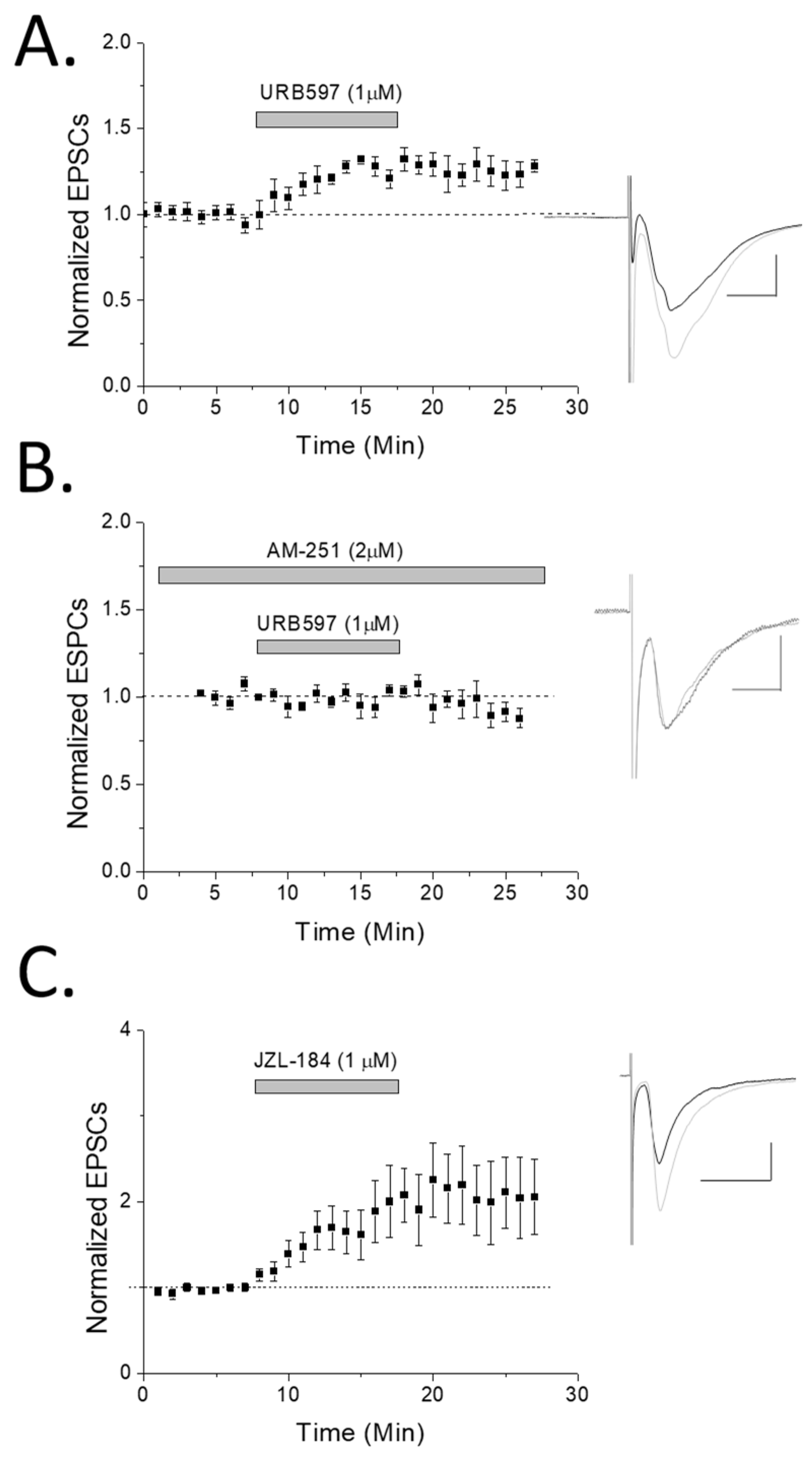
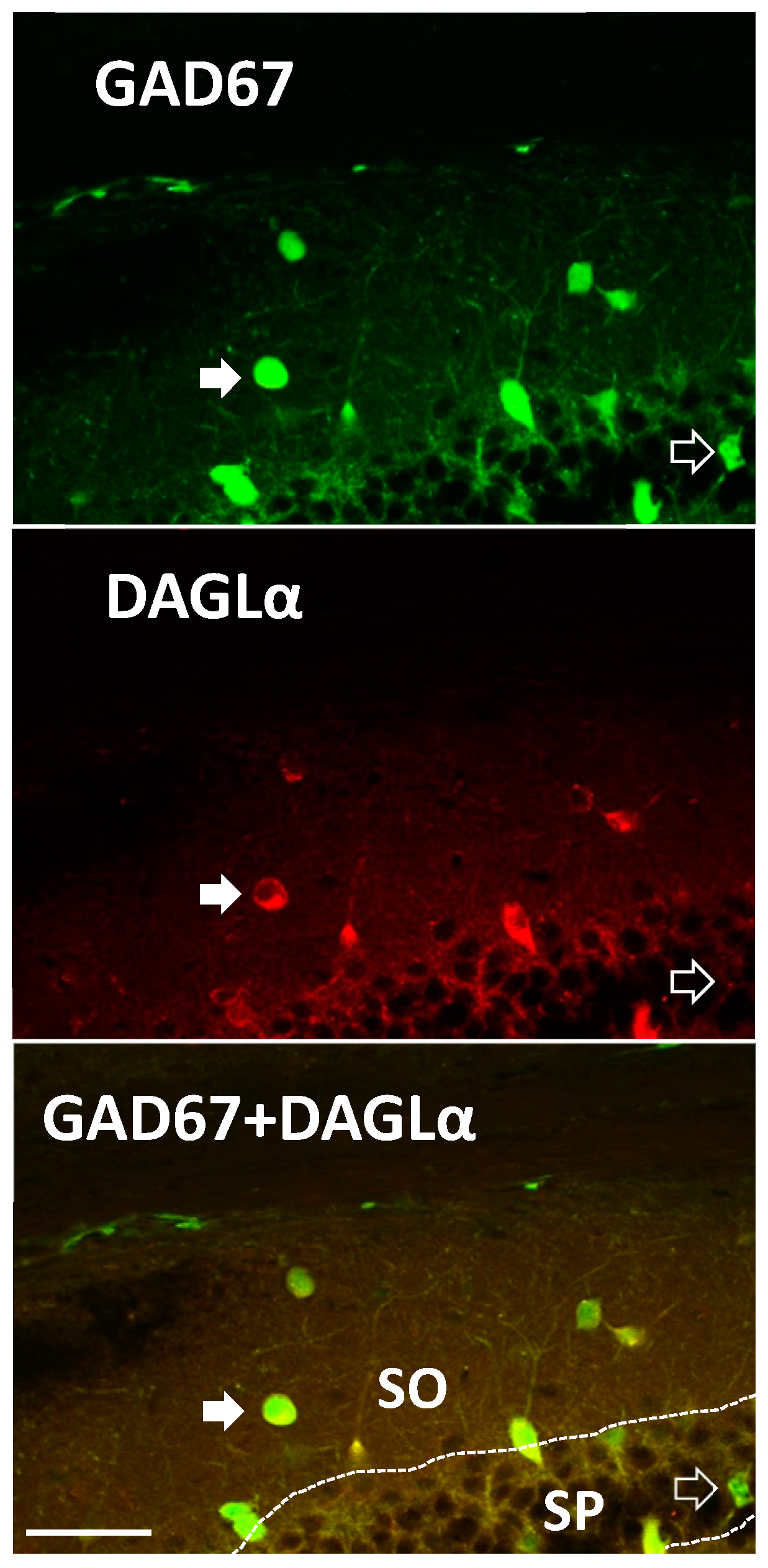
2. Results
3. Discussion
3.1. eCB Production
3.2. CB1 eCB Plasticity
4. Methods
4.1. Electrophysiology
4.2. Reverse Transcription and Pre-Amplification
4.3. Primer Design
4.4. Quantitative RT-PCR Reaction
4.5. Immunohistochemistry
5. Significance
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Bear, M.F. Homosynaptic Long-Term Depression in Area CA1 of Hippocampus and Effects of N-Methyl-D-Aspartate Receptor Blockade. Proc. Natl. Acad. Sci. USA 1992, 89, 4363–4367. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Takahashi, K.A.; Castillo, P.E. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 2006, 29, 37–76. [Google Scholar] [CrossRef]
- Egertova, M.; Simon, G.M.; Cravatt, B.F.; Elphick, M.R. Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: A new perspective on N-acylethanolamines as neural signaling molecules. J. Comp. Neurol. 2008, 506, 604–615. [Google Scholar] [CrossRef]
- Jung, K.M.; Mangieri, R.; Stapleton, C.; Kim, J.; Fegley, D.; Wallace, M.; Mackie, K.; Piomelli, D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol. Pharmacol. 2005, 68, 1196–1202. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M.; et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar] [CrossRef]
- Merrill, C.B.; McNeil, M.; Williamson, R.C.; Poole, B.R.; Nelson, B.; Sudweeks, S.; Edwards, J.G. Identification of mRNA for endocannabinoid biosynthetic enzymes within hippocampal pyramidal cells and CA1 stratum radiatum interneuron subtypes using quantitative real-time polymerase chain reaction. Neuroscience 2012, 218, 89–99. [Google Scholar] [CrossRef]
- Brown, A.J. Novel cannabinoid receptors. Br. J. Pharmacol. 2007, 152, 567–575. [Google Scholar] [CrossRef]
- Cristino, L.; Starowicz, K.; De Petrocellis, L.; Morishita, J.; Ueda, N.; Guglielmotti, V.; Di Marzo, V. Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience 2008, 151, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef]
- Freund, T.F.; Buzsaki, G. Interneurons of the hippocampus. Hippocampus 1996, 6, 347–470. [Google Scholar] [CrossRef]
- Oren, I.; Nissen, W.; Kullmann, D.M.; Somogyi, P.; Lamsa, K.P. Role of ionotropic glutamate receptors in long-term potentiation in rat hippocampal CA1 oriens-lacunosum moleculare interneurons. J. Neurosci. 2009, 29, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, E.; Kullmann, D.M. Long-term potentiation in hippocampal oriens interneurons: Postsynaptic induction, presynaptic expression and evaluation of candidate retrograde factors. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130133. [Google Scholar] [CrossRef]
- Huh, C.Y.; Amilhon, B.; Ferguson, K.A.; Manseau, F.; Torres-Platas, S.G.; Peach, J.P.; Scodras, S.; Mechawar, N.; Skinner, F.K.; Williams, S. Excitatory Inputs Determine Phase-Locking Strength and Spike-Timing of CA1 Stratum Oriens/Alveus Parvalbumin and Somatostatin Interneurons during Intrinsically Generated Hippocampal Theta Rhythm. J. Neurosci. 2016, 36, 6605–6622. [Google Scholar] [CrossRef]
- Maccaferri, G.; McBain, C.J. Long-term potentiation in distinct subtypes of hippocampal nonpyramidal neurons. J. Neurosci. 1996, 16, 5334–5343. [Google Scholar] [CrossRef]
- Schlingloff, D.; Kali, S.; Freund, T.F.; Hajos, N.; Gulyas, A.I. Mechanisms of sharp wave initiation and ripple generation. J. Neurosci. 2014, 34, 11385–11398. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.G.; Gibson, H.E.; Jensen, T.; Nugent, F.; Walther, C.; Blickenstaff, J.; Kauer, J.A. A novel non-CB1/TRPV1 endocannabinoid-mediated mechanism depresses excitatory synapses on hippocampal CA1 interneurons. Hippocampus 2012, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Le Duigou, C.; Kullmann, D.M. Group I mGluR Agonist-Evoked Long-Term Potentiation in Hippocampal Oriens Interneurons. J. Neurosci. 2011, 31, 5777–5781. [Google Scholar] [CrossRef]
- Lamsa, K.P.; Heeroma, J.H.; Somogyi, P.; Rusakov, D.A.; Kullmann, D.M. Anti-Hebbian Long-Term Potentiation in the Hippocampal Feedback Inhibitory Circuit. Science 2007, 315, 1262–1266. [Google Scholar] [CrossRef]
- Le Duigou, C.; Savary, E.; Kullmann, D.M.; Miles, R. Induction of Anti-Hebbian LTP in CA1 Stratum Oriens Interneurons: Interactions between Group I Metabotropic Glutamate Receptors and M1 Muscarinic Receptors. J. Neurosci. 2015, 35, 13542–13554. [Google Scholar] [CrossRef]
- Nicholson, E.; Kullmann, D.M. T-type calcium channels contribute to NMDA receptor independent synaptic plasticity in hippocampal regular-spiking oriens-alveus interneurons. J. Physiol. 2017. [Google Scholar] [CrossRef]
- Griguoli, M.; Cellot, G.; Cherubini, E. In hippocampal oriens interneurons anti-Hebbian long-term potentiation requires cholinergic signaling via alpha7 nicotinic acetylcholine receptors. J. Neurosci. 2013, 33, 1044–1049. [Google Scholar] [CrossRef]
- Wang, W.; Jia, Y.; Pham, D.T.; Palmer, L.C.; Jung, K.M.; Cox, C.D.; Rumbaugh, G.; Piomelli, D.; Gall, C.M.; Lynch, G. Atypical Endocannabinoid Signaling Initiates a New Form of Memory-Related Plasticity at a Cortical Input to Hippocampus. Cereb. Cortex (New York, N.Y. 1991) 2017, 28, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Trieu, B.H.; Palmer, L.C.; Jia, Y.; Pham, D.T.; Jung, K.M.; Karsten, C.A.; Merrill, C.B.; Mackie, K.; Gall, C.M.; et al. A Primary Cortical Input to Hippocampus Expresses a Pathway-Specific and Endocannabinoid-Dependent Form of Long-Term Potentiation. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Forro, T.; Valenti, O.; Lasztoczi, B.; Klausberger, T. Temporal organization of GABAergic interneurons in the intermediate CA1 hippocampus during network oscillations. Cerebr. Cortex (New York, N.Y. 1991) 2015, 25, 1228–1240. [Google Scholar] [CrossRef]
- Merrill, C.B.; Friend, L.N.; Newton, S.T.; Hopkins, Z.H.; Edwards, J.G. Ventral tegmental area dopamine and GABA neurons: Physiological properties and expression of mRNA for endocannabinoid biosynthetic elements. Sci. Rep. 2015, 5, 16176. [Google Scholar] [CrossRef]
- Gibson, H.E.; Edwards, J.G.; Page, R.S.; Van Hook, M.J.; Kauer, J.A. TRPV1 Channels Mediate Long-Term Depression at Synapses on Hippocampal Interneurons. Neuron 2008, 57, 746–759. [Google Scholar] [CrossRef]
- Tamamaki, N.; Yanagawa, Y.; Tomioka, R.; Miyazaki, J.; Obata, K.; Kaneko, T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 2003, 467, 60–79. [Google Scholar] [PubMed]
- Vasuta, C.; Artinian, J.; Laplante, I.; Hebert-Seropian, S.; Elayoubi, K.; Lacaille, J.C. Metaplastic Regulation of CA1 Schaffer Collateral Pathway Plasticity by Hebbian MGluR1a-Mediated Plasticity at Excitatory Synapses onto Somatostatin-Expressing Interneurons(1,2,3). eNeuro 2015, 2. [Google Scholar] [CrossRef]
- Lapointe, V.; Morin, F.; Ratte, S.; Croce, A.; Conquet, F.; Lacaille, J.C. Synapse-specific mGluR1-dependent long-term potentiation in interneurones regulates mouse hippocampal inhibition. J. Physiol. 2004, 555, 125–135. [Google Scholar] [CrossRef]
- Le Vasseur, M.; Ran, I.; Lacaille, J.C. Selective induction of metabotropic glutamate receptor 1- and metabotropic glutamate receptor 5-dependent chemical long-term potentiation at oriens/alveus interneuron synapses of mouse hippocampus. Neuroscience 2008, 151, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Katsumaru, H.; Kosaka, T.; Heizmann, C.W.; Hama, K. Immunocytochemical study of GABAergic neurons containing the calcium-binding protein parvalbumin in the rat hippocampus. Exp. Brain Res. 1988, 72, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.N.; Pertwee, R.G.; Riedel, G. Functions of cannabinoid receptors in the hippocampus. Neuropharmacology 2002, 42, 993–1007. [Google Scholar] [CrossRef]
- Lee, S.H.; Ledri, M.; Toth, B.; Marchionni, I.; Henstridge, C.M.; Dudok, B.; Kenesei, K.; Barna, L.; Szabo, S.I.; Renkecz, T.; et al. Multiple Forms of Endocannabinoid and Endovanilloid Signaling Regulate the Tonic Control of GABA Release. J. Neurosci. 2015, 35, 10039–10057. [Google Scholar] [CrossRef]
- Peterfi, Z.; Urban, G.M.; Papp, O.I.; Nemeth, B.; Monyer, H.; Szabo, G.; Erdelyi, F.; Mackie, K.; Freund, T.F.; Hajos, N.; et al. Endocannabinoid-mediated long-term depression of afferent excitatory synapses in hippocampal pyramidal cells and GABAergic interneurons. J. Neurosci. 2012, 32, 14448–14463. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, A.I.; Hajos, N.; Freund, T.F. Interneurons containing calretinin are specialized to control other interneurons in the rat hippocampus. J. Neurosci. 1996, 16, 3397–3411. [Google Scholar] [CrossRef]
- Freund, T.F.; Gulyas, A.I. Inhibitory control of GABAergic interneurons in the hippocampus. Can. J. Physiol. Pharmacol. 1997, 75, 479–487. [Google Scholar] [CrossRef]
- Freund, T.F.; Katona, I. Perisomatic inhibition. Neuron 2007, 56, 33–42. [Google Scholar] [CrossRef]
- Gloveli, T.; Dugladze, T.; Saha, S.; Monyer, H.; Heinemann, U.; Traub, R.D.; Whittington, M.A.; Buhl, E.H. Differential involvement of oriens/pyramidale interneurones in hippocampal network oscillations in vitro. J. Physiol. 2005, 562, 131–147. [Google Scholar] [CrossRef]
- Leao, R.N.; Mikulovic, S.; Leao, K.E.; Munguba, H.; Gezelius, H.; Enjin, A.; Patra, K.; Eriksson, A.; Loew, L.M.; Tort, A.B.; et al. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat. Neurosci. 2012, 15, 1524–1530. [Google Scholar] [CrossRef]
- Varma, N.; Carlson, G.C.; Ledent, C.; Alger, B.E. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J. Neurosci. 2001, 21, RC188. [Google Scholar] [CrossRef]
- Gee, C.E.; Lacaille, J.C. Group I metabotropic glutamate receptor actions in oriens/alveus interneurons of rat hippocampal CA1 region. Brain Res. 2004, 1000, 92–101. [Google Scholar] [CrossRef]
- van Hooft, J.A.; Giuffrida, R.; Blatow, M.; Monyer, H. Differential expression of group I metabotropic glutamate receptors in functionally distinct hippocampal interneurons. J. Neurosci. Off. J. Soc. Neurosc. 2000, 20, 3544–3551. [Google Scholar] [CrossRef]
- Topolnik, L.; Congar, P.; Lacaille, J.C. Differential regulation of metabotropic glutamate receptor- and AMPA receptor-mediated dendritic Ca2+ signals by presynaptic and postsynaptic activity in hippocampal interneurons. J. Neurosci. 2005, 25, 990–1001. [Google Scholar] [CrossRef]
- Hoffman, A.F.; Riegel, A.C.; Lupica, C.R. Functional localization of cannabinoid receptors and endogenous cannabinoid production in distinct neuron populations of the hippocampus. Eur. J. Neurosci. 2003, 18, 524–534. [Google Scholar] [CrossRef]
- Heifets, B.D.; Castillo, P.E. Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 2009, 71, 283–306. [Google Scholar] [CrossRef]
- Nyiri, G.; Szabadits, E.; Cserep, C.; Mackie, K.; Shigemoto, R.; Freund, T.F. GABAB and CB1 cannabinoid receptor expression identifies two types of septal cholinergic neurons. Eur. J. Neurosci. 2005, 21, 3034–3042. [Google Scholar] [CrossRef]
- Maccarrone, M.; Rossi, S.; Bari, M.; De Chiara, V.; Fezza, F.; Musella, A.; Gasperi, V.; Prosperetti, C.; Bernardi, G.; Finazzi-Agro, A.; et al. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat. Neurosci. 2008, 11, 152–159. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Castillo, P.E. Endocannabinoid-Mediated Metaplasticity in the Hippocampus. Neuron 2004, 43, 871–881. [Google Scholar] [CrossRef]
- Hajos, N.; Freund, T.F. Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology 2002, 43, 503–510. [Google Scholar] [CrossRef]
- McQuiston, A.R. Layer selective presynaptic modulation of excitatory inputs to hippocampal cornu Ammon 1 by mu-opioid receptor activation. Neuroscience 2008, 151, 209–221. [Google Scholar] [CrossRef]
- McQuiston, A.R.; Saggau, P. Mu-opioid receptors facilitate the propagation of excitatory activity in rat hippocampal area CA1 by disinhibition of all anatomical layers. J. Neurophysiol. 2003, 90, 1936–1948. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| eCB mRNA Expression in Oriens Interneurons | ||||
|---|---|---|---|---|
| Cell Marker | DAGLα | NAPE-PLD | 12-Lipo | Type I mGluRs |
| SOM; n = 14 | 6 | 4 | 1 | 5 |
| PV; n = 10 | 1 | 5 | 1 | 5 |
| CB; n = 3 | 0 | 1 | 1 | 0 |
| CR; n = 2 | 0 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friend, L.N.; Williamson, R.C.; Merrill, C.B.; Newton, S.T.; Christensen, M.T.; Petersen, J.; Wu, B.; Ostlund, I.; Edwards, J.G. Hippocampal Stratum Oriens Somatostatin-Positive Cells Undergo CB1-Dependent Long-Term Potentiation and Express Endocannabinoid Biosynthetic Enzymes. Molecules 2019, 24, 1306. https://doi.org/10.3390/molecules24071306
Friend LN, Williamson RC, Merrill CB, Newton ST, Christensen MT, Petersen J, Wu B, Ostlund I, Edwards JG. Hippocampal Stratum Oriens Somatostatin-Positive Cells Undergo CB1-Dependent Long-Term Potentiation and Express Endocannabinoid Biosynthetic Enzymes. Molecules. 2019; 24(7):1306. https://doi.org/10.3390/molecules24071306
Chicago/Turabian StyleFriend, Lindsey N., Ryan C. Williamson, Collin B. Merrill, Scott T. Newton, Michael T. Christensen, Jake Petersen, Bridget Wu, Isaac Ostlund, and Jeffrey G. Edwards. 2019. "Hippocampal Stratum Oriens Somatostatin-Positive Cells Undergo CB1-Dependent Long-Term Potentiation and Express Endocannabinoid Biosynthetic Enzymes" Molecules 24, no. 7: 1306. https://doi.org/10.3390/molecules24071306
APA StyleFriend, L. N., Williamson, R. C., Merrill, C. B., Newton, S. T., Christensen, M. T., Petersen, J., Wu, B., Ostlund, I., & Edwards, J. G. (2019). Hippocampal Stratum Oriens Somatostatin-Positive Cells Undergo CB1-Dependent Long-Term Potentiation and Express Endocannabinoid Biosynthetic Enzymes. Molecules, 24(7), 1306. https://doi.org/10.3390/molecules24071306