
An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl


Abstract
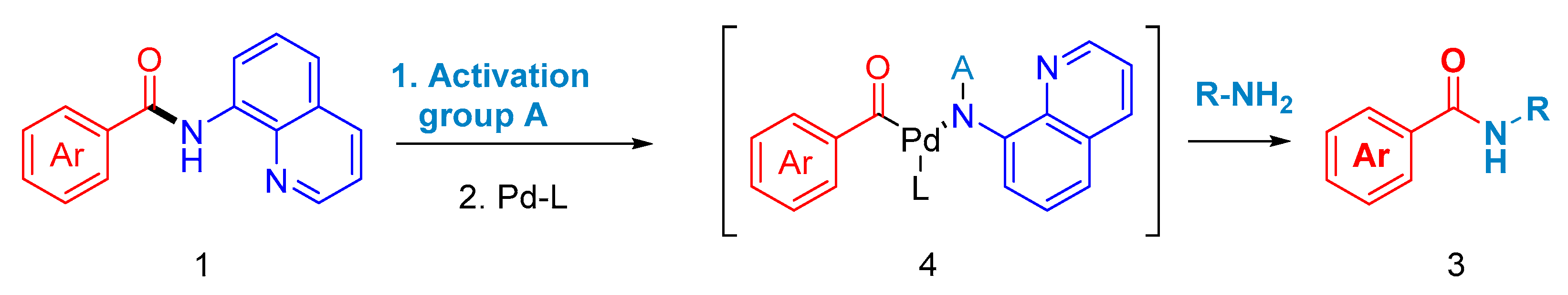
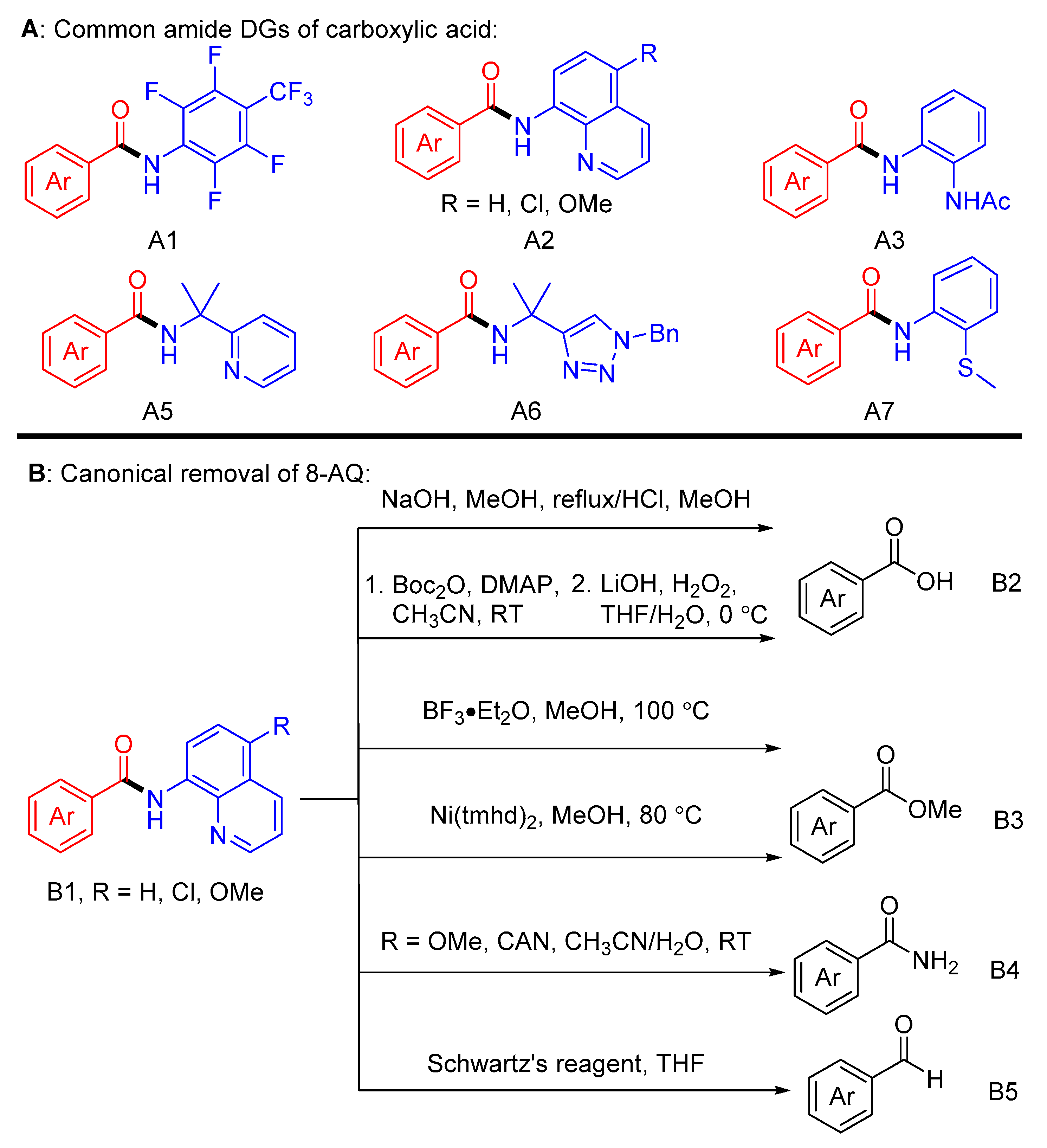
1. Introduction
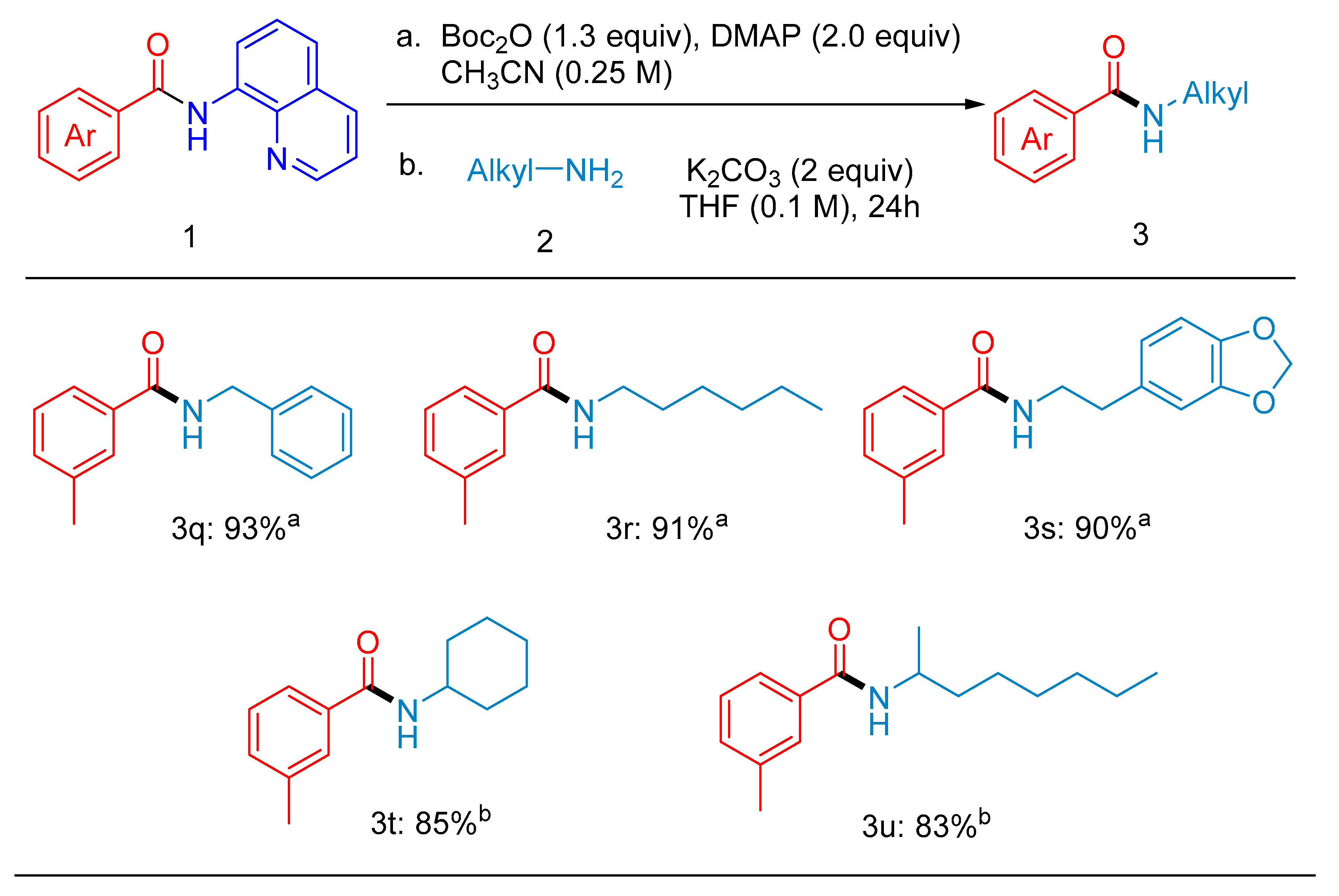
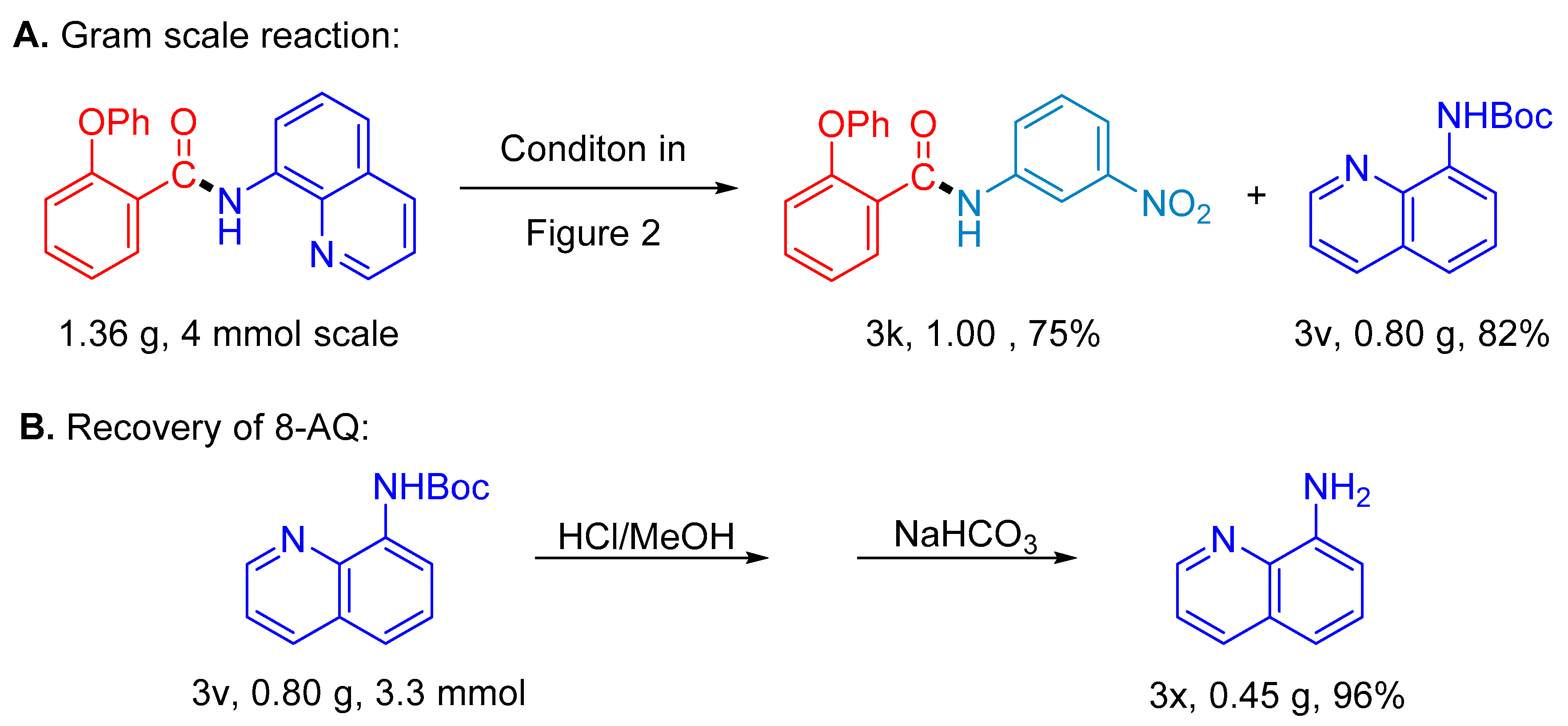
2. Results
3. Discussion
4. Experimental Section
4.1. Representative General Procedure for One-pot Transamidation of 8-Aminoquinoline Amides
4.2. Characterization Data for Products 3a–3u (Figure 2 and Figure 3)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shilov, A.E.; Shul’pin, G.B. Activation of C-H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium Catalyzed Transformations of Alkyl C−H Bonds. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C−H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Ren, Z.; Thompson, S.J.; Xu, Y.; Dong, G. Transition-Metal-Catalyzed C−H Alkylation Using Alkenes. Chem. Rev. 2017, 117, 9333–9403. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; White, M.C. A predictably selective aliphatic C-H oxidation reaction for complex molecule synthesis. Science 2007, 318, 783–787. [Google Scholar] [CrossRef]
- Qin, Y.; Zhu, L.; Luo, S. Organocatalysis in Inert C−H Bond Functionalization. Chem. Rev. 2017, 117, 9433–9520. [Google Scholar] [CrossRef]
- Abrams, D.J.; Provenchera, P.A.; Sorensen, E.J. Recent applications of C–H functionalization in complex natural product synthesis. Chem. Soc. Rev. 2018, 47, 8925–8967. [Google Scholar] [CrossRef]
- Shi, H.; Herron, A.N.; Shao, Y.; Shao, Q.; Yu, J.-Q. Enantioselective remote meta-C-H arylation and alkylation via a chiral transient mediator. Nature 2018, 558, 581–585. [Google Scholar] [CrossRef]
- Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 1993, 366, 529–531. [Google Scholar] [CrossRef]
- Kimura, N.; Kochi, T.; Kakiuchi, F. Iron-Catalyzed Regioselective Anti-Markovnikov Addition of C–H Bonds in Aromatic Ketones to Alkenes. J. Am. Chem. Soc. 2017, 139, 14849–14852. [Google Scholar] [CrossRef]
- Xiao, B.; Gong, T.-J.; Xu, J.; Liu, Z.-J.; Liu, L. Palladium-Catalyzed Intermolecular Directed C−H Amidation of Aromatic Ketones. J. Am. Chem. Soc. 2011, 133, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Fu, Y.; Xu, J.; Gong, T.-J.; Dai, J.-J.; Yi, J.; Liu, L. Pd(II)-Catalyzed C−H Activation/Aryl−Aryl Coupling of Phenol Esters. J. Am. Chem. Soc. 2010, 132, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Imi, K.; Davies, I.W. Elaboration of Conjugated Alkenes Initiated by Insertion into a Vinylic C-H Bond. J. Am. Chem. Soc. 1995, 117, 5371–5372. [Google Scholar] [CrossRef]
- Li, G.; Wan, L.; Zhang, G.; Leow, D.; Spangler, J.; Yu, J.-Q. Pd(II)-Catalyzed C–H Functionalizations Directed by Distal Weakly Coordinating Functional Groups. J. Am. Chem. Soc. 2015, 137, 4391–4397. [Google Scholar] [CrossRef]
- Huang, L.; Hackenberger, D.; Goossen, L.J. Iridium-Catalyzed ortho-Arylation of Benzoic Acids with Arenediazonium Salts. Angew. Chem. Int. Ed. 2015, 54, 12607–12611. [Google Scholar] [CrossRef]
- Biafora, A.; Krause, T.; Hackenberger, D.; Belitz, F.; Goossen, L.J. ortho-C−H Arylation of Benzoic Acids with Aryl Bromides and Chlorides Catalyzed by Ruthenium. Angew. Chem. Int. Ed. 2016, 55, 14752–14755. [Google Scholar] [CrossRef] [PubMed]
- Dastbaravardeh, N.; Toba, T.; Farmer, M.E.; Yu, J.-Q. Monoselective o-C–H Functionalizations of Mandelic Acid and α-Phenylglycine. J. Am. Chem. Soc. 2015, 137, 9877–9884. [Google Scholar] [CrossRef]
- Gao, P.; Guo, W.; Xue, J.J.; Zhao, Y.; Yuan, Y.; Xia, Y.Z.; Shi, Z.Z. Iridium(III)-Catalyzed Direct Arylation of C-H Bonds with Diaryliodonium Salts. J. Am. Chem. Soc. 2015, 137, 12231–12240. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ye, S.; Fanning, D.; Coon, T.; Schmidt, Y.; Krenitsky, P.; Stamos, D.; Yu, J.-Q. Orchestrated Triple C-H Activation Reactions Using Two Directing Groups: Rapid Assembly of Complex Pyrazoles. Angew. Chem. Int. Ed. 2015, 54, 2501–2504. [Google Scholar] [CrossRef] [PubMed]
- Boerth, J.A.; Ellman, J.A. A Convergent Synthesis of Functionalized Alkenyl Halides through Cobalt(III)-Catalyzed Three-Component C–H Bond Addition. Angew. Chem. Int. Ed. 2017, 56, 9976–9980. [Google Scholar] [CrossRef]
- Ikemoto, H.; Kanai, M.; Tanaka, R.; Yoshino, T.; Matsunaga, S.; Sakata, K.; Sakata, K.; Yoshino, T.; Matsunaga, S. Stereoselective Synthesis of Tetrasubstituted Alkenes via a Cp*CoIII-Catalyzed C–H Alkenylation/Directing Group Migration Sequence. Angew. Chem. Int. Ed. 2017, 56, 7156–7160. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.D.; Daugulis, O. Nonnatural Amino Acid Synthesis by Using Carbon–Hydrogen Bond Functionalization Methodology. Angew. Chem. Int. Ed. 2012, 51, 5188–5191. [Google Scholar] [CrossRef]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, G. Total Synthesis of Celogentin C by Stereoselective C–H Activation. Angew. Chem. Int. Ed. 2010, 49, 958–961. [Google Scholar] [CrossRef]
- Shabashov, D.; Daugulis, O. Auxiliary-Assisted Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 Carbon−Hydrogen Bonds. J. Am. Chem. Soc. 2010, 132, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Ano, Y.; Tobisu, M.; Chatani, N. Palladium-Catalyzed Direct Ethynylation of C(sp3)–H Bonds in Aliphatic Carboxylic Acid Derivatives. J. Am. Chem. Soc. 2011, 133, 12984–12986. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Gianatassio, R.; Baran, P.S. Sequential Csp3–H Arylation and Olefination: Total Synthesis of the Proposed Structure of Pipercyclobutanamide A. Angew. Chem. Int. Ed. 2012, 51, 7507–7510. [Google Scholar] [CrossRef]
- Shibata, K.; Chatani, N. Rhodium-catalyzed regioselective addition of the ortho C–H bond in aromatic amides to the C–C double bond in α,β-unsaturated γ-lactones and dihydrofurans. Chem. Sci. 2016, 7, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Natsui, S.; Chatani, N. Rhodium-Catalyzed Alkenylation of C–H Bonds in Aromatic Amides with Alkynes. Org. Lett. 2017, 19, 2234–2237. [Google Scholar] [CrossRef]
- Yang, Y.; Hou, W.; Qin, L.; Du, J.; Feng, H.; Zhou, B.; Li, Y. Rhodium-Catalyzed Directed Sulfenylation of Arene C–H Bonds. Chem.–Eur. J. 2014, 20, 416–420. [Google Scholar] [CrossRef]
- Shan, C.; Luo, X.; Qi, X.; Liu, S.; Li, Y.; Lan, Y. Mechanism of Ruthenium-Catalyzed Direct Arylation of C–H Bonds in Aromatic Amides: A Computational Study. Organometallics 2016, 35, 1440–1445. [Google Scholar] [CrossRef]
- Aihara, Y.; Tobisu, M.; Fukumoto, Y.; Chatani, N. Ni(II)-Catalyzed Oxidative Coupling between C(sp2)–H in Benzamides and C(sp3)–H in Toluene Derivatives. J. Am. Chem. Soc. 2014, 136, 15509–15512. [Google Scholar] [CrossRef]
- Aihara, Y.; Chatani, N. Nickel-Catalyzed Direct Arylation of C(sp3)–H Bonds in Aliphatic Amides via Bidentate-Chelation Assistance. J. Am. Chem. Soc. 2014, 136, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Aihara, Y.; Chatani, N. Nickel-Catalyzed Direct Alkylation of C–H Bonds in Benzamides and Acrylamides with Functionalized Alkyl Halides via Bidentate-Chelation Assistance. J. Am. Chem. Soc. 2013, 135, 5308–5311. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, Y.; Ge, H. Direct Aerobic Carbonylation of C(sp2)–H and C(sp3)–H Bonds through Ni/Cu Synergistic Catalysis with DMF as the Carbonyl Source. J. Am. Chem. Soc. 2015, 137, 4924–4927. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, Y.; Ge, H. Nickel-Catalyzed Site-Selective Alkylation of Unactivated C(sp3)–H Bonds. J. Am. Chem. Soc. 2014, 136, 1789–1792. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Yang, L.; Xia, C.; Li, F. Nickel-Catalyzed Alkynylation of a C(sp2)–H Bond Directed by an 8-Aminoquinoline Moiety. J. Org. Chem. 2015, 80, 6213–6221. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Chatani, N. Dicumyl Peroxide as a Methylating Reagent in the Ni-Catalyzed Methylation of Ortho C–H Bonds in Aromatic Amides. Org. Lett. 2016, 18, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, D.; Chen, H.; Wang, B.; Liu, Z.; Zhang, Y. Copper(II)/Silver(I)-Catalyzed Sequential Alkynylation and Annulation of Aliphatic Amides with Alkynyl Carboxylic Acids: Efficient Synthesis of Pyrrolidones. Adv. Synth. Catal. 2016, 358, 792–807. [Google Scholar] [CrossRef]
- Takamatsu, K.; Hirano, K.; Masahiro, M. Copper-Mediated Decarboxylative Coupling of Benzamides with ortho-Nitrobenzoic Acids by Directed C–H Cleavage. Angew. Chem. Int. Ed. 2017, 56, 5353–5357. [Google Scholar] [CrossRef]
- Liu, J.; Yu, L.; Zhuang, S.; Gui, Q.; Chen, X.; Wang, W.; Tan, Z. Copper-mediated ortho C–H sulfonylation of benzoic acid derivatives with sodium sulfinates. Chem. Commun. 2015, 51, 6418–6421. [Google Scholar] [CrossRef] [PubMed]
- Williamson, P.; Galvan, A.; Gaunt, M.J. Cobalt-catalysed C–H carbonylative cyclisation of aliphatic amides. Chem. Sci. 2017, 8, 2588–2591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, H.; Lin, C.; Liu, Z.; Wang, C.; Zhang, Y. Cobalt-Catalyzed Cyclization of Aliphatic Amides and Terminal Alkynes with Silver-Cocatalyst. J. Am. Chem. Soc. 2015, 137, 12990–12996. [Google Scholar] [CrossRef]
- Zhang, Z.-Z.; Han, Y.-Q.; Zhan, B.-B.; Wang, S.; Shi, B.-F. Synthesis of Bicyclo[n.1.0]alkanes by a Cobalt-Catalyzed Multiple C(sp3)–H Activation Strategy. Angew. Chem. Int. Ed. 2017, 56, 13145–13149. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed Directed C(sp2)–H and C(sp3)–H Functionalization with Trimethylaluminum. J. Am. Chem. Soc. 2015, 137, 7660–7663. [Google Scholar] [CrossRef]
- Shang, R.; Ilies, L.; Asako, S.; Nakamura, E. Iron-Catalyzed C(sp2)–H Bond Functionalization with Organoboron Compounds. J. Am. Chem. Soc. 2014, 136, 14349–14352. [Google Scholar] [CrossRef] [PubMed]
- Ilies, L.; Matsubara, T.; Ichikawa, S.; Asako, S.; Nakamura, E. Iron-Catalyzed Directed Alkylation of Aromatic and Olefinic Carboxamides with Primary and Secondary Alkyl Tosylates, Mesylates, and Halides. J. Am. Chem. Soc. 2014, 136, 13126–13129. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Ilies, R.; Matsumoto, A.; Nakamura, E. β-Arylation of Carboxamides via Iron-Catalyzed C(sp3)–H Bond Activation. J. Am. Chem. Soc. 2013, 135, 6030–6032. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Fleming, I. Comprehensive Organic Synthesis; Pergamon Press: Oxford, UK, 1991. [Google Scholar]
- Zhang, S.-Y.; Li, Q.; He, G.; Nack, W.A.; Chen, G. Stereoselective Synthesis of -Alkylated -Amino Acids via Palladium-Catalyzed Alkylation of Methylene C(sp3)−H Bonds. J. Am. Chem. Soc. 2013, 135, 12135–12141. [Google Scholar] [CrossRef]
- Deguchi, T.; Xin, H.-L.; Morimoto, H.; Ohshima, T. Direct Catalytic Alcoholysis of Unactivated 8-Aminoquinoline Amides. ACS Catal. 2017, 7, 3157–3161. [Google Scholar] [CrossRef]
- He, G.; Zhang, S.-Y.; Nack, W.A.; Chen, G. Use of a Readily Removable Auxiliary Group for the Synthesis of Pyrrolidones by the Palladium-Catalyzed Intramolecular Amination of Unactivated C(sp3)−H Bonds. Angew. Chem. Int. Ed. 2013, 52, 11124–11128. [Google Scholar] [CrossRef] [PubMed]
- Spletstoser, J.T.; White, J.M.; Tunoori, A.R.; Georg, G.I. Mild and Selective Hydrozirconation of Amides to Aldehydes Using Cp2Zr(H)Cl: Scope and Mechanistic Insight. J. Am. Chem. Soc. 2007, 129, 3408–3419. [Google Scholar] [CrossRef]
- Boit, T.B.; Weires, N.A.; Kim, J.; Garg, N.K. Nickel-Catalyzed Suzuki–Miyaura Coupling of Aliphatic Amides. ACS Catal. 2018, 8, 1003–1008. [Google Scholar] [CrossRef]
- Dander, J.E.; Baker, E.L.; Garg, N.K. Nickel-Catalyzed Transamidation of Aliphatic Amide Derivatives. Chem. Sci. 2017, 8, 6433–6438. [Google Scholar] [CrossRef] [PubMed]
- Weires, N.A.; Caspi, D.D.; Garg, N.K. Kinetic Modeling of the Nickel-Catalyzed Esterification of Amides. ACS Catal. 2017, 7, 4381–4385. [Google Scholar] [CrossRef]
- Medina, J.M.; Moreno, J.; Racine, S.; Du, S.; Garg, N.K. Mizoroki–Heck Cyclizations of Amide Derivatives for the Introduction of Quaternary Centers. Angew. Chem. Int. Ed. 2017, 56, 6567–6571. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Garg, N.K. Breaking Amides Using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Hie, L.; Baker, E.L.; Anthony, S.M.; Desrosiers, J.-N.; Senanayake, C.; Garg, N.K. Nickel-Catalyzed Esterification of Aliphatic Amides. Angew. Chem. Int. Ed. 2016, 55, 15129–15132. [Google Scholar] [CrossRef]
- Dander, J.E.; Weires, N.A.; Garg, N.K. Benchtop Delivery of Ni(cod)2 using Paraffin Capsules. Org. Lett. 2016, 18, 3934–3936. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.L.; Yamano, M.M.; Zhou, Y.; Anthony, S.M.; Garg, N.K. A Two-Step Approach to Achieve Secondary Amide Transamidation Enabled by Nickel Catalysis. Nat. Commun. 2016, 7, 1–5. [Google Scholar] [CrossRef]
- DerivativesSimmons, B.J.; Weires, N.A.; Dander, J.E.; Garg, N.K. Nickel-Catalyzed Alkylation of Amide. ACS Catal. 2016, 6, 3176–3179. [Google Scholar]
- Weires, N.A.; Baker, E.L.; Garg, N.K. Nickel-Catalysed Suzuki–Miyaura Coupling of Amides. Nat. Chem. 2016, 8, 75–79. [Google Scholar] [CrossRef]
- Hie, L.; Nathel, N.F.F.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.-F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of Amides to Esters by the Nickel-Catalysed Activation of Amide C–N Bonds. Nature 2015, 524, 79–83. [Google Scholar] [CrossRef]
- Ra Liu, C.; Shi, S.; Liu, Y.; Liu, R.; Lalancette, R.; Szostak, R.; Szostak, M. The Most Twisted Acyclic Amides: Structures and Reactivity. Org. Lett. 2018, 20, 7771–7774. [Google Scholar] [CrossRef]
- Shi, S.; Nolan, S.P.; Szostak, M. Well-Defined Palladium(II)-NHC (NHC = N-Heterocyclic Carbene) Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective Acyl CO–X (X = N, O) Cleavage. Acc. Chem. Res. 2018, 51, 2589–2599. [Google Scholar] [CrossRef]
- Li, G.; Szostak, M. Highly Selective Transition-Metal-Free Transamidation of Amides and Amidation of Esters at Room Temperature. Nature Commun. 2018, 9, 4165. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, G.; Shi, S.; Meng, G.; Lalancette, R.; Szostak, R.; Szostak, M. Acyl and Decarbonylative Suzuki Coupling of N-Acetyl Amides: Electronic Tuning of Twisted, Acyclic Amides in Catalytic Carbon−Nitrogen Bond Cleavage. ACS Catal. 2018, 8, 9131–9139. [Google Scholar] [CrossRef]
- Li, G.; Lei, P.; Szostak, M. Transition-Metal-Free Esterification of Amides via Selective N−C Cleavage under Mild Conditions. Org. Lett. 2018, 20, 5622–5625. [Google Scholar] [CrossRef] [PubMed]
- Szostak, R.; Szostak, M. N-Acyl-Glutarimides: Resonance and Proton Affinities of Rotationally-Inverted Twisted Amides Relevant to N−C(O) Cross-Coupling. Org. Lett. 2018, 20, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Reversible Twisting of Primary Amides via Ground State N−C(O) Destabilization: Highly Twisted Rotationally Inverted Acyclic Amides. J. Am. Chem. Soc. 2018, 140, 727–734. [Google Scholar] [CrossRef]
- Meng, G.; Szostak, M. Site-Selective C−H/C−N Activation by Cooperative Catalysis: Primary Amides as Arylating Reagents in Directed C−H Arylation. ACS Catal. 2017, 7, 7251–7256. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Pd–PEPPSI: A General Pd–NHC Precatalyst for Buchwald–Hartwig Cross-Coupling of Esters and Amides (Transamidation) under the Same Reaction Conditions. Chem. Commun. 2017, 53, 10584–10587. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Meng, G.; Shi, S.; Ling, Y.; An, J.; Szostak, R.; Szostak, M. Suzuki–Miyaura Cross-Coupling of Amides and Esters at Room Temperature: Correlation with Barriers to Rotation around C–N and C–O Bonds. Chem. Sci. 2017, 8, 6525–6530. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, R.; Szostak, M. Suzuki−Miyaura Cross-Coupling of N-Acylpyrroles and Pyrazoles: Planar, Electronically Activated Amides in Catalytic N–C Cleavage. Org. Lett. 2017, 19, 3596–3599. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Lei, P.; Szostak, M. A General Method for Two-Step Transamidation of Secondary Amides Using Commercially Available, Air- and Moisture-Stable Palladium/NHC (N-Heterocyclic Carbene) Complexes. Org. Lett. 2017, 19, 2158–2161. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, S.; Achtenhagen, M.; Liu, R.; Szostak, M. Metal-Free Transamidation of Secondary Amides via Selective N–C Cleavage under Mild Conditions. Org. Lett. 2017, 19, 1614–1617. [Google Scholar] [CrossRef]
- Lei, P.; Meng, G.; Szostak, M. General Method for the Suzuki-Miyaura Cross-Coupling of Amides Using Commercially Available, Air- and Moisture-Stable Palladium/NHC (NHC = N-Heterocyclic Carbene) Complexes. ACS Catal. 2017, 7, 1960–1965. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Nickel-Catalyzed Diaryl Ketone Synthesis by N–C Cleavage: Direct Negishi Cross-Coupling of Primary Amides by Site- Selective N,N-Di-Boc Activation. Org. Lett. 2016, 18, 5872–5875. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Shi, S.; Szostak, M. Palladium-Catalyzed Suzuki−Miyaura Cross-Coupling of Amides via Site-Selective N−C Bond Cleavage by Cooperative Catalysis. ACS Catal. 2016, 6, 7335–7339. [Google Scholar] [CrossRef]
- Pace, V.; Holzer, W.; Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Structures of Highly Twisted Amides Relevant to Amide N−C Cross-Coupling: Evidence for Ground-State Amide Destabilization. Chem. Eur. J. 2016, 22, 14494–14498. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Meng, G.; Szostak, M. Synthesis of Biaryls via Nickel Catalyzed Suzuki–Miyaura Coupling of Amides by Carbon–Nitrogen Cleavage. Angew. Chem. Int. Ed. 2016, 55, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Sterically-Controlled Pd-Catalyzed Chemoselective Ketone Synthesis via N–C Cleavage in Twisted Amides. Org. Lett. 2015, 17, 4364–4367. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Gagliardi, A.; Leggio, A.; Leotta, V.; Romio, E.; Liguori, A. N-Urethane protection of amines and amino acids in an ionic liquid. RSC Adv. 2015, 5, 63407–63420. [Google Scholar] [CrossRef]
- Nardi, M.; Cano, N.H.; Costanzo, P.; Oliverio, M.; Sindona, G.; Procopio, A. Aqueous MW eco-friendly protocol for amino group protection. RSC Adv. 2015, 5, 18751. [Google Scholar] [CrossRef]
- Durga, T.V.; Rambabu, A.; Reddy, M.S.; Babu, B.H. Ionic Liquid as Efficient Reaction Medium for N-tert-Boc Protection of Amines. Asian J. Chem. 2017, 29, 1313–1316. [Google Scholar]
- Sunitha, S.; Kanjilal, S.; Reddy, P.S.; Prasad, R.B.N. An efficient and chemoselective Brønsted acidic ionic liquid-catalyzed N-Boc protection of amines. Tetrahedron Lett. 2008, 49, 2527–2532. [Google Scholar] [CrossRef]
- Schwarzer, M.C.; Konno, R.; Hojo, T.; Ohtsuki, A.; Nakamura, K.; Yasutome, A.; Takahashi, H.; Shimasaki, T.; Tobisu, M.; Chatani, N.; et al. Combined Theoretical and Experimental Studies of Nickel-Catalyzed Cross-Coupling of Methoxyarenes with Arylboronic Esters via C–O Bond Cleavage. J. Am. Chem. Soc. 2017, 139, 10347–10358. [Google Scholar] [CrossRef] [PubMed]
- Schaub, T.; Fischer, P.; Steffen, A.; Braun, T.; Radius, U.; Mix, A. C−F Activation of Fluorinated Arenes using NHC-Stabilized Nickel(0) Complexes: Selectivity and Mechanistic Investigations. J. Am. Chem. Soc. 2008, 130, 9304–9317. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: All samples are available from authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Yi, J.; Xu, H.; Li, S.; Yuan, R. An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl. Molecules 2019, 24, 1234. https://doi.org/10.3390/molecules24071234
Wu W, Yi J, Xu H, Li S, Yuan R. An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl. Molecules. 2019; 24(7):1234. https://doi.org/10.3390/molecules24071234
Chicago/Turabian StyleWu, Wengang, Jun Yi, Huipeng Xu, Shuangjun Li, and Rongxin Yuan. 2019. "An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl" Molecules 24, no. 7: 1234. https://doi.org/10.3390/molecules24071234
APA StyleWu, W., Yi, J., Xu, H., Li, S., & Yuan, R. (2019). An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl. Molecules, 24(7), 1234. https://doi.org/10.3390/molecules24071234