
Cardiac-Specific Cre Induces Age-Dependent Dilated Cardiomyopathy (DCM) in Mice


Abstract
1. Introduction
2. Results
2.1. The Change in Lifespan of αMHC-cre Mice
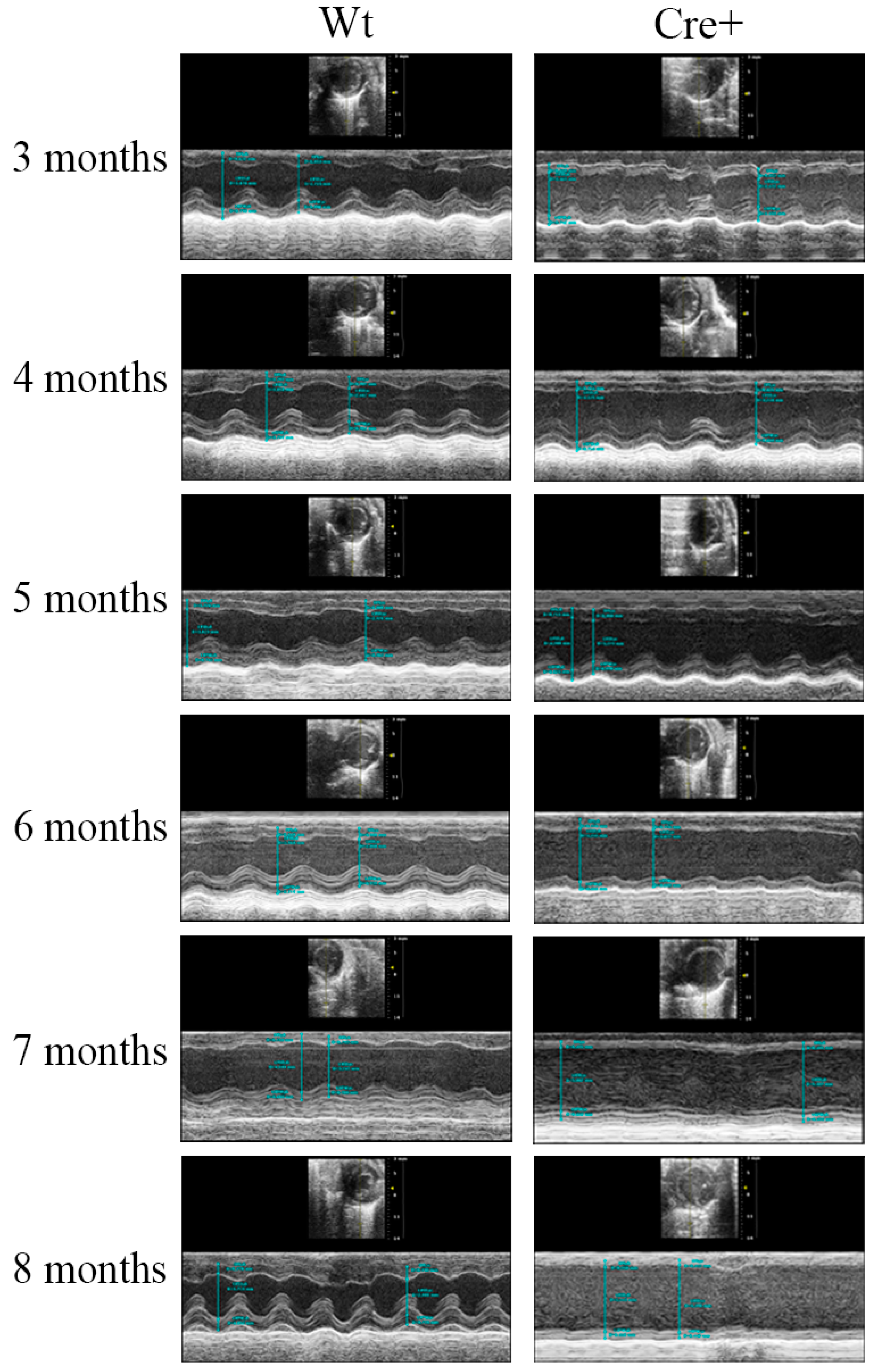
2.2. αMHC-cre Mice Presented with an Age-Dependent Dilated Cardiomyopathy
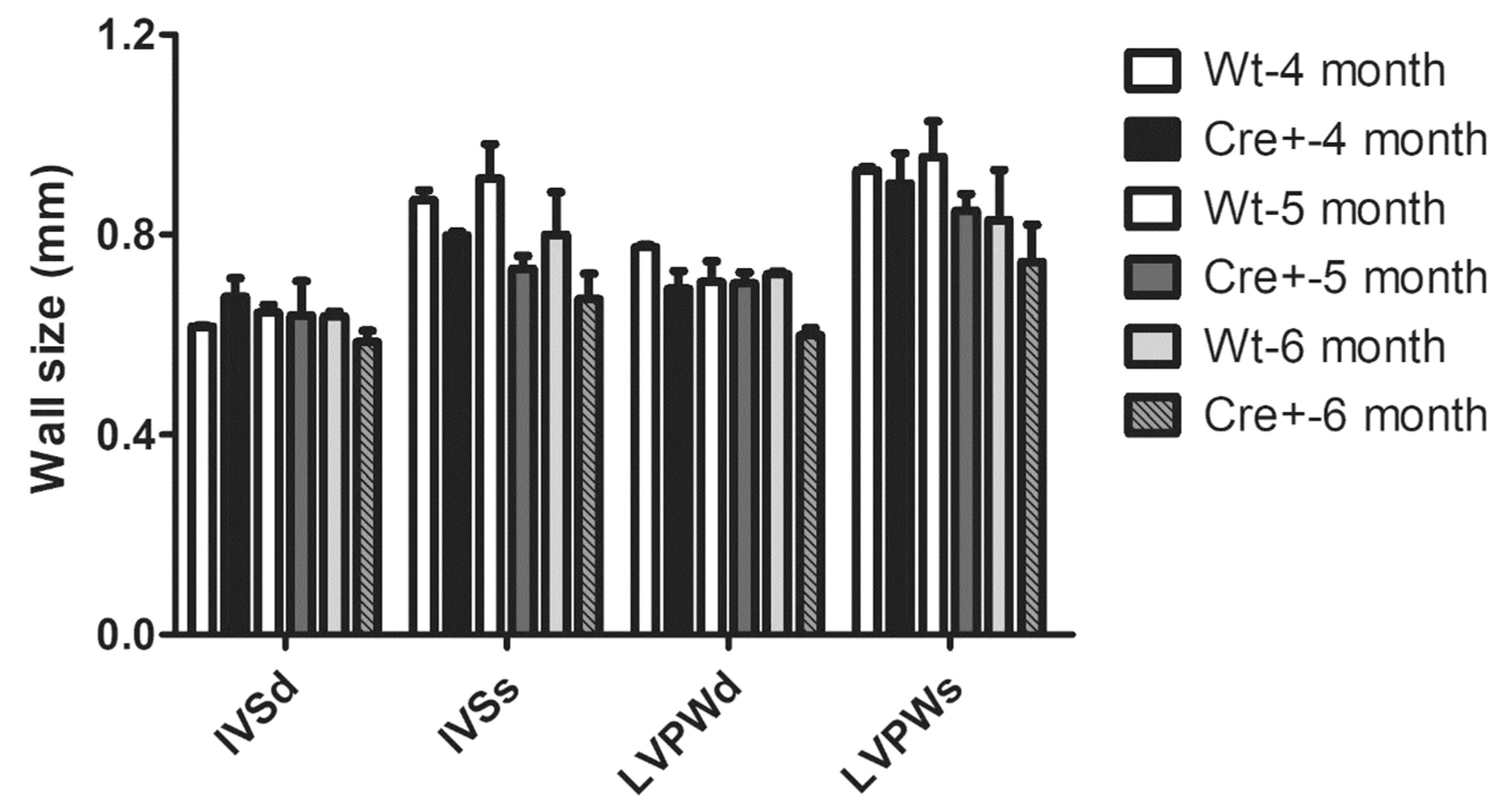
2.3. Lack of Hypertrophy and Robust DCM in αMHC-cre Mice
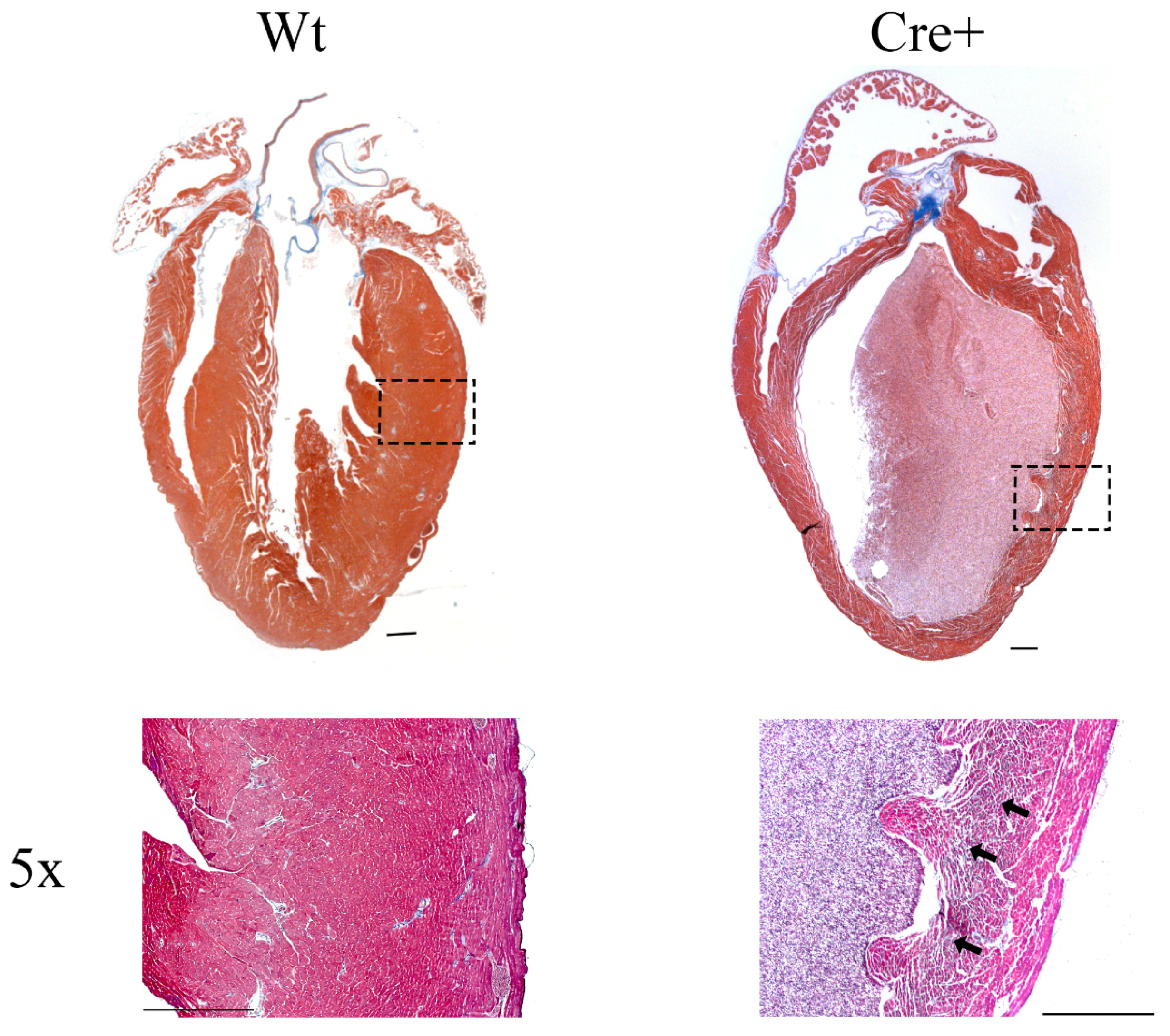
2.4. αMHC-cre Mice Exhibited Dilated Hearts and Fibrosis
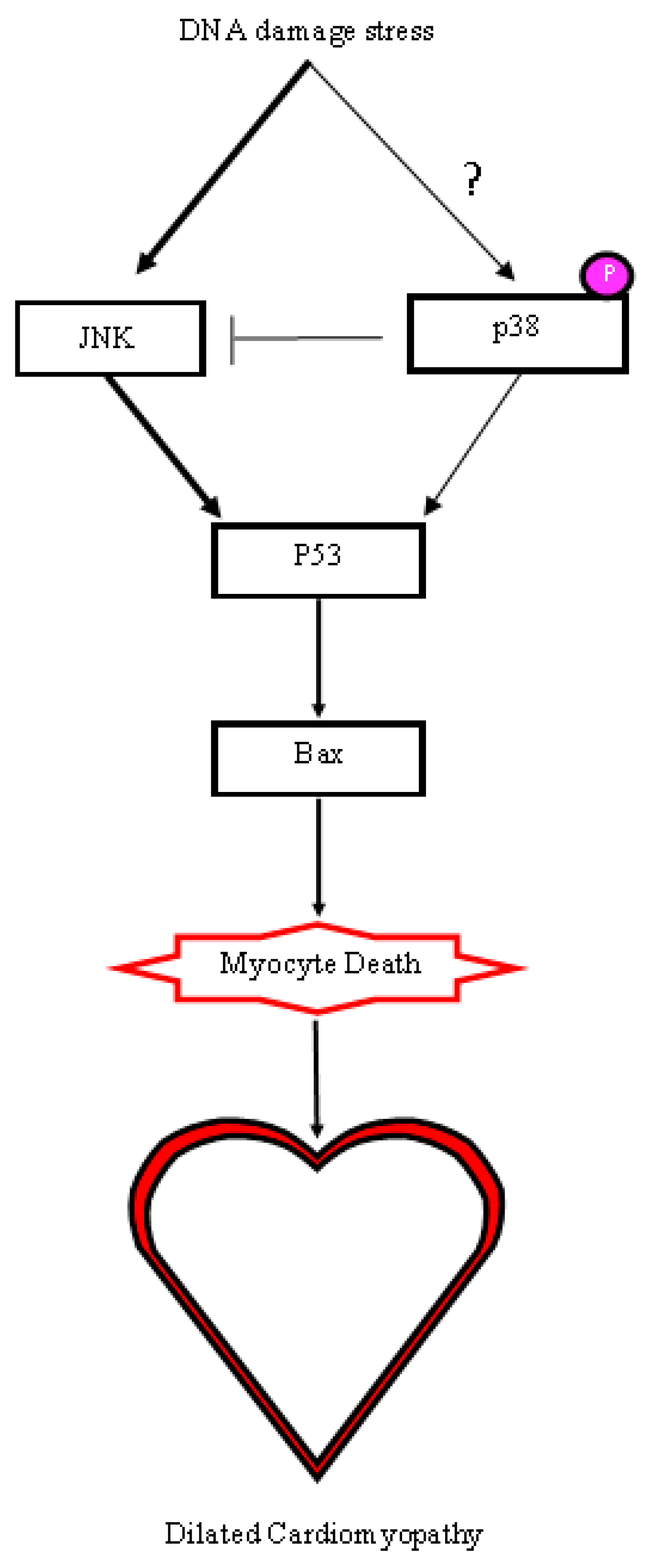
2.5. A DNA Damage Response Presented in αMHC-cre Myocardium
3. Discussion
4. Materials and Methods
4.1. Maintenance of αMHC-cre Mouse Colonies
4.2. Protein Isolation from Mouse Hearts
4.3. SDS-PAGE and Western Blots
4.4. Echocardiography
4.5. Histological Analysis
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Houdebine, L. Transgenic animal models in biomedical research. Methods Mol. Biol. 2007, 360, 163–202. [Google Scholar] [PubMed]
- Hardouin, S.; Nagy, A. Mouse models for human disease. Clin. Genet. 2000, 57, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, N.; Brown, S. The mouse ascending: Perspectives for human-disease models. Nat. Cell. Biol. 2007, 9, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Gene targeting in ES cells. Annu. Rev. Neurosci. 1995, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bradley, A. Engineering chromosomal rearrangements in mice. Nat. Rev. Genet. 2001, 2, 780–790. [Google Scholar] [CrossRef]
- Hochman, L.; Segev, N.; Sternberg, N.; Cohen, G. Site-specific recombinational circularization of bacteriophage P1 DNA. Virology 1983, 131, 11–17. [Google Scholar] [CrossRef]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef]
- Branda, C.; Dymecki, S. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev. Cell. 2004, 6, 7–28. [Google Scholar] [CrossRef]
- Hoess, R.; Wierzbicki, A.; Abremski, K. The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res. 1986, 14, 2287–2300. [Google Scholar] [CrossRef]
- Tora, L.; White, J.; Brou, C.; Tasset, D.; Webster, N.; Scheer, E.; Chambon, P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 1989, 59, 477–487. [Google Scholar] [CrossRef]
- Gagniuc, P.; Ionescu-Tirgoviste, C. Eukaryotic genomes may exhibit up to 10 generic classes of gene promoters. BMC Genom. 2012, 13, 512. [Google Scholar] [CrossRef] [PubMed]
- Littlewood, T.; Hancock, D.; Danielian, P.; Parker, M.; Evan, G. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995, 23, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Orban, P.; Chui, D.; Marth, J. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6861–6865. [Google Scholar] [CrossRef] [PubMed]
- Agah, R.; Frenkel, P.; French, B.; Michael, L.; Overbeek, P.; Schneider, M. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Investig. 1997, 100, 169–179. [Google Scholar]
- Davis, J.; Maillet, M.; Miano, J.; Molkentin, J. Lost in transgenesis: A user’s guide for genetically manipulating the mouse in cardiac research. Circ. Res. 2012, 111, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Pugach, E.; Richmond, P.; Azofeifa, J.; Dowell, R.; Leinwand, L. Prolonged Cre expression driven by the α-myosin heavy chain promoter can be cardiotoxic. J. Mol. Cell. Cardiol. 2015, 86, 54–61. [Google Scholar] [CrossRef]
- Schmidt, E.; Taylor, D.; Prigge, J.; Barnett, S.; Capecchi, M. Illegitimate Cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc. Natl. Acad. Sci. USA 2000, 97, 13702–13707. [Google Scholar] [CrossRef]
- Janbandhu, V.; Moik, D.; Fässler, R. Cre recombinase induces DNA damage and tetraploidy in the absence of loxP sites. Cell Cycle 2013, 13, 462–470. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Guimarães, M.; Groth, A.; Calos, M. Mammalian genomes contain active recombinase recognition sites. Gene 2000, 244, 47–54. [Google Scholar] [CrossRef]
- Ito, M.; Yamanouchi, K.; Naito, K.; Calos, M.; Tojo, H. Site-specific integration of transgene targeting an endogenous lox-like site in early mouse embryos. J. Appl. Genet. 2011, 52, 89–94. [Google Scholar] [CrossRef]
- Koitabashi, N.; Bedja, D.; Zaiman, A.; Pinto, Y.; Zhang, M.; Gabrielson, K.; Takimoto, E.; Kass, D. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circ. Res. 2009, 105, 12–15. [Google Scholar] [CrossRef]
- Bersell, K.; Choudhury, S.; Mollova, M.; Polizzotti, B.D.; Ganapathy, B.; Walsh, S.; Wadugu, B.; Arab, S.; Kühn, B. Moderate and high amounts of tamoxifen in αMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis. Models Mech. 2013, 6, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, T.; Foltz, C.; Karlsson, E.; Linton, C.; Smith, J. Health evaluation of experimental laboratory mice. Curr. Protoc. Mouse Biol. 2012, 12, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Rhaleb, N.; Kurihara, N.; Kim, H.; Carretero, O. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am. J. Physiol. 1999, 277, 1967–1974. [Google Scholar] [CrossRef]
- Luk, A.; Ahn, E.; Soor, G.; Butany, J. Dilated cardiomyopathy: A review. J. Clin. Pathol. 2009, 62, 219–225. [Google Scholar] [CrossRef]
- Loonstra, A.; Vooijs, M.; Beverloo, H.; Allak, B.; van Drunen, E.; Kanaar, R.; Berns, A.; Jonkers, J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9209–9214. [Google Scholar] [CrossRef]
- Prives, C.; Hall, P. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Raingeaud, J.; Gupta, S.; Rogers, J.; Dickens, M.; Han, J.; Ulevitch, R.; Davis, R. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Lalier, L.; Cartron, P.; Juin, P.; Nedelkina, S.; Manon, S.; Bechinger, B.; Vallette, F. Bax activation and mitochondrial insertion during apoptosis. Apoptosis 2007, 12, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- MacLellan, W.; Schneider, M. Death by design. Programmed cell death in cardiovascular biology and disease. Circ. Res. 1997, 81, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 28, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Miura, H.; Kondo, Y.; Matsuda, M.; Aoki, K. Cell-to-Cell heterogeneity in p38-mediated cross-inhibition of JNK causes stochastic cell death. Cell Rep. 2018, 24, 2658–2668. [Google Scholar] [CrossRef]
- Williams, A.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect Med. 2016, 6, 5. [Google Scholar] [CrossRef]
- Picco, V.; Pagès, G. Linking JNK activity to the DNA damage response. Genes Cancer 2013, 4, 360–368. [Google Scholar] [CrossRef]
- Donauer, J.; Schreck, I.; Liebel, U.; Weiss, C. Role and interaction of p53, BAX and the stress-activated protein kinases p38 and JNK in benzo(a)pyrene-diolepoxide induced apoptosis in human colon carcinoma cells. Arch. Toxicol. 2012, 86, 329. [Google Scholar] [CrossRef]
- Burchfield, J.; Xie, M.; Hill, J. Pathological ventricular mechanisms. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, M. Decade in review—Cardiomyopathies: Cardiomyopathy on the move. Nat. Rev. Cardiol. 2014, 11, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Zechner, D.; Thuerauf, D.; Hanford, D.; McDonough, P.; Glembotski, C. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J. Cell. Biol. 1997, 6, 115–127. [Google Scholar] [CrossRef]
- Liao, P.; Georgakopoulos, D.; Kovacs, A.; Zheng, M.; Lerner, D.; Pu, H.; Saffitz, J.; Chien, K.; Xiao, R.; Kass, D.; et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. USA 2001, 98, 12283–12288. [Google Scholar] [CrossRef]
- Lemke, L.; Bloem, L.; Fouts, R.; Esterman, M.; Sandusky, G.; Vlahos, C. Decreased p38 MAPK activity in end-stage failing human myocardium: p38 MAPK alpha is the predominant isoform expressed in human heart. J. Mol. Cell. Cardiol. 2001, 33, 1527–1540. [Google Scholar] [CrossRef]
- Haq, S.; Choukroun, G.; Lim, H.; Tymitz, K.; del Monte, F.; Gwathmey, J.; Grazette, L.; Michael, A.; Hajjar, R.; Force, T.; et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 2001, 103, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Su, B.; Sah, V.; Brown, J.; Han, J.; Chien, K. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J. Biol. Chem. 1998, 273, 5423–5426. [Google Scholar] [CrossRef]
- Liang, Q.; Molkentin, J. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: Dichotomy between cultured myocytes and animal models. J. Mol. Cell. Cardiol. 2003, 35, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.; Towbin, J.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.; Seidman, C.; Young, J. Contemporary definitions and classification of the cardiomyopathies. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Arabacilar, P.; Marber, M. The case for inhibiting p38 mitogen-activated protein kinase in heart failure. Front Pharm. 2015, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Buerger, A.; Rozhitskaya, O.; Sherwood, M.; Dorfman, A.; Bisping, E.; Abel, E.D.; Pu, W.; Izumo, S.; Jay, P. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J. Card. Fail. 2006; 12, 392–398. [Google Scholar]
- Harding, P.; Yang, X.; Yang, J.; Shesely, E.; He, Q.; LaPointe, M. Gene expression profiling of dilated cardiomyopathy in older male EP4 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, S.; Leu, N.; Pavlov, I.; Han, X.; Ribeiro PA, X.T.B.R.; Saha, S.; Wang, J.; Cornachione, A.; Mai, W.; Yates, J.; et al. Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J. Mol. Cell Cardiol. 2012, 53, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Kratsios, P.; Huth, M.; Temmerman, L.; Salimova, E.; Al Banchaabouchi, M.; Sgoifo, A.; Manghi, M.; Suzuki, K.; Rosenthal, N.; Mourkioti, F. Antioxidant amelioration of dilated cardiomyopathy caused by conditional deletion of NEMO/IKKgamma in cardiomyocytes. Circ. Res. 2010, 106, 133–144. [Google Scholar] [CrossRef]
- Xiong, S.; Van Pelt, C.; Elizondo-Fraire, A.; Fernandez-Garcia, B.; Lozano, G. Loss of Mdm4 Results in p53-Dependent Dilated Cardiomyopathy. Circulation 2007, 115, 2925–2930. [Google Scholar] [CrossRef]
- Oakley, R.; Ren, R.; Cruz-Topete, D.; Bird, G.; Myers, P.; Boyle, M.; Schneider, M.; Willis, M.; Cidlowski, J. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. USA 2013, 110, 17035–17040. [Google Scholar] [CrossRef]
- Rowe, G.; Asimaki, A.; Graham, E.; Martin, K.; Margulies, K.; Das, S.; Saffitz, J.; Arany, Z. Development of dilated cardiomyopathy and impaired calcium homeostasis with cardiac-specific deletion of ESRRβ. Am. J. Physiol. Heart Circ. Physiol 2017, 312, 662–671. [Google Scholar] [CrossRef]
- Huang, J.; Min Lu, M.; Cheng, L.; Yuan, L.; Zhu, X.; Stout, A.; Chen, M.; Li, J.; Parmacek, M. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc. Natl. Acad. Sci. USA 2009, 106, 18734–18739. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Xiong, D.; Dorner, A.; Youn, T.; Yung, A.; Liu, T.; Gu, Y.; Dalton, N.; Wright, A.; Evans, S.; et al. Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J. Clin. Investig. 2008, 118, 2758–2770. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, J.; Kalinowski, A.; Liu, M.; Zhang, S.; Gao, Q.; Chai, G.; Ji, L.; Iwamoto, Y.; Li, E.; Schneider, M.; et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc. Natl. Acad. Sci. USA 2003, 100, 12929–12934. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; García-Prieto, J.; Baker, M.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, 6365. [Google Scholar] [CrossRef]
- Collins, H.; He, L.; Zou, L.; Litovsky, S.; Yang, Q.; Young, M.; Marchase, R.; Chatham, J. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am. J. Physiol. Heart Circ. Physiol 2014, 306, 1231–1239. [Google Scholar] [CrossRef]
- Liu, L.; Trent, C.; Fang, X.; Son, N.; Jiang, H.; Blaner, W.; Hu, Y.; Yin, Y.; Farese, R.J.; Homma, S.; et al. Cardiomyocyte-specific loss of diacylglycerol acyltransferase 1 (DGAT1) reproduces the abnormalities in lipids found in severe heart failure. J. Biol. Chem. 2014, 289, 29881–29891. [Google Scholar] [CrossRef] [PubMed]
- Levkau, B.; Schäfers, M.; Wohlschlaeger, J.; von Wnuck Lipinski, K.; Keul, P.; Hermann, S.; Kawaguchi, N.; Kirchhof, P.; Fabritz, L.; Stypmann, J.; et al. Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation 2008, 117, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of any compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 months | Wt | Cre+ | p-value | 4 months | Wt | Cre+ | p-value |
| IVS;d (mm) | 0.630 ± 0.025 | 0.620 ± 0.027 | 0.291692478 | IVS;d (mm) | 0.645 ± 0.028 | 0.675 ± 0.033 | 0.788462538 |
| IVS;s (mm) | 0.841 ± 0.024 | 0.752 ± 0.042 | 0.088168344 | IVS;s (mm) | 0.840 ± 0.031 | 0.799 ± 0.006 | 0.063367682 |
| LVID;d (mm) | 3.988 ± 0.120 | 3.891 ± 0.062 | 0.844773361 | LVID;d (mm) | 3.945 ± 0.130 | 3.816 ± 0.170 | 0.854738647 |
| LVID;s (mm) | 2.965 ± 0.104 | 3.062 ± 0.078 | 0.533414051 | LVID;s (mm) | 2.983 ± 0.210 | 2.923 ± 0.180 | 0.816015832 |
| LVPW;d (mm) | 0.752 ± 0.039 | 0.703 ± 0.009 | 0.421564695 | LVPW;d (mm) | 0.741 ± 0.033 | 0.692 ± 0.031 | 0.163169522 |
| LVPW;s (mm) | 0.950 ± 0.058 | 0.865 ± 0.023 | 0.186036258 | LVPW;s (mm) | 0.917 ± 0.021 | 0.903 ± 0.052 | 0.612779363 |
| EF (%) | 51.078 ± 1.225 | 43.930 ± 1.415 | 0.050885727 | EF (%) | 49.190 ± 4.900 | 47.703 ± 2.932 | 0.626142978 |
| FS (%) | 25.670 ± 0.730 | 21.351 ± 0.789 | 0.040507965 | FS (%) | 24.692 ± 2.920 | 23.568 ± 1.676 | 0.592817029 |
| LV Mass (mg) | 77.478 ± 6.617 | 69.760 ± 0.624 | 0.462915868 | LV Mass (mg) | 76.123 ± 4.930 | 70.596 ± 4.086 | 0.474495439 |
| LV Vol;d (µL) | 69.792 ± 4.975 | 65.625 ± 2.444 | 0.828743538 | LV Vol;d (µL) | 68.114 ± 5.262 | 63.086 ± 6.373 | 0.875915463 |
| LV Vol;s (µL) | 34.230 ± 2.917 | 36.890 ± 2.231 | 0.540609259 | LV Vol;s (µL) | 35.345 ± 5.924 | 33.372 ± 4.70 | 0.832144194 |
| Heart Rate (bpm) | 475.063 ± 31.085 | 426.813 ± 25.030 | 0.24795013 | Heart Rate (bpm) | 442.375 ± 15.083 | 488.813 ± 18.431 | 0.131686783 |
| 5 months | Wt | Cre+ | p-value | 6 months | Wt | Cre+ | p-value |
| IVS;d (mm) | 0.703 ± 0.055 | 0.621 ± 0.033 | 0.221718131 | IVS;d (mm) | 0.645 ± 0.0270 | 0.623 ± 0.025 | 0.582227989 |
| IVS;s (mm) | 0.867 ± 0.037 | 0.760 ± 0.032 | 0.066845485 | IVS;s (mm) | 0.770 ± 0.046 | 0.773 ± 0.036 | 0.944541382 |
| LVID;d (mm) | 4.015 ± 0.070 | 4.24 ± 0.022 | 0.011199612 | LVID;d (mm) | 3.860 ± 0.086 | 4.267 ± 0.050 | 0.000984789 |
| LVID;s (mm) | 3.010 ± 0.094 | 3.428 ± 0.029 | 0.007459428 | LVID;s (mm) | 3.111 ± 0.115 | 3.480 ± 0.073 | 0.015556863 |
| LVPW;d (mm) | 0.771 ± 0.045 | 0.720 ± 0.019 | 0.292980414 | LVPW;d (mm) | 0.741 ± 0.013 | 0.670 ± 0.022 | 0.037445862 |
| LVPW;s (mm) | 0.941 ± 0.033 | 0.853 ± 0.036 | 0.123190118 | LVPW;s (mm) | 0.881 ± 0.058 | 0.840 ± 0.038 | 0.533692627 |
| EF (%) | 46.214 ± 3.352 | 39.801 ± 0.561 | 0.070796387 | EF (%) | 40.113 ± 4.339 | 38.503 ± 2.380 | 0.728792586 |
| FS (%) | 22.836 ± 2.019 | 19.160 ± 0.307 | 0.081406368 | FS (%) | 19.377 ± 2.510 | 18.545 ± 1.313 | 0.75158218 |
| LV Mass (mg) | 85.180 ± 7.413 | 82.324 ± 3.134 | 0.711666602 | LV Mass (mg) | 73.251 ± 2.367 | 79.583 ± 3.594 | 0.228762289 |
| LV Vol;d (µL) | 70.721 ± 2.901 | 80.372 ± 0.981 | 0.010393045 | LV Vol;d (µL) | 64.510 ± 3.442 | 81.820 ± 2.206 | 0.000959293 |
| LV Vol;s (µL) | 38.018 ± 2.791 | 48.402 ± 0.994 | 0.006305021 | LV Vol;s (µL) | 38.590 ± 3.453 | 50.370 ± 2.421 | 0.015009289 |
| Heart Rate (bpm) | 468.229 ± 19.224 | 437.563 ± 22.691 | 0.470433394 | Heart Rate (bpm) | 456.333 ± 23.479 | 457.083 ± 21.667 | 0.985755943 |
| 7 months | Wt | Cre+ | p-value | 8 months | Wt | Cre+ | p-value |
| IVS;d (mm) | 0.652 ± 0.046 | 0.530 ± 0.017 | 0.004871736 | IVS;d (mm) | 0.672 ± 0.038 | 0.525 ± 0.020 | 0.002876701 |
| IVS;s (mm) | 0.852 ± 0.075 | 0.655 ± 0.028 | 0.004820388 | IVS;s (mm) | 0.897 ± 0.038 | 0.583 ± 0.023 | 2.01935E-05 |
| LVID;d (mm) | 4.153 ± 0.107 | 4.710 ± 0.085 | 0.00075479 | LVID;d (mm) | 3.992 ± 0.087 | 5.0120 ± 0.733 | 0.002527764 |
| LVID;s (mm) | 3.100 ± 0.078 | 4.015 ± 0.146 | 0.000380906 | LVID;s (mm) | 2.911 ± 0.181 | 4.613 ± 0.218 | 0.000506287 |
| LVPW;d (mm) | 0.731 ± 0.035 | 0.612 ± 0.0260 | 0.010122283 | LVPW;d (mm) | 0.729 ± 0.043 | 0.607 ± 0.029 | 0.037276373 |
| LVPW;s (mm) | 0.960 ± 0.063 | 0.735 ± 0.033 | 0.001783791 | LVPW;s (mm) | 0.899 ± 0.048 | 0.658 ± 0.047 | 0.009337212 |
| EF (%) | 50.140 ± 2.792 | 31.463 ± 3.171 | 0.000971708 | EF (%) | 53.049 ± 5.311 | 17.836 ± 4.746 | 0.001064173 |
| FS (%) | 25.264 ± 1.830 | 14.966 ± 1.571 | 0.000618349 | FS (%) | 27.203 ± 3.350 | 8.287 ± 2.190 | 0.000655345 |
| LV Mass (mg) | 83.423 ± 9.092 | 80.60 ± 2.389 | 0.667096037 | LV Mass (mg) | 78.378 ± 3.758 | 90.433 ± 6.474 | 0.242979928 |
| LV Vol;d (µL) | 76.765 ± 4.640 | 103.311 ± 4.489 | 0.001168324 | LV Vol;d (µL) | 69.821 ± 3.655 | 120.755 ± 9.005 | 0.00328618 |
| LV Vol;s (µL) | 38.055 ± 2.364 | 71.912 ± 6.740 | 0.002004722 | LV Vol;s (µL) | 33.135 ± 5.253 | 100.210 ± 10.261 | 0.001357712 |
| Heart Rate (bpm) | 458.750 ± 33.700 | 488.292 ± 20.507 | 0.509364526 | Heart Rate (bpm) | 426.833 ± 45.470 | 503.890 ± 12.685 | 0.308473653 |
| Antibody | Manufacturer | Dilution |
|---|---|---|
| Phospho-p38 | Cell Signaling Technology (9215) | 1:1000 |
| p38 | Santa Cruz (sc-81621) | 1:250 |
| p53 | Santa Cruz (sc-126) | 1:250 |
| JNK | Cell Signaling Technology (9252) | 1:1000 |
| BAX | Cell Signaling Technology (2772) | 1:1000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehmani, T.; Salih, M.; Tuana, B.S. Cardiac-Specific Cre Induces Age-Dependent Dilated Cardiomyopathy (DCM) in Mice. Molecules 2019, 24, 1189. https://doi.org/10.3390/molecules24061189
Rehmani T, Salih M, Tuana BS. Cardiac-Specific Cre Induces Age-Dependent Dilated Cardiomyopathy (DCM) in Mice. Molecules. 2019; 24(6):1189. https://doi.org/10.3390/molecules24061189
Chicago/Turabian StyleRehmani, Taha, Maysoon Salih, and Balwant S. Tuana. 2019. "Cardiac-Specific Cre Induces Age-Dependent Dilated Cardiomyopathy (DCM) in Mice" Molecules 24, no. 6: 1189. https://doi.org/10.3390/molecules24061189
APA StyleRehmani, T., Salih, M., & Tuana, B. S. (2019). Cardiac-Specific Cre Induces Age-Dependent Dilated Cardiomyopathy (DCM) in Mice. Molecules, 24(6), 1189. https://doi.org/10.3390/molecules24061189