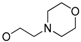3.1. Chemistry
1H-NMR and 13C-NMR spectra were recorded on a Bruker AVANCE 400 or 500 spectrometer (Karlsruhe, Germany). Chemical shifts of protons are reported in parts per million downfield from tetramethylsilane. Peaks are labeled as single (s), broad singlet (br), doublet (d), triplet (t), double doublet (dd), doublet of triplets (dt), or multiplet (m). The high-resolution mass spectra were analyzed on a SHIMADZU LCMS-IT-TOF mass spectrometer. The purity of the synthesized compounds was determined by high-performance liquid chromatography (HPLC) (Agilent, Palo Alto, CA, USA) with a TC-C18 column (250 mm × 4.6 mm, 5 μm) using methanol (1/1000 diethylamine)/water mobile phase (0.50 mL/min). Melting points were determined in open capillary tubes on a MPA100 Optimelt automated melting point system (Stanford Research Systems, San Francisco, CA, USA,). All chemicals were purchased from Sigma-Aldrich and Alfa Aesar chemical companies (Shanghai, China) and were used without further purification.
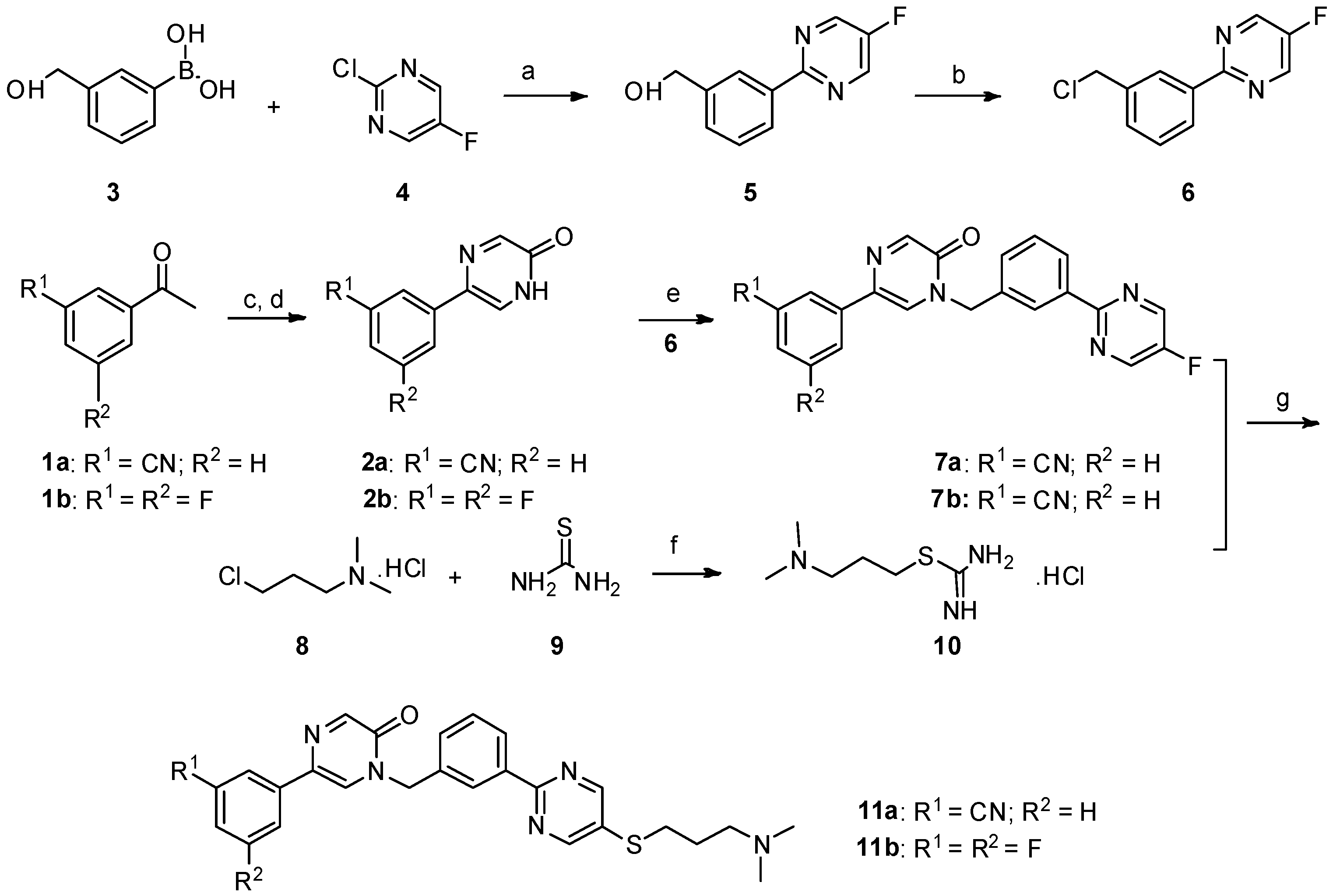
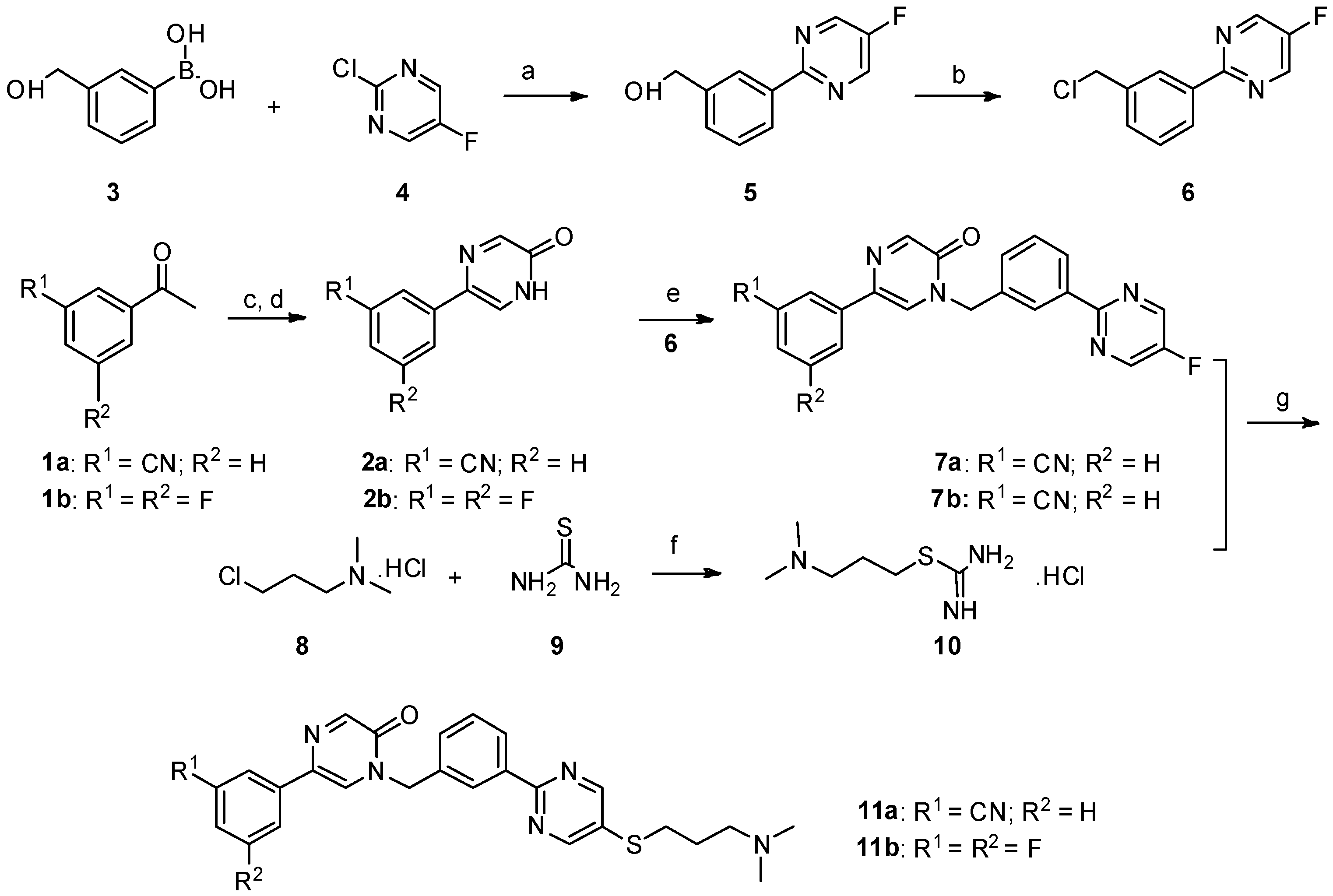
Synthesis of 5-(3-cyanophenyl)-1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)pyrazin-2(1H)-one (11a). In a 200 mL two-necked round-bottomed flask provided with a magnetic stirrer and condenser, 50 mmol of thiourea
9 were dissolved in 60 mL of absolute ethanol. Compound
8 (3.161 mg, 20 mmol) was added in one portion in the thiourea solution. After heating at reflux for 12 h, the reaction mixture was concentrated by rotary evaporation under reduced pressure. The desired compound
10 was obtained as a white solid and was further used without purification [
18].
NaOH (135 mg, 3.375 mmol) in 0.5 mL water was added to a mixture of 7a (287.25 mg, 0.75 mmol) and 10 (444 mg) in DMF (5 mL) under nitrogen, and the mixture was stirred at room temperature for 15 min. Then, the mixture was stirred at 60 °C under nitrogen for 8 h. The reaction mixture was allowed to cool to room temperature. The aqueous phase was extracted with dichloromethane (30 mL × 3). The combined organic layer was washed with H2O (15 mL) and brine (10 mL), and then dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash chromatography over silica gel (DCM/MeOH = 40:1–10:1) to give 5-(3-cyanophenyl)-1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)pyrazin-2(1H)-one (11a) (224 mg, 62%) as a white solid. m.p. 106.3–107.9 °C. 1H-NMR (400 MHz, CDCl3) δ 8.74 (s, 1H), 8.46–8.39 (m, 2H), 8.32 (s, 1H), 8.01 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.64 (s, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.55–7.47 (m, 3H), 5.27 (s, 2H), 3.03 (t, J = 7.2 Hz, 2H), 2.43 (t, J = 7.0 Hz, 2H), 2.24 (s, 6H), 1.91–1.76 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ 161.18, 157.41, 155.35, 149.17, 138.08, 136.67, 134.95, 131.70, 131.28, 130.82, 129.79, 129.70, 129.08, 128.52, 128.04, 125.10, 118.62, 113.06, 57.87, 52.29, 45.34, 31.60, 27.04. HRMS (ESI) calculated for C27H26N6OS [M + H]+: 483.1962, found: 483.1947. Purity: 98.3% (by HPLC).
5-(3,5-difluorophenyl)-1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)pyrazin-2(1H)-one (11b). Compound 11b was prepared via a similar procedure of 11a. White solid. m.p. 143.7–145.2 °C. Yield: 65%. 1H-NMR (500 MHz, CDCl3) δ 8.74(s, 2H), 8.46–8.39 (m, 2H), 8.30 (s, 1H), 7.56 (s, 1H), 7.54–7.46 (m, 2H), 7.25–7.18 (m, 2H), 6.82–6.65 (m, 1H), 5.25 (s, 2H), 3.03 (t, J = 7.2 Hz, 2H), 2.42 (t, J = 7.0 Hz, 2H), 2.24 (s, 6H), 1.92–1.76 (m, 2H). 13C-NMR (126 MHz, CDCl3) δ 164.52 (JCF = 247.5 Hz), 163.42 (JCF = 246.25 Hz), 161.21, 157.45, 155.37, 148.97, 138.74 (JCF = 10.0 Hz), 138.10, 134.92, 131.64 (J CF = 3.0 Hz), 130.81, 130.76, 129.76, 128.49, 128.00, 125.03, 107.79 (JCF = 26.25 Hz), 107.78 (JCF = 13.75 Hz), 103.21 (JCF = 26.25 Hz), 57.89, 52.24, 45.38, 31.65, 27.11. HRMS (ESI) calculated for C26H25N5OF2S [M + H]+: 494.1821, found: 494.1823. Purity: 99.7% (by HPLC).
5-(3-cyanophenyl)-1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)pyrimidin-2(1H)-one (18). Compound 18 was prepared via a similar procedure of 11a. White oil. Yield: 77.1%. 1H-NMR (500 MHz, CDCl3) δ 8.82 (d, J = 3.2 Hz, 1H), 8.69 (s, 2H), 8.42 (s, 1H), 8.38 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 3.2 Hz, 1H), 7.65 (s, 1H), 7.60 (t, J = 7.1 Hz, 2H), 7.55–7.46 (m, 3H), 5.28 (s, 2H), 3.01 (t, J = 7.2 Hz, 2H), 2.40 (t, J = 7.0 Hz, 2H), 2.21 (s, 6H), 1.86–1.76 (m, 2H). 13C-NMR (126 MHz, CDCl3) δ 164.70, 161.23, 157.43, 155.73, 145.11, 138.15, 135.10, 134.64, 131.48, 131.11, 130.93, 130.32, 130.14, 129.83, 129.37, 128.58, 128.18, 118.25, 116.63, 57.95, 54.44, 45.43, 31.67, 27.17. HRMS (ESI) calculated for C27H26N6OS [M + H]+: 483.1962, found: 483.1942. Purity: 98.3% (by HPLC).
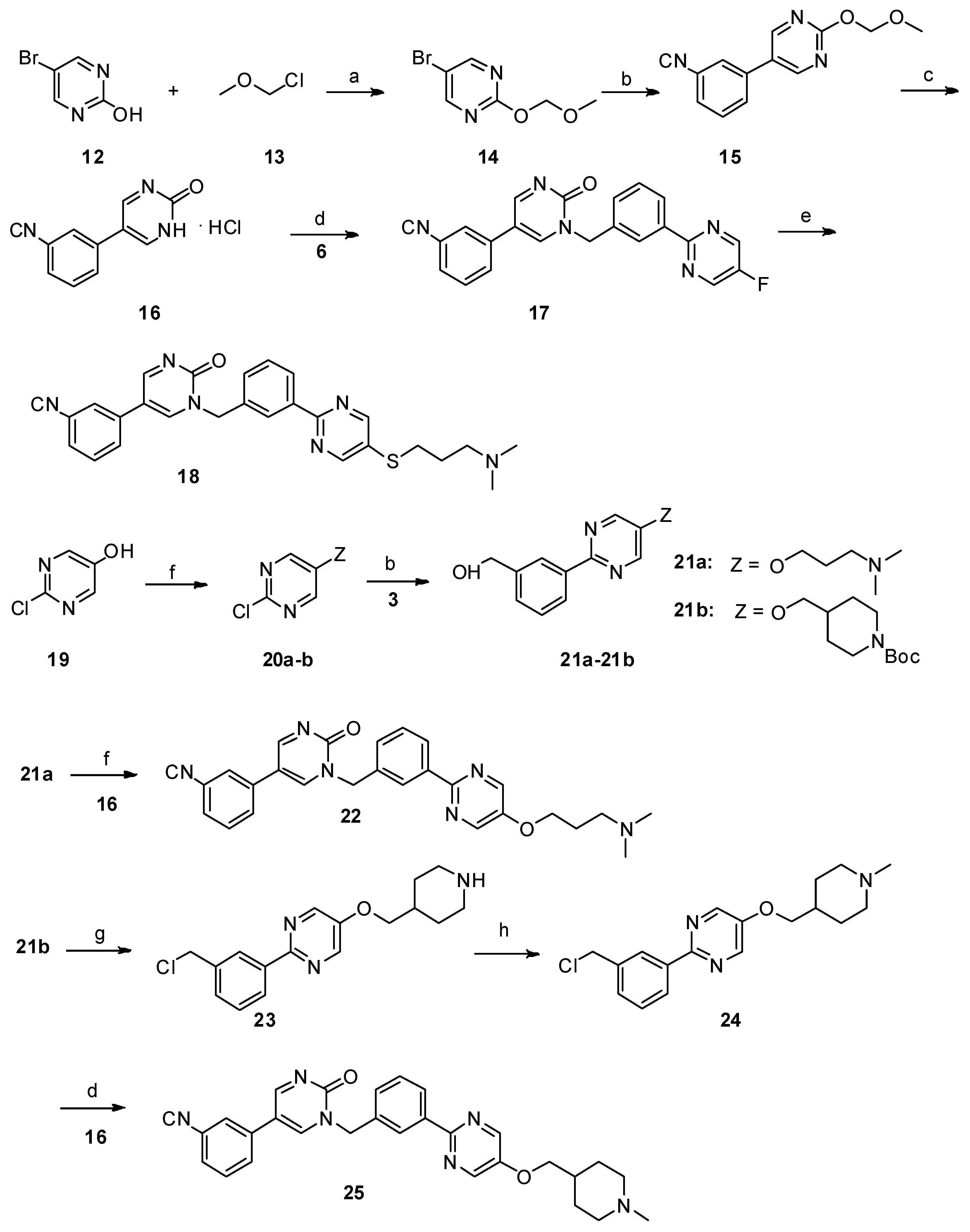
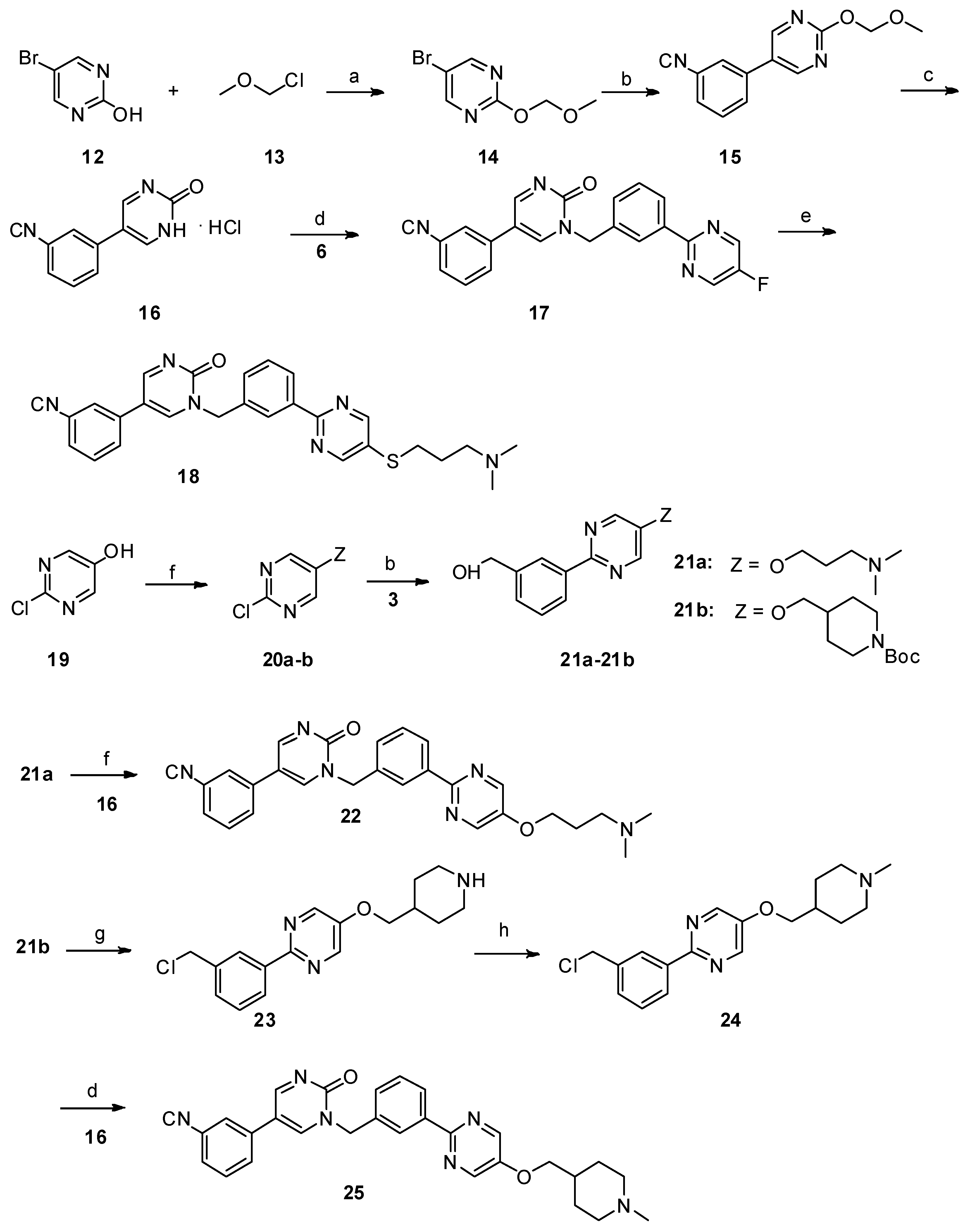
Synthesis of 5-(3-cyanophenyl)-1-(3-(5-(3-(dimethylamino)propoxy)pyrimidin-2-yl)benzyl)pyrimidin-2(1H)-one (22). Compound
21a (157.85 mg, 0.55 mmol) and triphenylphosphine (328 mg, 1.25 mmol) were added successively to a suspension of
16 (116.75 mg, 0.5 mmol) in THF (5 mL) under nitrogen. A solution of diisopropyl azodicarboxylate (104.49 mg, 0.6 mmol) in THF (1 mL) was then slowly added dropwise with ice cooling. The resultant solution was stirred at room temperature for 12 h. The aqueous phase was extracted with dichloromethane (30 mL × 3). The combined organic layer was washed with H
2O (20 mL) and brine (10 mL), and then dried over anhydrous Na
2SO
4, filtered, and evaporated in vacuo. The residue was purified by flash chromatography over silica gel (DCM/MeOH = 40:1–10:1) to give
22 [
14]. Yellow solid. m.p. 122.3–123.6 °C. Yield: 45%.
1H-NMR (500 MHz, CDCl
3)
δ 8.84 (d,
J = 3.0 Hz, 1H), 8.45 (s, 2H), 8.40–8.33 (m, 2H), 7.91 (d,
J = 3.0 Hz, 1H), 7.66 –7.57 (m, 3H), 7.56–7.47 (m, 3H), 5.29 (s, 2H), 4.18 (t,
J = 6.2 Hz, 2H), 2.53 (t,
J = 6.2 Hz, 2H), 2.31 (s, 6H), 2.07–2.00 (m, 2H).
13C-NMR (126 MHz, CDCl
3)
δ 164.56, 156.67, 155.65, 151.81, 144.80, 143.87, 138.53, 134.73, 134.60, 131.43, 130.25, 130.20, 130.09, 129.76, 129.26, 128.14, 127.78, 118.14, 116.54, 113.67, 67.04, 55.84, 54.31, 45.37, 27.17. HRMS (ESI) calculated for C
27H
26N
6O
2 [M + H]
+: 467.2190, found: 467.2185. Purity: 96.784% (by HPLC).
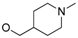
Synthesis of 5-(3-cyanophenyl)-1-(3-(5-((1-methylpiperidin-4-yl)methoxy)pyrimidin-2-yl)benzyl)pyrimidin-2(1H)-one (25). 2-(3-(chloromethyl)phenyl)-5-((1-methylpiperidin-4-yl)methoxy)pyrimidine
24 (99 mg, 0.3 mmol, 1 eq.) and potassium carbonate (124 mg, 0.9 mmol, 3 eq.) were added to a suspension of
16 (77 mg, 0.33 mmol) in dry DMF (2 mL), and the mixture was stirred at 80 °C for 12 h. The reaction mixture was allowed to cool to room temperature. The aqueous phase was extracted with dichloromethane (10 mL × 3). The combined organic layer was washed with H
2O (5 mL) and brine (10 mL), and then dried over anhydrous Na
2SO
4, filtered, and evaporated in vacuo. The residue was purified by flash chromatography over silica gel (DCM/MeOH = 30:1–10:1) to give compound
25 [
15]. White solid. m.p. 116.7–118.2 °C. Yield: 48%.
1H-NMR (500 MHz, CDCl
3)
δ 8.84 (d,
J = 2.9 Hz, 1H), 8.44 (s, 2H), 8.39–8.33 (m, 2H), 7.89 (d,
J = 2.6 Hz, 1H), 7.65–7.60 (m, 2H), 7.60–7.56 (m, 1H), 7.56–7.47 (m, 3H), 5.29 (s, 2H), 3.96 (d,
J = 5.6 Hz, 2H), 2.98 (d,
J = 11.1 Hz, 2H), 2.34 (s, 3H), 2.04 (t,
J = 11.8 Hz, 2H), 1.89–1.81 (m, 3H), 1.54–1.46 (m, 2H).
13C-NMR (126 MHz, CDCl
3)
δ 164.56, 156.70, 155.63, 151.85, 144.72, 143.82, 138.52, 134.73, 134.61, 131.43, 130.25, 130.22, 130.08, 129.77, 129.26, 128.15, 127.77, 118.13, 116.53, 113.68, 73.28, 55.16, 54.30, 46.21, 35.17, 28.69. HRMS (ESI) calculated for C
29H
28N
6O
2 [M + H]
+: 493.2347, found: 493.2351. Purity: 97.8% (by HPLC).
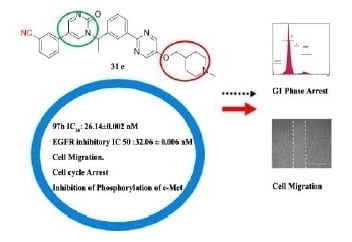
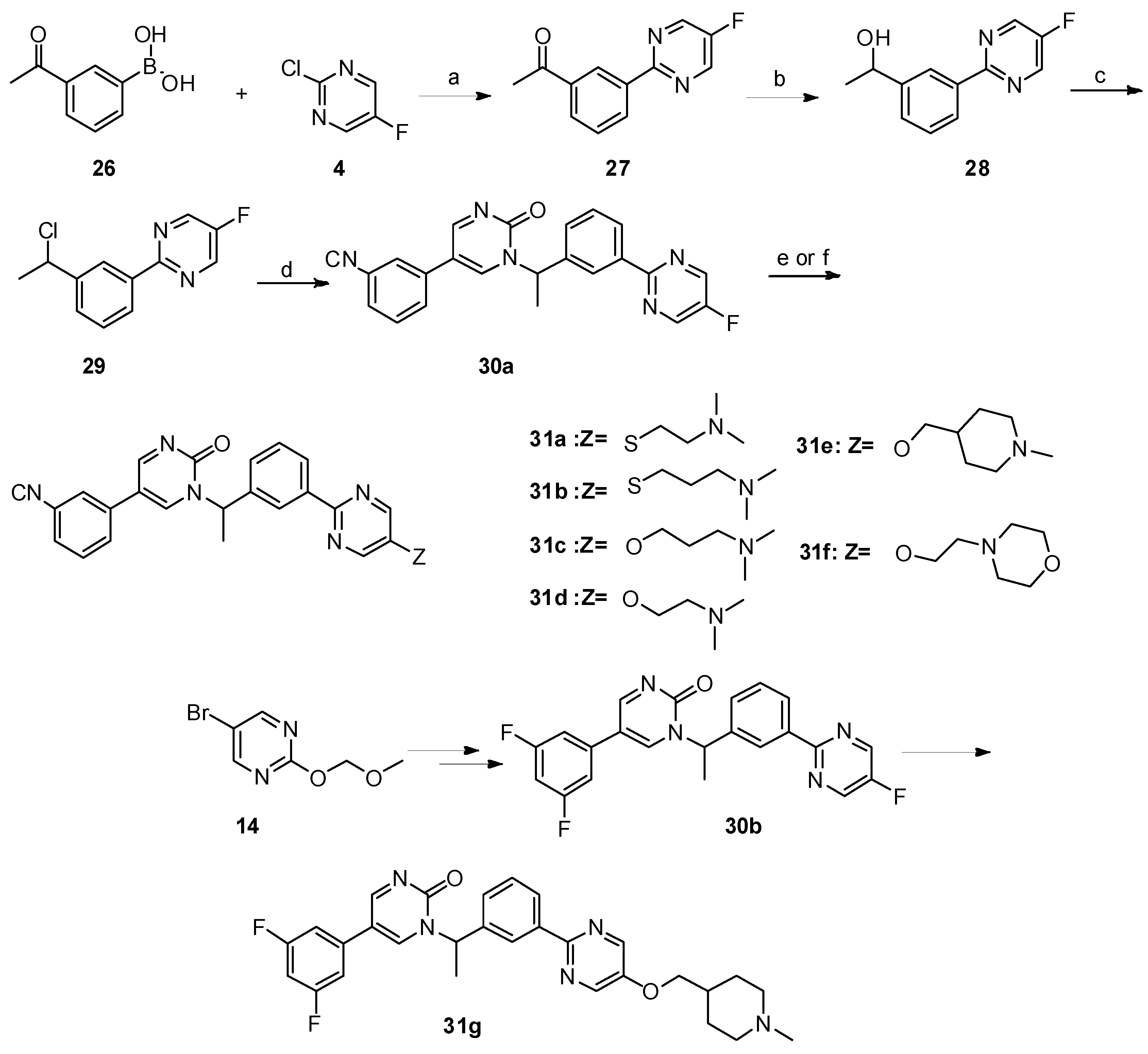
5-(3-cyanophenyl)-1-(1-(3-(5-((2-(dimethylamino)ethyl)thio)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31a). Compound 31a was prepared via a similar procedure of 11a. Yellow oil. Yield: 66%. 1H NMR (400 MHz, CDCl3) δ 8.78 (d, J = 3.3 Hz, 1H), 8.74 (s, 2H), 8.48 (s, 1H), 8.42 (d, J = 7.5 Hz, 1H), 7.71 (d, J = 3.3 Hz, 1H), 7.63–7.53 (m, 3H), 7.52–7.45 (m, 3H), 6.35 (q, J = 6.9 Hz, 1H), 3.08 (t, J = 7.1 Hz, 2H), 2.60 (t, J = 7.1 Hz, 2H), 2.28 (s, 6H), 1.90 (d, J = 7.0 Hz, 3H). 13C-NMR (126 MHz, CDCl3) δ 164.01, 161.33, 157.54, 155.44, 142.40, 138.86, 138.26, 134.89, 131.45, 130.97, 130.31, 130.15, 129.84, 129.32, 128.64, 126.92, 118.22, 116.76, 113.72, 58.36, 56.32, 45.37, 32.02, 19.19. HRMS (ESI) calculated for C27H26N6OS [M + H]+: 483.1962, found: 483.1949. Purity: 98% (by HPLC).
5-(3-cyanophenyl)-1-(1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31b). Compound 31b was prepared via a similar procedure of 11a. Yellow oil. Yield: 75%. 1H NMR (500 MHz, CDCl3) δ 8.78 (d, J = 3.2 Hz, 1H), 8.73 (s, 2H), 8.49 (s, 1H), 8.42 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 3.2 Hz, 1H), 7.63–7.58 (m, 1H), 7.57–7.46 (m, 5H), 6.36 (q, J = 6.9 Hz, 1H), 3.03 (t, J = 7.2 Hz, 2H), 2.42 (t, J = 7.0 Hz, 3H), 2.23 (s, 6H), 1.91 (d, J = 7.0 Hz, 3H), 1.88–1.77 (m, 2H). 13C-NMR (126 MHz, CDCl3) δ 164.04, 161.27, 157.50, 155.49, 142.43, 138.82, 138.32, 134.88, 131.49, 131.02, 130.33, 130.31, 130.18, 129.86, 129.34, 128.66, 126.92, 118.24, 116.83, 113.74, 57.97, 56.37, 45.46, 31.70, 27.18, 19.21. HRMS (ESI) calculated for C28H28N6OS [M + H]+: 497.2118, found: 497.2108. Purity: 97.1% (by HPLC).
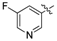
Synthesis of 5-(3-cyanophenyl)-1-(1-(3-(5-(3-(dimethylamino)propoxy)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31c). A solution of 3-(dimethylamino)propan-1-ol (113.3 mg, 1.1 mmol, 2 eq.) in dry DMF( 5 mL) was added to a suspension of NaH (43.56 mg, 3.3 eq.) in dry DMF (5 mL) at 0 °C. After 30 mins, 5-(3-cyanophenyl)-1-(1-(3-(5-fluoropyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1
H)-one
30a (210 mg, 0.55 mmol, 1 eq.) was added. After the reaction mixture was stirred for 5 h, the reaction was quenched with water (15 mL) and the mixture was extracted with dichloromethane (20 mL × 3). The combined organic layer was dried over anhydrous Na
2SO
4 and filtered. After the solvent was removed in vacuo, the crude product was purified by column chromatography (DCM/MeOH = 50:1–10:1) to afford
31c as a white solid [
19]. m.p. 68.3–69.9 °C. Yield: 62%.
1H-NMR (400 MHz, CDCl
3)
δ 8.78 (d,
J = 3.4 Hz, 1H), 8.46 (s, 2H), 8.42 (s, 1H), 8.35 (d,
J = 7.7 Hz, 1H), 7.73 (d,
J = 3.4 Hz, 1H), 7.64–7.58 (m, 1H), 7.56–7.42 (m, 5H), 6.35 (q,
J = 7.0 Hz, 1H), 4.21 (t,
J = 6.1 Hz, 2H), 2.75 (t,
J = 7.3 Hz, 2H), 2.47 (s, 6H), 2.24–2.09 (p, 2H), 1.90 (d,
J = 7.0 Hz, 3H).
13C-NMR (101 MHz, CDCl
3)
δ 163.98, 156.99, 155.48, 151.75, 144.00, 142.53, 138.71, 138.62, 134.92, 131.44, 130.32, 130.19, 129.77, 129.44, 129.35, 128.22, 126.55, 118.28, 116.76, 113.69, 66.75, 56.44, 55.79, 44.79, 26.49, 19.24. HRMS (ESI) calculated for C
28H
28N
6O
2 [M + HCOO]
−: 525.2256, found: 525.2238. Purity: 98.3% (by HPLC).
5-(3-cyanophenyl)-1-(1-(3-(5-(2-(dimethylamino)ethoxy)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31d). Compound 31d was prepared via a similar procedure of 31c. White solid. m.p. 53.3-55.0 °C. Yield: 74%. 1H-NMR (400 MHz, CDCl3) δ 8.80 (d, J = 3.3 Hz, 1H), 8.51 (s, 2H), 8.45 (s, 1H), 8.38 (d, J = 7.7 Hz, 1H), 7.72 (d, J = 3.4 Hz, 1H), 7.64–7.58 (m, 1H), 7.57–7.42 (m, 5H), 6.38 (q, J = 7.0 Hz, 1H), 4.24 (t, J = 5.4 Hz, 2H), 2.83 (t, J = 5.4 Hz, 2H), 2.40 (s, 6H), 1.91 (d, J = 7.0 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 163.94, 156.94, 155.45, 151.78, 144.05, 142.52, 138.67, 138.59, 134.89, 131.40, 130.27, 130.15, 129.73, 129.44, 129.32, 128.19, 126.53, 118.24, 116.72, 113.66, 66.88, 58.10, 56.40, 45.90, 19.18. HRMS (ESI) calculated for C27H26N6O2 [M + HCOO]−: 511.2099, found: 511.2074. Purity: 98% (by HPLC).
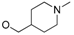
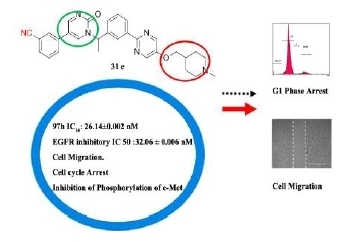
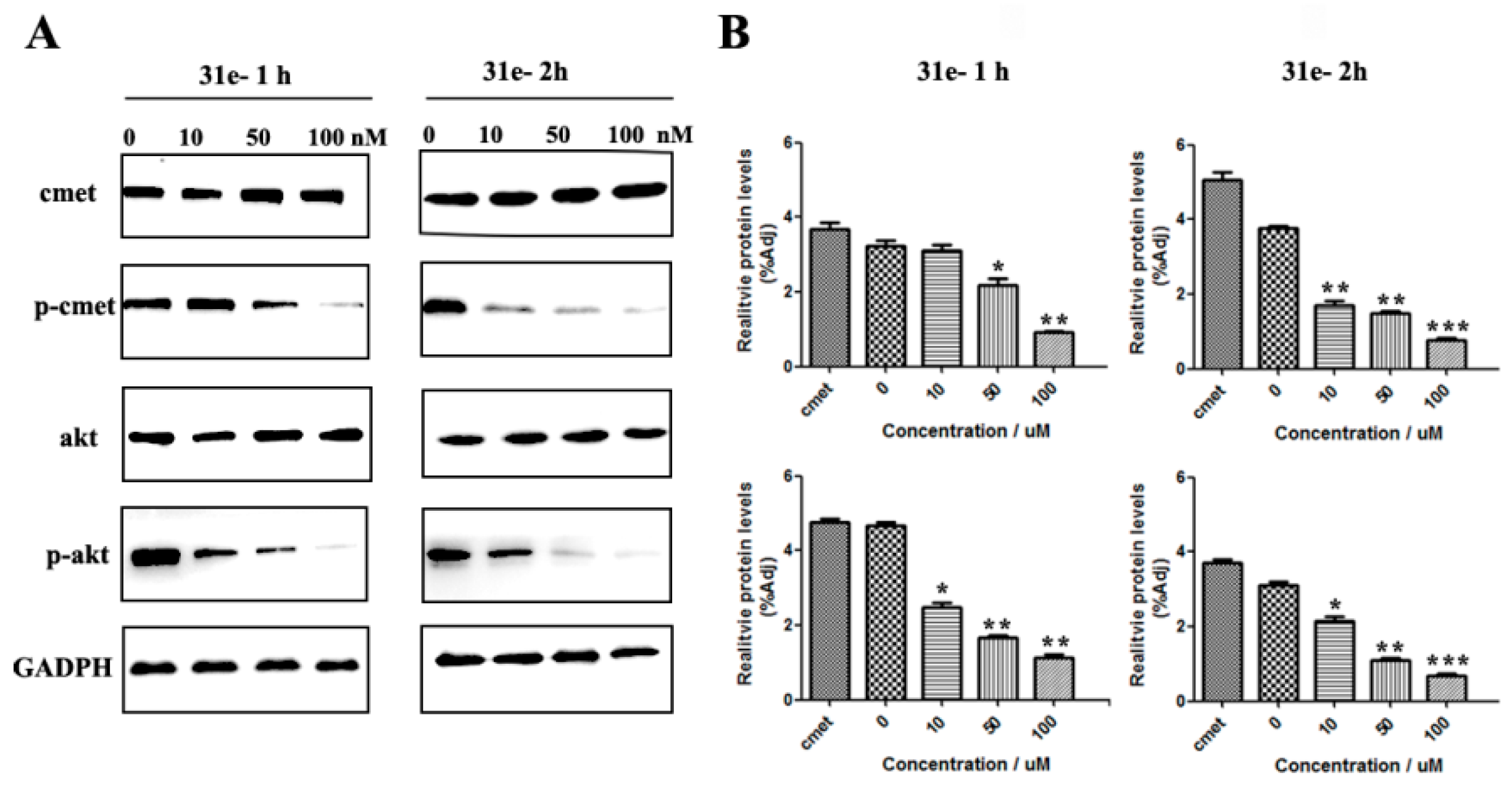
5-(3-cyanophenyl)-1-(1-(3-(5-((1-methylpiperidin-4-yl)methoxy)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31e). Compound 31e was prepared via a similar procedure of 31c. Yellow solid. m.p. 98.2–99.9 °C. Yield: 76%. 1H-NMR (500 MHz, DMSO-d6) δ 9.04 (d, J = 3.2 Hz, 1H), 8.72 (d, J = 3.2 Hz, 1H), 8.64 (s, 2H), 8.33 (s, 1H), 8.27–8.18 (m, 2H), 8.03 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.59–7.45 (m, 2H), 6.02 (q, J = 7.0 Hz, 1H), 4.04 (d, J = 5.8 Hz, 2H), 2.80 (d, J = 11.1 Hz, 2H), 2.16 (s, 3H), 1.92 (d, J = 7.2 Hz, 3H), 1.87 (d, J = 11.3 Hz, 2H), 1.78–1.67 (m, 3H), 1.36–1.27 (m, 2H). 13C-NMR (101 MHz, CDCl3) δ 163.97, 156.82, 155.49, 151.98, 143.94, 142.52, 138.66, 134.92, 131.44, 130.30, 130.17, 129.77, 129.44, 129.34, 128.21, 126.52, 118.26, 116.75, 113.71, 73.39, 56.42, 55.30, 46.35, 35.26, 28.81, 19.21. HRMS (ESI) calculated for C30H30N6O2 [M + HCOO]−: 551.2412, found: 551.2402. Purity: 98.9% (by HPLC).
5-(3-cyanophenyl)-1-(1-(3-(5-(2-morpholinoethoxy)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31f). Compound 31f was prepared via a similar procedure of 31c. White solid. m.p. 82.3–83.9 °C. Yield: 68%. 1H-NMR (400 MHz, CDCl3) δ 8.78 (d, J = 3.4 Hz, 1H), 8.47 (s, 2H), 8.43 (s, 1H), 8.35 (d, J = 7.7 Hz, 1H), 7.72 (d, J = 3.4 Hz, 1H), 7.62–7.56 (m, 1H), 7.56–7.42 (m, 5H), 6.35 (q, J = 7.0 Hz, 1H), 4.25 (t, J = 5.5 Hz, 2H), 3.79–3.66 (m, 4H), 2.85 (t, J = 5.5 Hz, 2H), 2.68–2.50 (m, 4H), 1.89 (d, J = 7.0 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 163.95, 157.05, 155.45, 151.72, 144.13, 142.50, 138.70, 138.56, 134.91, 131.42, 130.28, 130.15, 129.75, 129.46, 129.33, 128.21, 126.56, 118.25, 116.72, 113.68, 66.87, 66.77, 57.53, 56.39, 54.17, 19.21. HRMS (ESI) calculated for C29H28N6O3 [M + HCOO]−: 553.2205, found: 553.2181. Purity: 99.6% (by HPLC).
5-(3,5-difluorophenyl)-1-(1-(3-(5-((1-methylpiperidin-4-yl)methoxy)pyrimidin-2-yl)phenyl)ethyl)pyrimidin-2(1H)-one (31g). Compound 31g was prepared via a similar procedure of 31c. White solid. m.p. 86.9–88.6 °C. Yield: 55%. 1H-NMR (500 MHz, CDCl3) δ 8.76 (d, J = 3.2 Hz, 1H), 8.43 (s, 2H), 8.42 (s, 2H), 8.35 (d, J = 7.7 Hz, 1H), 7.67 (d, J = 3.3 Hz, 1H), 7.51 (t, J = 7.7 Hz, 1H), 7.47–7.39 (m, 1H), 6.82–6.69 (m, 3H), 6.35 (q, J = 6.9 Hz, 1H), 3.95 (d, J = 5.7 Hz, 2H), 3.00 (d, J = 11.2 Hz, 2H), 2.35 (s, 3H), 2.08 (t, J = 11.5 Hz, 2H), 1.93–1.77 (m, 6H), 1.60–1.49 (m, 2H). 13C-NMR (126 MHz, CDCl3) δ 163.92, 163.665(JCF = 248.75 Hz), 163.565(JCF = 248.75 Hz), 156.88, 155.54, 151.94, 143.92, 142.42, 138.69, 138.65, 136.67 (JCF = 10.0 Hz), 129.74, 129.36, 128.18, 126.59, 116.74 (JCF = 2.2 Hz),108.83 (JCF = 12.5 Hz), 108.825(JCF = 26.25 Hz), 103.41 (JCF = 25.2 Hz), 73.28, 56.39, 55.20, 46.15, 35.17, 28.61, 19.18. HRMS (ESI) calculated for C29H29N5O2F2 [M + HCOO]−: 562.2271, found: 562.2248. Purity: 99.4% (by HPLC).
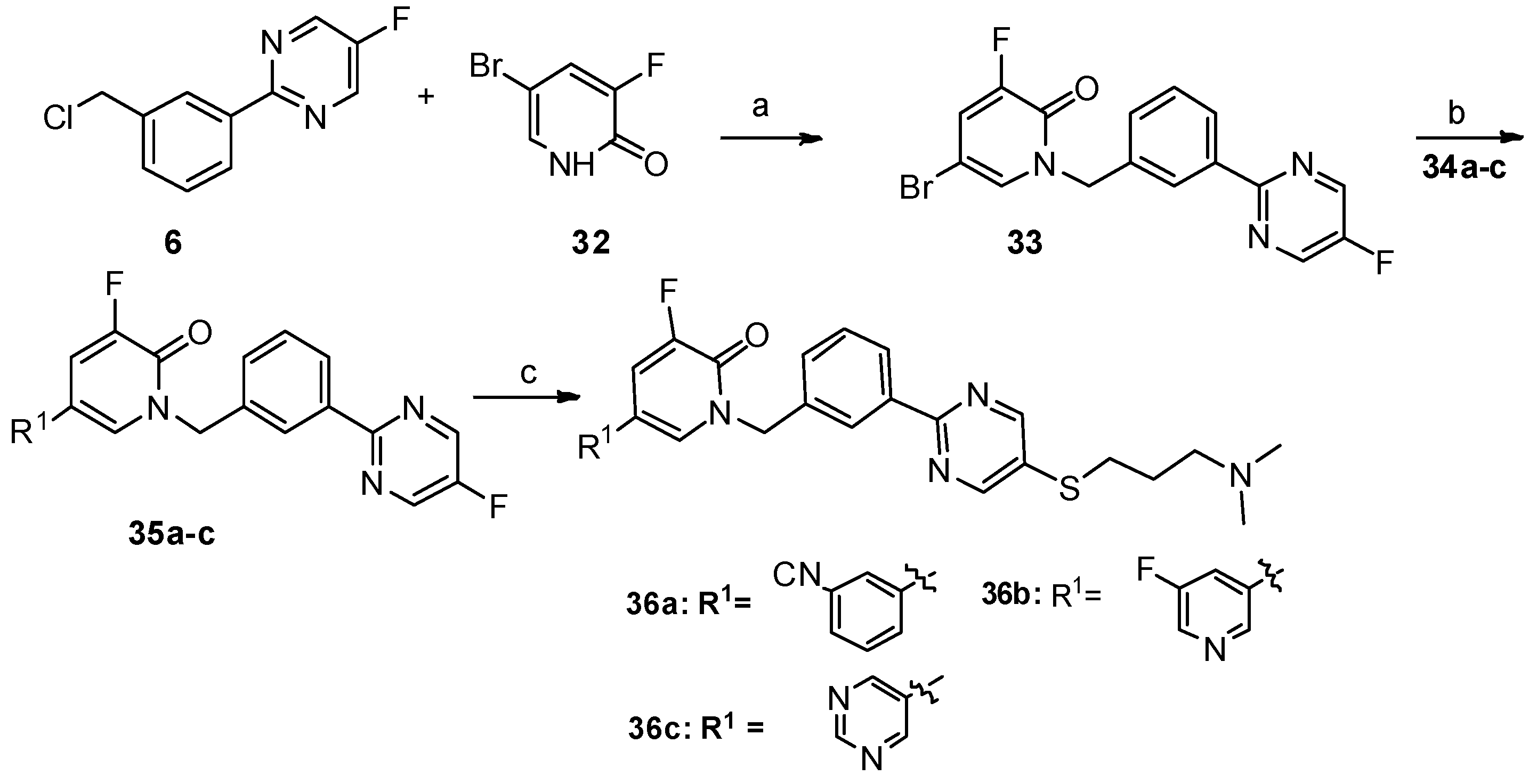
5-(3-cyanophenyl)-1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)-3-fluoropyridin-2(1H)-one (36a). Compound 36a was prepared via a similar procedure of 11a. Yellow solid. m.p. 116.3–117.9 °C. Yield: 72%. 1H-NMR (400 MHz, CDCl3) δ 8.74 (s, 1H), 8.43 (s, 1H), 8.41–8.37 (m, 1H), 7.64 (s, 1H), 7.62–7.52 (m, 3H), 7.52–7.49 (m, 2H), 7.45–7.41 (m, 1H), 7.37 (dd, J = 9.8, 2.3 Hz, 1H), 5.36 (s, 2H), 3.03 (t, J = 7.2 Hz, 2H), 2.45 (t, J = 7.0 Hz, 2H), 2.26 (s, 6H), 1.93–1.80 (m, 2H). 13C-NMR (126 MHz, CDCl3) δ 161.35, 157.48, 155.62 (JCF = 26.25 Hz), 152.58 (JCF = 251.25 Hz), 137.92, 137.05, 135.88, 131.10, 130.72, 130.68, 130.23, 130.16(JCF = 3.75 Hz), 130.06, 129.66, 129.42, 128.25, 127.85, 119.54 (JCF = 17.5 Hz), 118.29, 116.57 (JCF = 5.0 Hz), 113.46, 57.86, 52.50, 45.31, 31.65, 27.03. HRMS (ESI) calculated for C28H26N5OFS [M + H]+: 500.1915, found: 500.1898. Purity: 98.1% (by HPLC).
1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)-5,5′-difluoro-[3,3′-bipyridin]-6(1H)-one (36b). Compound 36b was prepared via a similar procedure of 11a. White solid. m.p. 128.3–129.9 °C. Yield: 75%. 1H-NMR (400 MHz, CDCl3) δ 8.74 (s, 2H), 8.47–8.36 (m, 4H), 7.51 (d, J = 5.1 Hz, 2H), 7.45–7.42 (m, 1H), 7.40–7.33 (m, 2H), 5.37 (s, 2H), 3.04 (t, J = 7.2 Hz, 2H), 2.44 (t, J = 7.0 Hz, 2H), 2.25 (s, 6H), 1.91–1.79 (m, 2H). 13C-NMR (126 MHz, CDCl3) δ 161.29, 159.52 (JCF = 257.50 Hz), 157.52, 157.45, 155.59 (JCF = 26.25 Hz), 152.66 (JCF = 252.50 Hz), 142.80 (JCF = 3.75 Hz), 137.96, 137.20 (JCF = 23.75Hz), 135.75, 130.77, 130.73, 130.36 (JCF = 5.0 Hz), 129.65, 128.26, 127.88, 120.01 (JCF = 18.75 Hz), 119.33 (JCF = 18.75 Hz), 113.96 (JCF = 6.25 Hz), 57.91, 52.48, 45.40, 31.69, 27.16. HRMS (ESI) calculated for C26H25N5OF2S [M + H]+: 494.1821, found: 494.1807. Purity: 99.7% (by HPLC).
1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)benzyl)-3-fluoro-5-(pyrimidin-5-yl)pyridin-2(1H)-one (36c). Compound 36c was prepared via a similar procedure of 11a. White solid. 128.9–129.8 °C. Yield: 78%. 1H-NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 8.75 (s, 2H), 8.74 (s, 2H), 8.45–8.37 (m, 2H), 7.51 (d, J = 5.1 Hz, 2H), 7.44 (s, 1H), 7.36 (dd, J = 9.5, 2.3 Hz, 1H), 5.37 (s, 2H), 3.04 (t, J = 7.2 Hz, 2H), 2.45 (t, J = 7.0 Hz, 2H), 2.26 (s, 6H), 1.93–1.80 (m,2H). 13C-NMR (126 MHz, CDCl3) δ 161.06, 157.52, 157.25, 155.40 (JCF = 25.7 Hz), 153.68, 151.64, 137.82, 135.46, 131.62 (JCF = 10.2 Hz), 130.63, 130.16 (JCF = 4.6 Hz), 129.50, 128.71 (JCF = 12.5 Hz), 128.14, 127.73, 118.68 (JCF = 18.2 Hz), 111.65 (JCF = 5.5 Hz), 57.73, 52.35, 45.21, 31.49, 26.96. HRMS (ESI) calculated for C25H25N6OFS [M + H]+: 477.1867, found: 477.1857. Purity: 98.9% (by HPLC).
5-(3-cyanophenyl)-1-(1-(3-(5-((3-(dimethylamino)propyl)thio)pyrimidin-2-yl)phenyl)ethyl)-3-fluoropyridin-2(1H)-one (39). Compound 39 was prepared via a similar procedure of 11a. Colorless oil. Yield: 82%. 1H-NMR (400 MHz, CDCl3) δ 8.75 (s, 2H), 8.49 (s, 1H), 8.40 (d, J = 7.6 Hz, 1H), 7.60–7.43 (m, 6H), 7.32 (dd, J = 9.8, 2.3 Hz, 1H), 7.22 (s, 1H), 6.62 (q, J = 7.0 Hz, 1H), 3.04 (t, J = 7.2 Hz, 2H), 2.47 (t, J = 7.0 Hz, 2H), 2.27 (s, 6H), 1.91–1.81 (m, 5H). 13C-NMR (101 MHz, CDCl3) δ 161.36, 157.45, 155.42(JCF = 25.0 Hz), 152.15(JCF = 251.0 Hz), 139.61, 137.97, 137.32, 130.98, 130.76, 130.21, 130.03, 130.01, 129.53, 129.40, 128.20, 127.26 (JCF = 5.0 Hz), 126.64, 119.00 (JCF = 18.0 Hz), 118.33, 116.57 (JCF = 6.0 Hz), 113.37, 57.88, 53.78, 45.34, 31.65, 27.08, 19.23. HRMS (ESI) calculated for C29H28N5OFS [M + H]+: 514.2071, found: 514.2066. Purity: 99% (by HPLC).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}