Role of Natural Products in Modulating Histone Deacetylases in Cancer

 ,
, 
Abstract
:1. Introduction
1.1. HDAC’s Role in Cancer
1.1.1. Pro-Cancer Effects
1.1.2. Anti-Cancer Effects
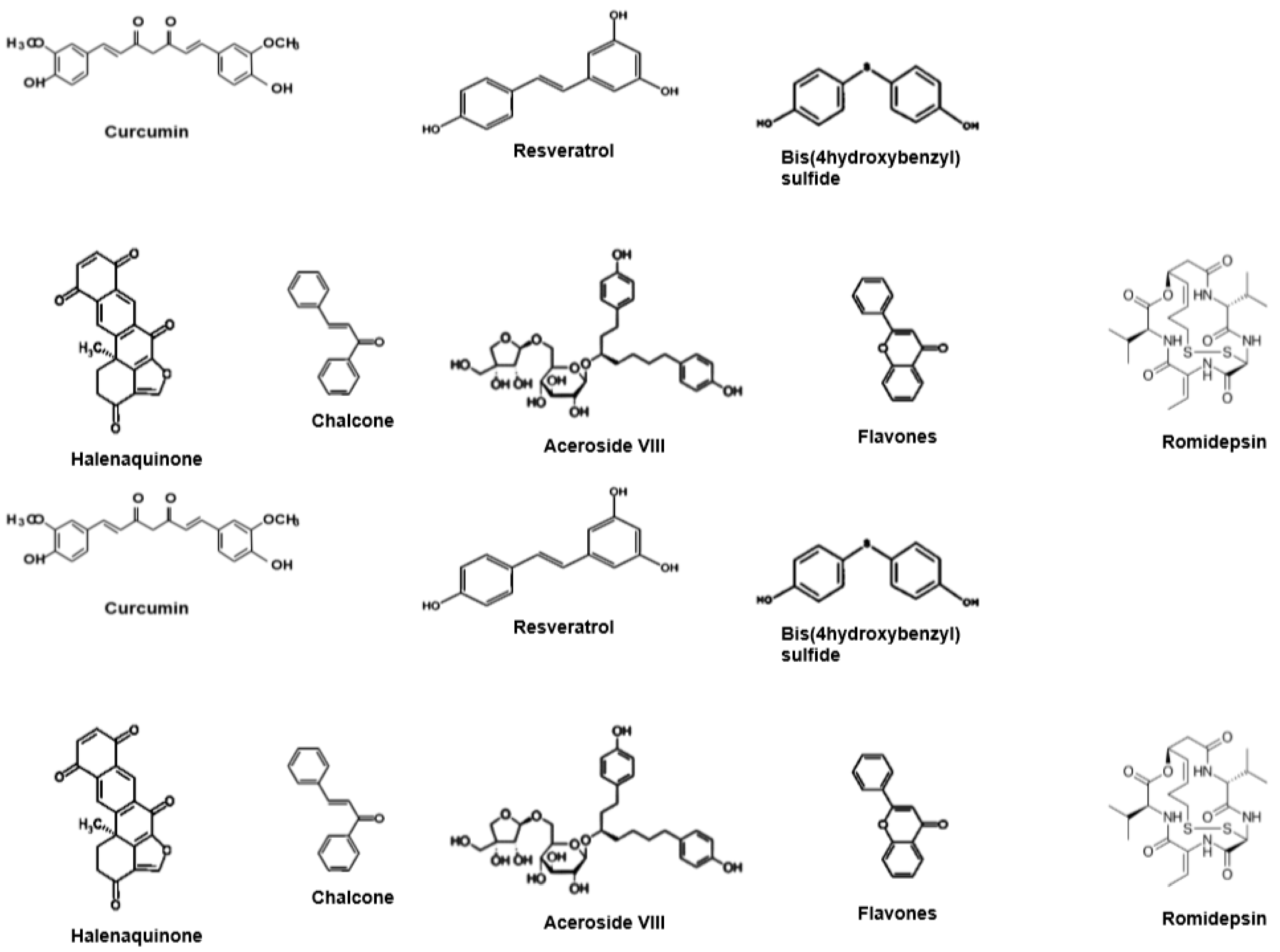
1.2. HDACis from Natural Products
1.2.1. Resveratrol (RVT)
1.2.2. Curcumin
1.2.3. Marine Products
Actinomycetes Strains
Marine Polycyclic Quinone-Type, Halenaquinone
Other Products
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| pAkt | Phosphorylated Protein kinase B |
| Bax | Bcl-2-associated X protein |
| c-Myc | Proto-oncogene |
| DSBs | Double-Strand DNA Breaks |
| ERα | Estrogen receptor-α acetyl-H3. |
| EGFR | Epidermal Growth Factor |
| HAT protein | Histone acetyltransferase |
| HDAC | Histone deacetylases |
| Hsp90 | Chaperone |
| I κBα | Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor α (I κB α) |
| MTA1 | Metastasis associated protein |
| Mec1 | Serine/threonine-protein kinase |
| Notch 1 | Notch homolog 1, translocation-associated (Drosophila) |
| PAX1 | Paired box gene1 |
| PARP | Poly ADP ribose polymerase |
| p27 & p21 | Cyclin-dependent kinase inhibitor |
| pERK | phosphorylated Extracellular signal-regulated kinases |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| Raf-1 | Proto-oncogene serine/threonine-protein kinase |
| ROS | Reactive oxygen species. STAT3: Signal transducer and activator of transcription 3 |
| SIRT | Sirtuin |
| TNFα | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| UHRF1 | Ubiquitin-like with PHD and RING Finger domains 1. |
References
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Lee, J.H.; Chai, E.Z.; Kanchi, M.M.; Kar, S.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Ramar, P.S.; Looi, C.Y.; et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. Semin. Cancer Biol. 2016, 40–41, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Kannaiyan, R.; Sethi, G. Targeting cell signaling and apoptotic pathways by dietary agents: Role in the prevention and treatment of cancer. Nutr. Cancer 2011, 63, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Deorukhkar, A.; Krishnan, S.; Sethi, G.; Aggarwal, B.B. Back to basics: How natural products can provide the basis for new therapeutics. Expert Opin. Investig. Drugs 2007, 16, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef]
- Valdez, B.C.; Brammer, J.E.; Li, Y.; Murray, D.; Liu, Y.; Hosing, C.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Romidepsin targets multiple survival signaling pathways in malignant T cells. Blood Cancer J. 2015, 5, e357. [Google Scholar] [CrossRef] [PubMed]
- Havas, A.P.; Rodrigues, K.B.; Bhakta, A.; Demirjian, J.A.; Hahn, S.; Tran, J.; Scavello, M.; Tula-Sanchez, A.A.; Zeng, Y.; Schmelz, M.; et al. Belinostat and vincristine demonstrate mutually synergistic cytotoxicity associated with mitotic arrest and inhibition of polyploidy in a preclinical model of aggressive diffuse large B cell lymphoma. Cancer Biol. Ther. 2016, 17, 1240–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414–11426. [Google Scholar] [CrossRef]
- Kaypee, S.; Sudarshan, D.; Shanmugam, M.K.; Mukherjee, D.; Sethi, G.; Kundu, T.K. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol. Ther. 2016, 162, 98–119. [Google Scholar] [CrossRef]
- Selvi, R.B.; Swaminathan, A.; Chatterjee, S.; Shanmugam, M.K.; Li, F.; Ramakrishnan, G.B.; Siveen, K.S.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Inhibition of p300 lysine acetyltransferase activity by luteolin reduces tumor growth in head and neck squamous cell carcinoma (HNSCC) xenograft mouse model. Oncotarget 2015, 6, 43806–43818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Sethi, G. Role of epigenetics in inflammation-associated diseases. Sub-Cell. Biochem. 2013, 61, 627–657. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Kook, H. Posttranslational modifications of histone deacetylases: Implications for cardiovascular diseases. Pharmacol. Ther. 2014, 143, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Jeon, E.J.; Li, Q.L.; Lee, Y.H.; Choi, J.K.; Kim, W.J.; Lee, K.Y.; Bae, S.C. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J. Biol. Chem. 2004, 279, 29409–29417. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Pontiki, E.; Hadjipavlou-Litina, D. Histone deacetylase inhibitors (HDACIs). Structure--activity relationships: History and new QSAR perspectives. Med. Res. Rev. 2012, 32, 1–165. [Google Scholar] [CrossRef]
- Vaquero, A.; Sternglanz, R.; Reinberg, D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007, 26, 5505–5520. [Google Scholar] [CrossRef] [Green Version]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Holbert, M.A.; Marmorstein, R. Structure and activity of enzymes that remove histone modifications. Curr. Opin. Struct. Biol. 2005, 15, 673–680. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenet. 2012, 4, 5. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Krusche, C.A.; Wulfing, P.; Kersting, C.; Vloet, A.; Bocker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and -3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005, 90, 15–23. [Google Scholar] [CrossRef]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Wisnieski, F.; Calcagno, D.Q.; Leal, M.F.; Santos, L.C.; Gigek, C.O.; Chen, E.S.; Demachki, S.; Artigiani, R.; Assumpcao, P.P.; Lourenco, L.G.; et al. CDKN1A histone acetylation and gene expression relationship in gastric adenocarcinomas. Clin. Exp. Med. 2017, 17, 121–129. [Google Scholar] [CrossRef]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef]
- Meisenberg, C.; Ashour, M.E.; El-Shafie, L.; Liao, C.; Hodgson, A.; Pilborough, A.; Khurram, S.A.; Downs, J.A.; Ward, S.E.; El-Khamisy, S.F. Epigenetic changes in histone acetylation underpin resistance to the topoisomerase I inhibitor irinotecan. Nucleic Acids Res. 2017, 45, 1159–1176. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Wang, X.Q.; Bai, H.M.; Li, S.T.; Sun, H.; Min, L.Z.; Tao, B.B.; Zhong, J.; Li, B. Knockdown of HDAC1 expression suppresses invasion and induces apoptosis in glioma cells. Oncotarget 2017, 8, 48027–48040. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Lee, K.S.; Park, S.Y.; Kim, J.H.; Kang, E.Y.; Kim, S.W.; Eom, K.Y.; Kim, J.S.; Kim, I.A. Potential Prognostic Value of Histone Deacetylase 6 and Acetylated Heat-Shock Protein 90 in Early-Stage Breast Cancer. J. Breast Cancer 2015, 18, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Marquard, L.; Gjerdrum, L.M.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Ralfkiaer, E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 2008, 53, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.F.; Weng, C.J.; Sethi, G.; Hu, D.N. Natural bioactives and phytochemicals serve in cancer treatment and prevention. Evid.-Based Complement. Altern. Med. eCAM 2013, 2013, 698190. [Google Scholar] [CrossRef]
- Tang, C.H.; Sethi, G.; Kuo, P.L. Novel medicines and strategies in cancer treatment and prevention. BioMed Res. Int. 2014, 2014, 474078. [Google Scholar] [CrossRef]
- Hsieh, Y.S.; Yang, S.F.; Sethi, G.; Hu, D.N. Natural bioactives in cancer treatment and prevention. BioMed Res. Int. 2015, 2015, 182835. [Google Scholar] [CrossRef] [PubMed]
- Yarla, N.S.; Bishayee, A.; Sethi, G.; Reddanna, P.; Kalle, A.M.; Dhananjaya, B.L.; Dowluru, K.S.; Chintala, R.; Duddukuri, G.R. Targeting arachidonic acid pathway by natural products for cancer prevention and therapy. Semin. Cancer Biol. 2016, 40–41, 48–81. [Google Scholar] [CrossRef] [PubMed]
- Hasanpourghadi, M.; Looi, C.Y.; Pandurangan, A.K.; Sethi, G.; Wong, W.F.; Mustafa, M.R. Phytometabolites Targeting the Warburg Effect in Cancer Cells: A Mechanistic Review. Curr. Drug Targets 2017, 18, 1086–1094. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sethi, G.; Baladandayuthapani, V.; Krishnan, S.; Shishodia, S. Targeting cell signaling pathways for drug discovery: An old lock needs a new key. J. Cell. Biochem. 2007, 102, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Hwang, S.T.; Sethi, G.; Fan, L.; Arfuso, F.; Ahn, K.S. Potential Anti-Inflammatory and Anti-Cancer Properties of Farnesol. Molecules 2018, 23, 2827. [Google Scholar] [CrossRef] [PubMed]
- Merarchi, M.; Sethi, G.; Fan, L.; Mishra, S.; Arfuso, F.; Ahn, K.S. Molecular Targets Modulated by Fangchinoline in Tumor Cells and Preclinical Models. Molecules 2018, 23, 2538. [Google Scholar] [CrossRef]
- Sethi, G.; Shanmugam, M.K.; Warrier, S.; Merarchi, M.; Arfuso, F.; Kumar, A.P.; Bishayee, A. Pro-Apoptotic and Anti-Cancer Properties of Diosgenin: A Comprehensive and Critical Review. Nutrients 2018, 10, 645. [Google Scholar] [CrossRef]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef]
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. Semin. Cancer Biol. 2016, 40–41, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Nguyen, A.H.; Kumar, A.P.; Tan, B.K.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrimali, D.; Shanmugam, M.K.; Kumar, A.P.; Zhang, J.; Tan, B.K.; Ahn, K.S.; Sethi, G. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013, 341, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Kumar, A.; Li, K.; Tzivion, G.; Levenson, A.S. Resveratrol regulates PTEN/Akt pathway through inhibition of MTA1/HDAC unit of the NuRD complex in prostate cancer. Biochim. Biophys. Acta 2015, 1853, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Venturelli, S.; Berger, A.; Bocker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Tollefsbol, T.O. A Novel Combinatorial Epigenetic Therapy Using Resveratrol and Pterostilbene for Restoring Estrogen Receptor-alpha (ERalpha) Expression in ERalpha-Negative Breast Cancer Cells. PLoS ONE 2016, 11, e0155057. [Google Scholar] [CrossRef] [PubMed]
- Parashar, G.; Capalash, N. Promoter methylation-independent reactivation of PAX1 by curcumin and resveratrol is mediated by UHRF1. Clin. Exp. Med. 2016, 16, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shu, W.; Chen, W.; Wu, Q.; Liu, H.; Cui, G. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin. Pharmacol. Toxicol. 2007, 101, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lin, P.Y.; Chiu, Y.C.; Huang, J.S.; Kuo, Y.T.; Wu, J.C.; Chen, C.C. Curcumin-Mediated HDAC Inhibition Suppresses the DNA Damage Response and Contributes to Increased DNA Damage Sensitivity. PLoS ONE 2015, 10, e0134110. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Gomez-Quiroz, L.; Arreguin Camacho, L.O.; Pinna, F.; Lee, Y.H.; Kitade, M.; Dominguez, M.P.; Castven, D.; Breuhahn, K.; Conner, E.A.; et al. Curcumin effectively inhibits oncogenic NF-kappaB signaling and restrains stemness features in liver cancer. J. Hepatol. 2015, 63, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Krauthauser, C.; Maduskuie, V.; Fawcett, P.T.; Olson, J.M.; Rajasekaran, S.A. Curcumin-induced HDAC inhibition and attenuation of medulloblastoma growth in vitro and in vivo. BMC Cancer 2011, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Graham, K.; Lanza-Jacoby, S. Curcumin enhances the anticancer effects of trichostatin a in breast cancer cells. Mol. Carcinog. 2013, 52, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Giommarelli, C.; Zuco, V.; Favini, E.; Pisano, C.; Dal Piaz, F.; De Tommasi, N.; Zunino, F. The enhancement of antiproliferative and proapoptotic activity of HDAC inhibitors by curcumin is mediated by Hsp90 inhibition. Cell. Mol. Life Sci. CMLS 2010, 67, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Chen, J.; Shi, Y.; Jia, J.; Zhang, Y. Curcumin-induced histone hypoacetylation: The role of reactive oxygen species. Biochem. Pharmacol. 2005, 69, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Yar Saglam, A.S.; Yilmaz, A.; Onen, H.I.; Alp, E.; Kayhan, H.; Ekmekci, A. HDAC inhibitors, MS-275 and salermide, potentiates the anticancer effect of EF24 in human pancreatic cancer cells. EXCLI J. 2016, 15, 246–255. [Google Scholar] [CrossRef]
- Abdelfattah, M.S.; Elmallah, M.I.Y.; Faraag, A.H.I.; Hebishy, A.M.S.; Ali, N.H. Heliomycin and tetracinomycin D: Anthraquinone derivatives with histone deacetylase inhibitory activity from marine sponge-associated Streptomyces sp. SP9. 3 Biotech 2018, 8, 282. [Google Scholar] [CrossRef]
- Varghese, T.A.; Jayasri, M.A.; Suthindhiran, K. Marine Actinomycetes as potential source for histone deacetylase inhibitors and epigenetic modulation. Lett. Appl. Microbiol. 2015, 61, 69–76. [Google Scholar] [CrossRef]
- Shih, S.P.; Lee, M.G.; El-Shazly, M.; Juan, Y.S.; Wen, Z.H.; Du, Y.C.; Su, J.H.; Sung, P.J.; Chen, Y.C.; Yang, J.C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.W.; Lee, D.H.; Shin, D.H.; Kim, S.H.; Kwon, S.H. Aceroside VIII is a new natural selective HDAC6 inhibitor that synergistically enhances the anticancer activity of HDAC inhibitor in HT29 cells. Planta Med. 2015, 81, 222–227. [Google Scholar] [CrossRef]
- Son, I.H.; Lee, S.I.; Yang, H.D.; Moon, H.I. Bis(4-hydroxybenzyl)sulfide: A sulfur compound inhibitor of histone deacetylase isolated from root extract of Pleuropterus ciliinervis. Molecules 2007, 12, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Schnekenburger, M.; Zloh, M.; Golais, F.; Diederich, M.; Tasdemir, D. Natural chalcones as dual inhibitors of HDACs and NF-kappaB. Oncol. Rep. 2012, 28, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Bontempo, P.; Mita, L.; Miceli, M.; Doto, A.; Nebbioso, A.; De Bellis, F.; Conte, M.; Minichiello, A.; Manzo, F.; Carafa, V.; et al. Feijoa sellowiana derived natural Flavone exerts anti-cancer action displaying HDAC inhibitory activities. Int. J. Biochem. Cell Biol. 2007, 39, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.; Crozier, A. Plant foods and herbal sources of resveratrol. J. Agric. Food Chem. 2002, 50, 3337–3340. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.G.; D’Orazio, J.A.; Pearson, K.J. Resveratrol and cancer: Focus on in vivo evidence. Endocr.-Relat. Cancer 2014, 21, R209–R225. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomed. Int. J. Phytother. Phytopharmacol. 2016, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Kunnumakkara, A.B.; Sethi, G.; Diagaradjane, P.; Anand, P.; Pandey, M.K.; Gelovani, J.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int. J. Cancer 2010, 127, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Sethi, G.; Vadhan-Raj, S.; Bueso-Ramos, C.; Takada, Y.; Gaur, U.; Nair, A.S.; Shishodia, S.; Aggarwal, B.B. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood 2007, 109, 2293–2302. [Google Scholar] [CrossRef]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. J. Lab. Clin. Med. 2015, 165, 36–47. [Google Scholar] [CrossRef]
- Zbuk, K.M.; Eng, C. Cancer phenomics: RET and PTEN as illustrative models. Nat. Rev. Cancer 2007, 7, 35–45. [Google Scholar] [CrossRef]
- Singh, A.; Patel, V.K.; Jain, D.K.; Patel, P.; Rajak, H. Panobinostat as Pan-deacetylase Inhibitor for the Treatment of Pancreatic Cancer: Recent Progress and Future Prospects. Oncol. Ther. 2016, 4, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helland, O.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjorge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed]
- Bots, M.; Verbrugge, I.; Martin, B.P.; Salmon, J.M.; Ghisi, M.; Baker, A.; Stanley, K.; Shortt, J.; Ossenkoppele, G.J.; Zuber, J.; et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014, 123, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuto, A.; Kirschbaum, M.; Buettner, R.; Kujawski, M.; Cermak, J.M.; Atadja, P.; Jove, R. SIRT1 activation enhances HDAC inhibition-mediated upregulation of GADD45G by repressing the binding of NF-kappaB/STAT3 complex to its promoter in malignant lymphoid cells. Cell Death Dis. 2013, 4, e635. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, A.; Shahzad, R.; Lee, Y.S. Curcumin: A Potent Modulator of Multiple Enzymes in Multiple Cancers. Enzymes 2014, 36, 149–174. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef]
- Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Anand, P.; Guha, S.; Aggarwal, B.B. Curcumin circumvents chemoresistance in vitro and potentiates the effect of thalidomide and bortezomib against human multiple myeloma in nude mice model. Mol. Cancer Ther. 2009, 8, 959–970. [Google Scholar] [CrossRef]
- Kamat, A.M.; Sethi, G.; Aggarwal, B.B. Curcumin potentiates the apoptotic effects of chemotherapeutic agents and cytokines through down-regulation of nuclear factor-kappaB and nuclear factor-kappaB-regulated gene products in IFN-alpha-sensitive and IFN-alpha-resistant human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 1022–1030. [Google Scholar] [CrossRef]
- Shishodia, S.; Sethi, G.; Aggarwal, B.B. Curcumin: Getting back to the roots. Ann. N. Y. Acad. Sci. 2005, 1056, 206–217. [Google Scholar] [CrossRef]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Sandur, S.K.; Ichikawa, H.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Sethi, G.; Aggarwal, B.B. Role of pro-oxidants and antioxidants in the anti-inflammatory and apoptotic effects of curcumin (diferuloylmethane). Free Radic. Biol. Med. 2007, 43, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Dicato, M.; Diederich, M. Curcumin as a regulator of epigenetic events. Mol. Nutr. Food Res. 2013, 57, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Kan, Y.Y.; Liou, Y.L.; Wang, H.J.; Chen, C.Y.; Sung, L.C.; Chang, C.F.; Liao, C.I. PAX1 methylation as a potential biomarker for cervical cancer screening. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2014, 24, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M.; Omran, Z.; Zamzami, M.A.; Al-Malki, A.L.; Choudhry, H.; Mousli, M.; Bronner, C. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J. Exp. Clin. Cancer Res. CR 2016, 35, 174. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.J.; Ison, C.A. Haemophilus, Gardnerella and other bacilli. In Mackie and McCartney Practical Medical Microbiology, 14th ed.; Collee, J.G., Fraser, A.G., Marmion, B.P., Simmons, A., Eds.; Churchill Livingstone: New York, NY, USA, 1996; pp. 329–341. [Google Scholar]
- Ray, P.; Sharma, J.; Marak, R.S.; Singhi, S.; Taneja, N.; Garg, R.K.; Sharma, M. Chromobacterium violaceum septicaemia from north India. Indian J. Med. Res. 2004, 120, 523–526. [Google Scholar] [PubMed]
- Davis, E.S. Chromobacterium. In Infectious Diseases and Medical Microbiology, 2nd ed.; Braude, A.I., Davis, C.E., Fierer, J., Eds.; WB Saunders: Philadelphia, PA, USA, 1986; pp. 358–361. [Google Scholar]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physicochemical and biological properties, and antitumor activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef] [PubMed]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cancer Types | Pathways/Molecules Altered | Concentration Range Tested | IC50 | References | |
|---|---|---|---|---|---|
| Resveratrol | Prostate cancer (DU145) | Akt↓, MTA1/HDAC I, II, IV complex↓, PTEN↑ | 5–100 µM | N.D. | [54] |
| Hepatoma cancer (HepG2, Hep3B and HuH7) | HDAC I, II, IV↓ | 5–100 µM | 32 µM (HepG2) | [55,56] | |
| 200 µM (Hep3B) | |||||
| 29 µM (HuH7) | |||||
| Resveratrol + pterostilbene | Breast cancer (MDA-MB-157) | ERα↑, acetyl-H3↑, acetyl-H3 lysine9↑, acetyl-H4↑, HDAC↓, DNMT↓ | 15µM resveratrol + 5µM pterostilbene | N.D. | [57] |
| Curcumin | HeLa cells | PAX1 ↑ | N.D. | N.D. | [58] |
| SiHa cells | UHRF1 ↓ | ||||
| Curcumin | B-non-Hodgkin lymphoma cell line | HDAC I↓, HDAC III↓, Notch 1↓, IκBα↑, p300↓ | 3.125–50 µM | 25 µM | [59] |
| RPD3 mutants of yeast cells | HDAC↑, Mec1↓, Rad52↓, DSB repair↓ | 50–200 µM | N.D. | [60] | |
| Hepatocellular carcinoma | HDAC I/II↑ NF-κB↓ | N.D. | [61] | ||
| desmoplastic cerebellar medulloblastoma /DAOY tumor xenografts and Smo/Smo mice | HDAC VI↓ G2/M ↓ cleavage of caspase-3↑ tubulin acetylation↑ | 10–40 µM | N.D. | [62] | |
| Curcumin + Trichostatin | Breast cancer (SkBr3 and 435eB) | HDAC I/II↓ pERK↓ pAkt↓ p21 and p27↑ p53↓ Cyclin D1↓ cleavage of caspase-3↑ | 10–20µM | N.D. | [63] |
| Curcumin + vorinostat/panobinostat | Hsp90 acetylation↑ EGFR↓ Raf-1↓ Akt↓ survivin↓ | [64] | |||
| Curcumin + Trichostatin A | Human hepatoma | histone acetylation↓ HAT protein↓ ROS↑ | [65] | ||
| EF24 + Entinostat or Salermide | Human pancreatic cancer (BxPC-3) | acetylation of histone H3 and H4↑ cells in G1 phase↑ | [66] | ||
| Heliomycin | Cervical cancer (HeLa) | HDAC III↓ | 29.8 µM | [67] | |
| Tetracenomycin D | Cervical cancer (HeLa) | HDAC II↓ | 10.9 µM | [67] | |
| Nocardiopsis sp | Cervical cancer (HeLa) | HDAC↓ | 5.9 µM | [68] | |
| Streptomyces sp | Cervical cancer (HeLa) | HDAC↓ | 7.2 µM | [68] | |
| Halenaquinone | Lymphoblastic leukemia (Molt 4) | Oxidative Stress↑ Bax↑ PARP cleavage↑ caspase activation↑ cytochrome c↑ HDAC↓ Topoisomerase I & II↓ | 0.18 µM | [69] | |
| Human chronic myelogenous leukemia (K562) | p-Akt↓ NF-κB↓ HDAC↓ Bcl-2↓ hexokinase II↓ | 0.48 µM | [69] | ||
| Breast adenocarcinoma (MDA-MB-231) | p-PTEN↓ p-GSK3β↓ p-PDK1↓ ROS↑ | 8 µM | [69] | ||
| Colon adenocarcinoma (DLD-1) | 6.76 µM | [69] | |||
| Aceroside VIII | Colon cancer (HT29) | HDAC VI ↓ | [70] | ||
| Aceroside VIII + A452 | Colon cancer (HT29) | HDAC VI↓ acetylated α-tubulin↑ | [70] | ||
| Bis (4-hydroxybenzyl)sulfide (1) | Breast cancer (MDA-MB-231) | HDACs↓ | 1.45 µM | [71] | |
| Prostate cancer (PC3) | HDACs↓ | 7.86 µM | [72] | ||
| Chalcones: Butein | Human | HDACs I, II, and IV↓ TNFα↓ NF-κB↓ | 0–1000 µM | 60 µM | [72] |
| Philadelphia | |||||
| chromosome | |||||
| positive chronic myelogenous | |||||
| leukemia | |||||
| (K562) | |||||
| Flavone | Human myeloid leukemia | HDAC↓ caspase↑ p16↑ p21↑ TRAIL↑ | [73] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merarchi, M.; Sethi, G.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Ahn, K.S. Role of Natural Products in Modulating Histone Deacetylases in Cancer. Molecules 2019, 24, 1047. https://doi.org/10.3390/molecules24061047
Merarchi M, Sethi G, Shanmugam MK, Fan L, Arfuso F, Ahn KS. Role of Natural Products in Modulating Histone Deacetylases in Cancer. Molecules. 2019; 24(6):1047. https://doi.org/10.3390/molecules24061047
Chicago/Turabian StyleMerarchi, Myriam, Gautam Sethi, Muthu K. Shanmugam, Lu Fan, Frank Arfuso, and Kwang Seok Ahn. 2019. "Role of Natural Products in Modulating Histone Deacetylases in Cancer" Molecules 24, no. 6: 1047. https://doi.org/10.3390/molecules24061047
APA StyleMerarchi, M., Sethi, G., Shanmugam, M. K., Fan, L., Arfuso, F., & Ahn, K. S. (2019). Role of Natural Products in Modulating Histone Deacetylases in Cancer. Molecules, 24(6), 1047. https://doi.org/10.3390/molecules24061047