
Glycopeptides and -Mimetics to Detect, Monitor and Inhibit Bacterial and Viral Infections: Recent Advances and Perspectives


Abstract
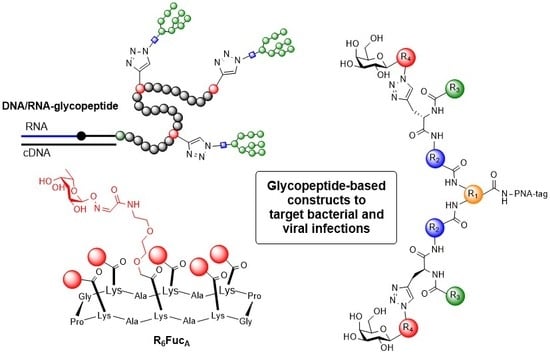
{kind=link}
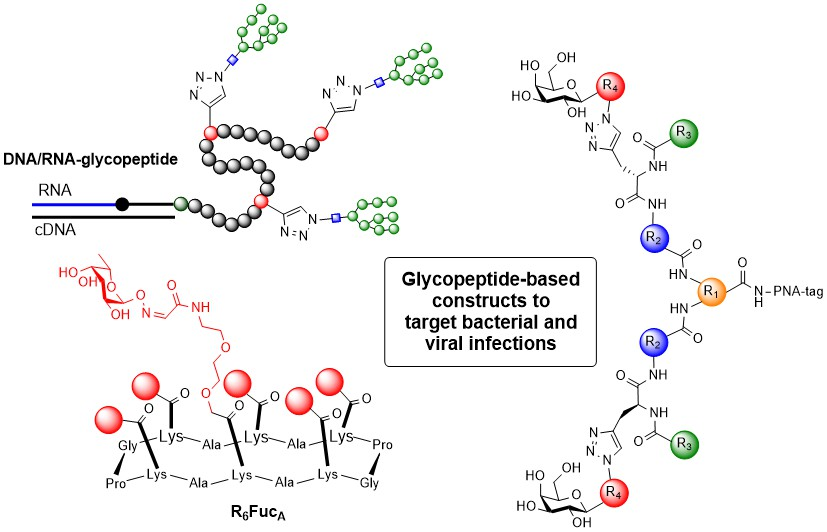
{kind=link}
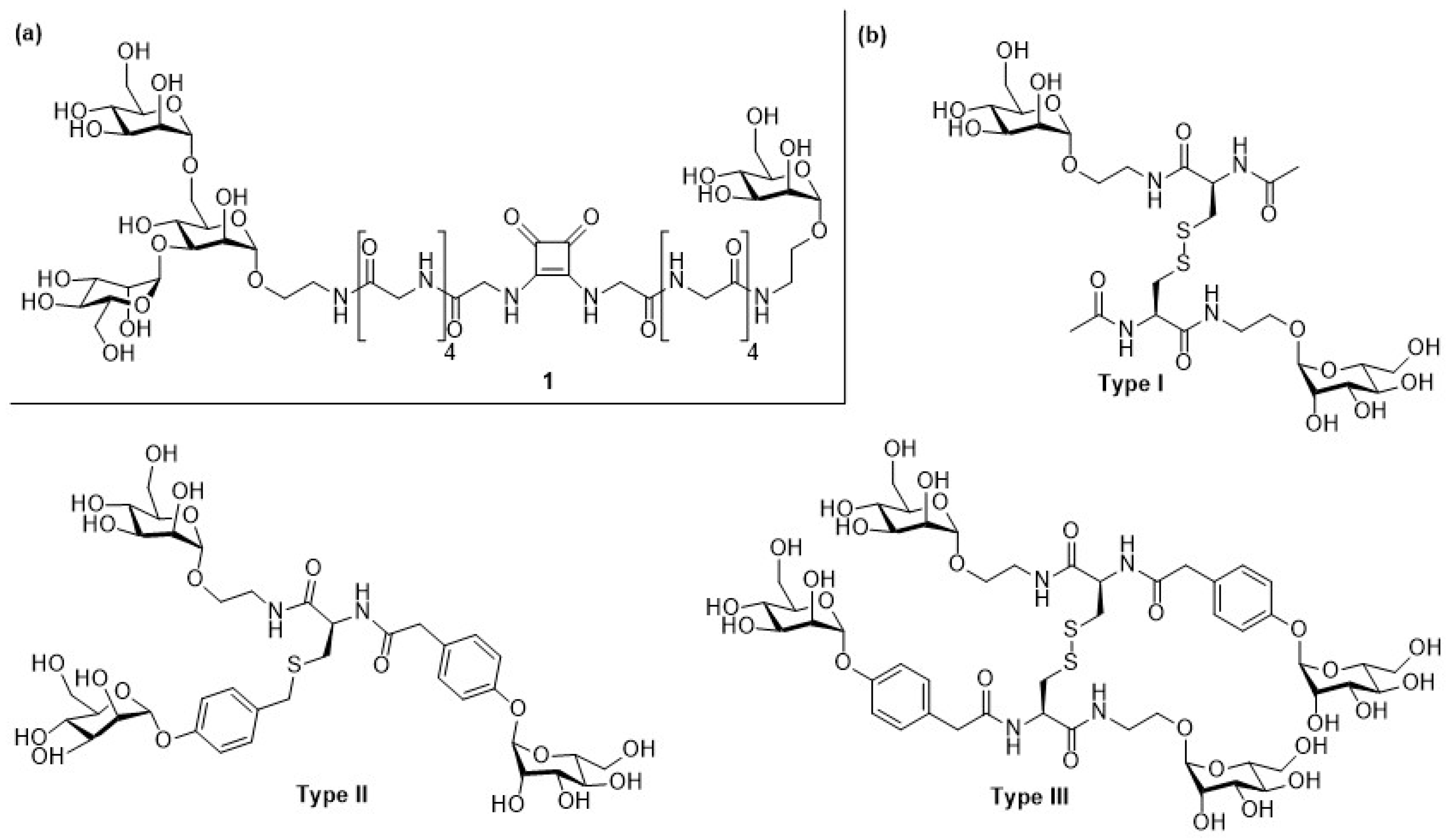
{kind=link}
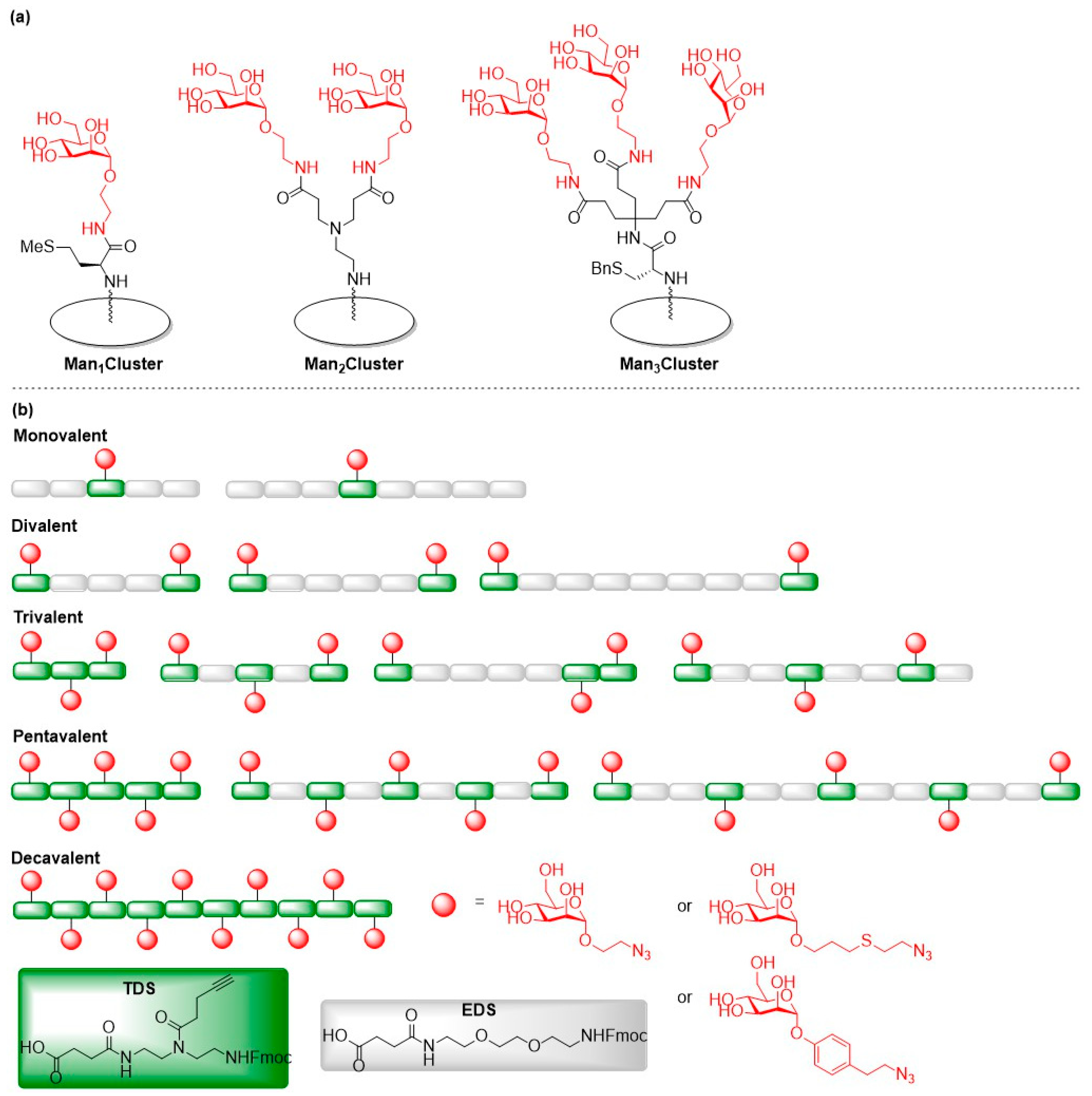
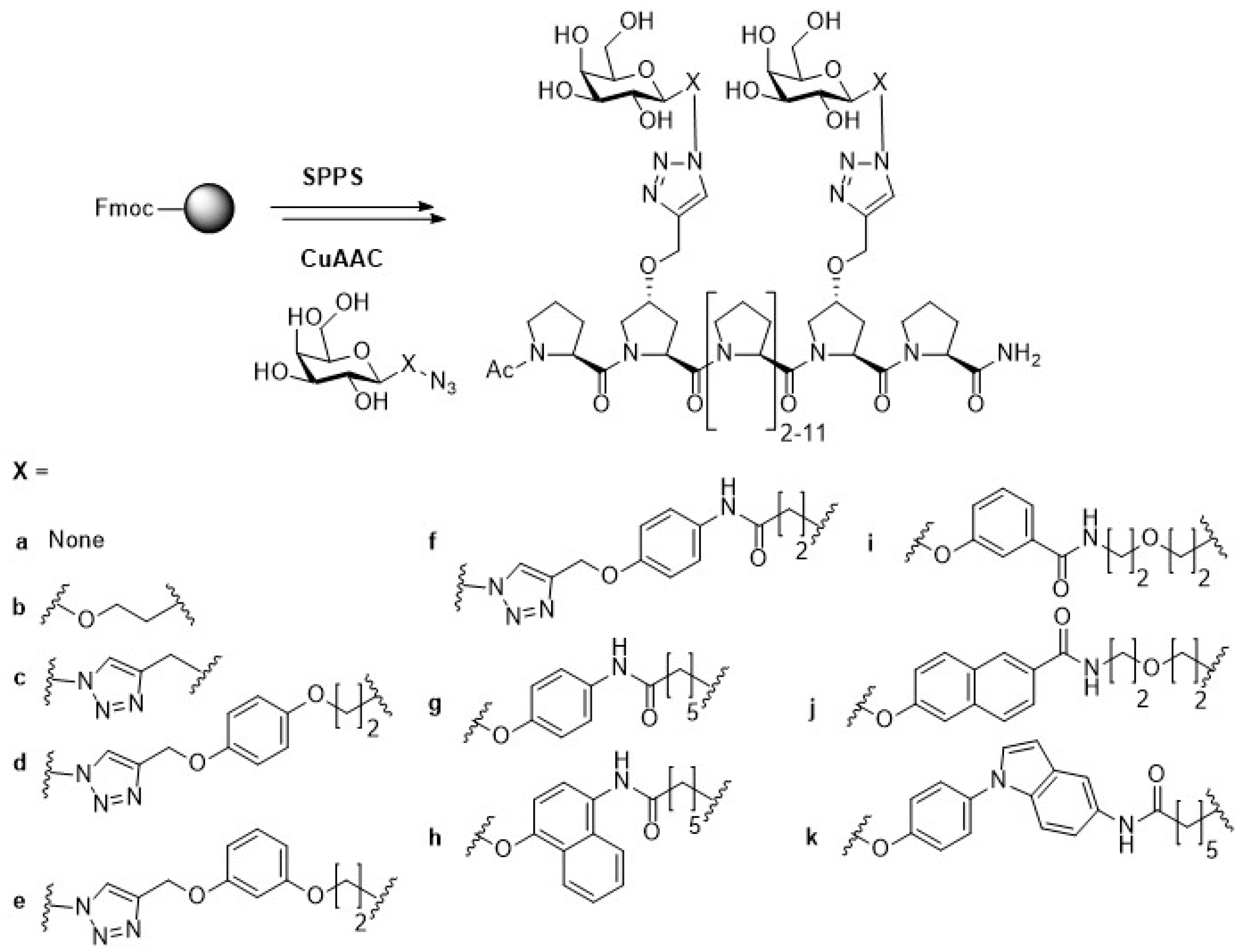{kind=link}
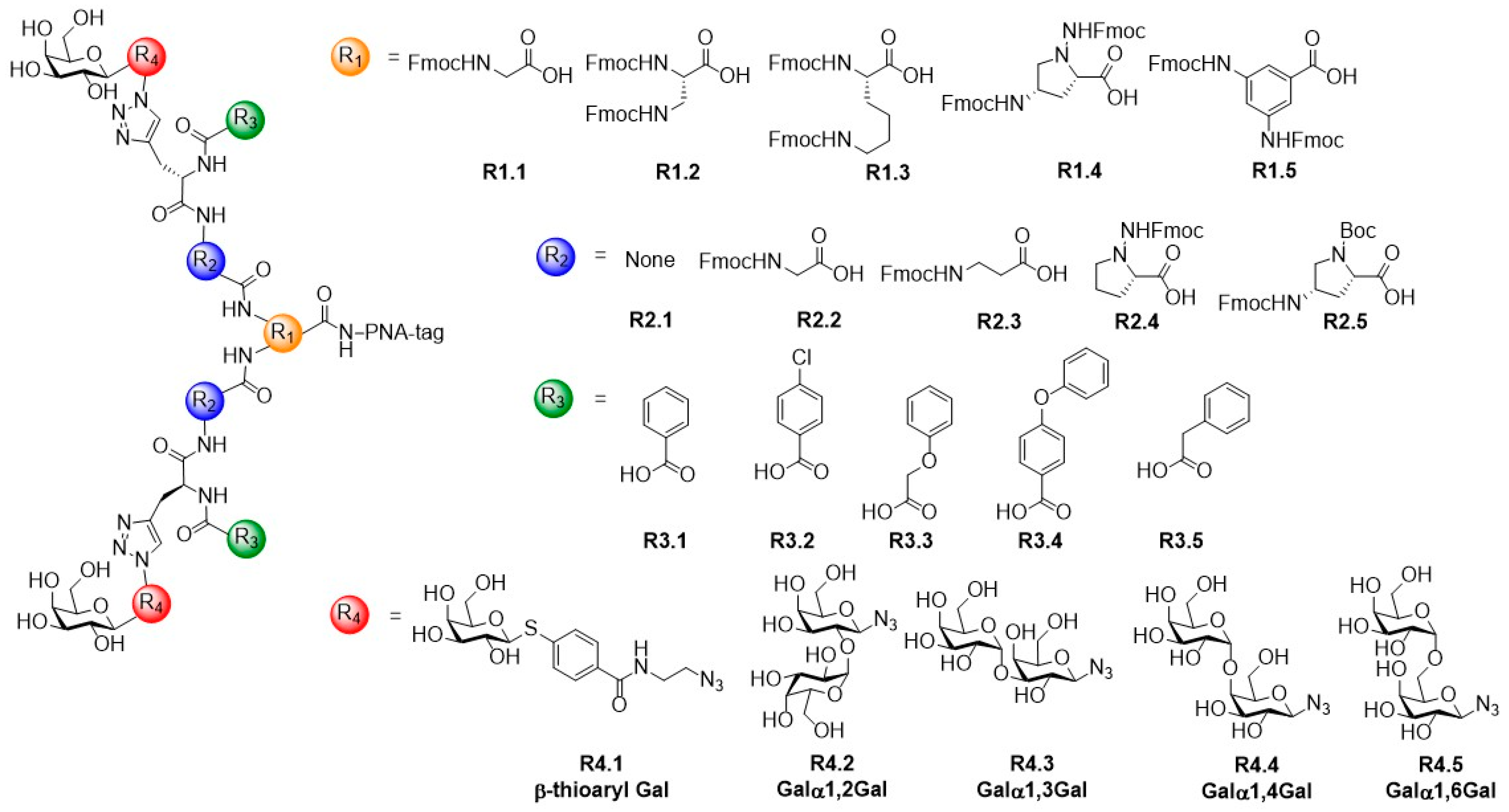{kind=link}
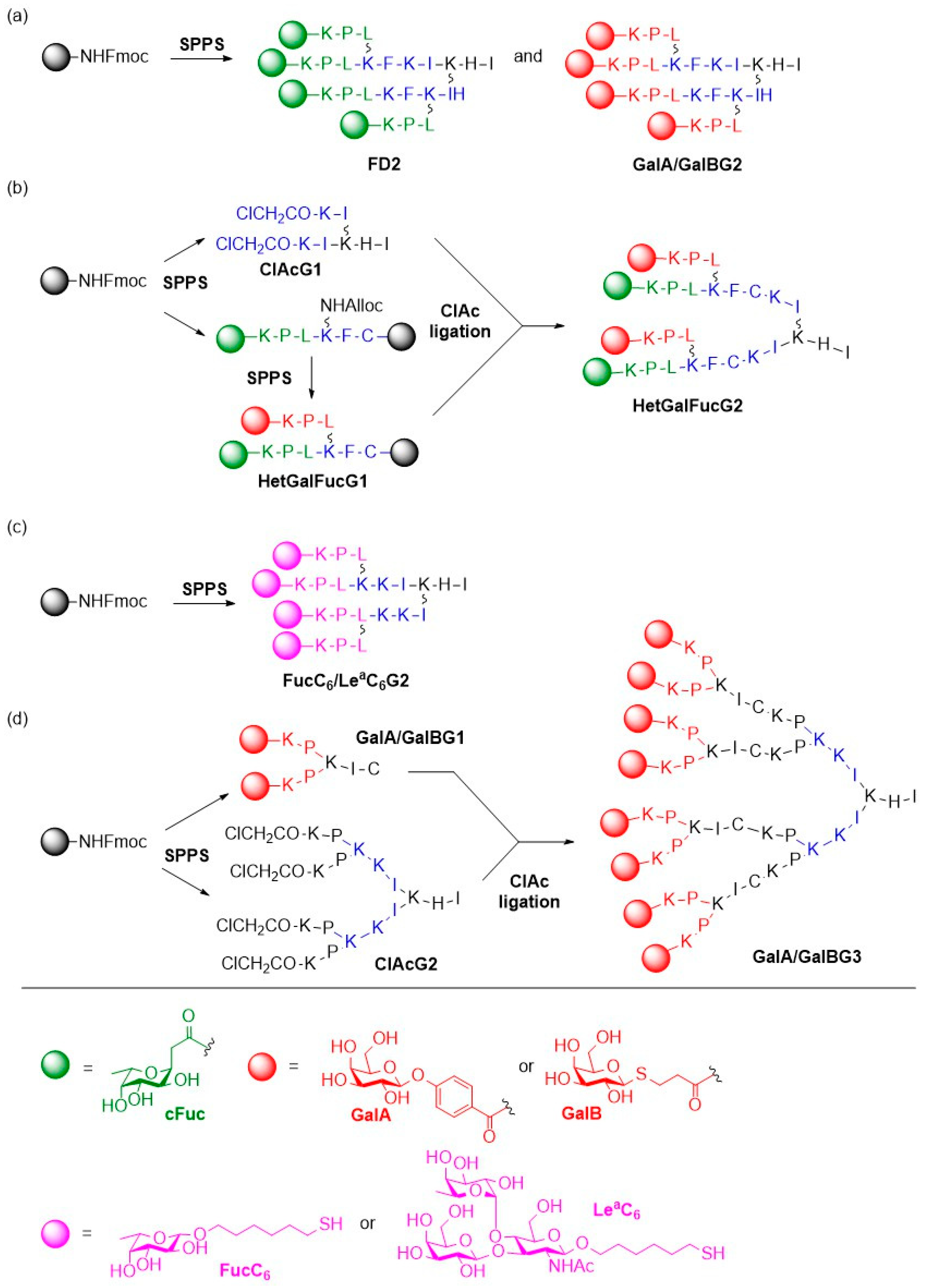{kind=link}
{kind=link}
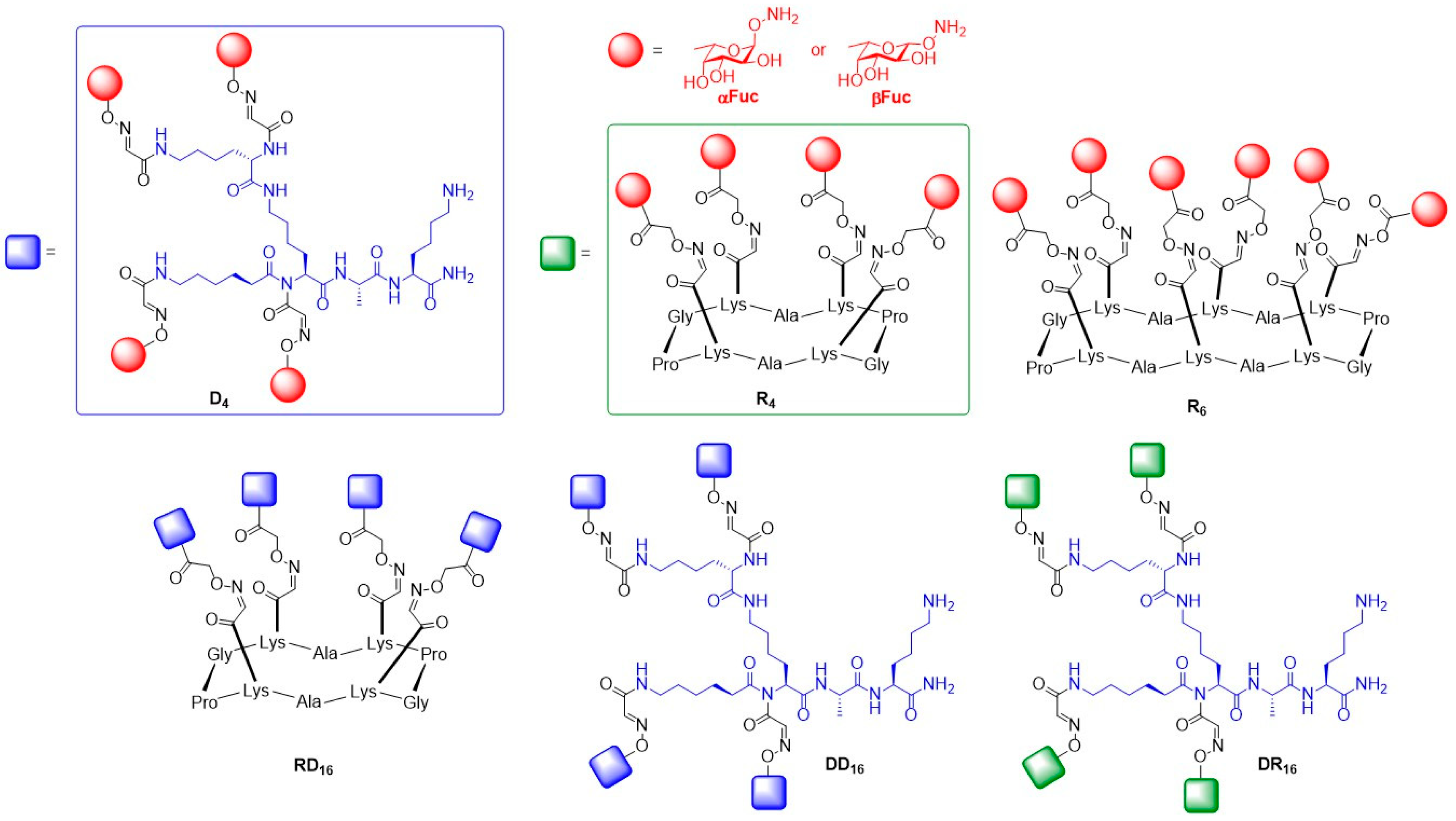{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bacterial Infections
2.1. Escherichia coli (E. coli)
2.2. Pseudomonas aeruginosa
2.3. Burkholderia cepacia
2.4. Mycobacterium tuberculosis
3. Viral Infections
3.1. Human Immunodeficiency Virus Type-1 (HIV-1)
3.2. Epstein-Barr Virus (EBV)
3.3. Herpes Simplex Virus (HSV)
3.4. Influenza A Virus (IAV)
3.5. Norovirus (NoV)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bhatia, S.; Dimde, M.; Haag, R. Multivalent glycoconjugates as vaccines and potential drug candidates. Med. Chem. Commun. 2014, 5, 862–878. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar] [CrossRef]
- Dam, T.K.; Brewer, C.F. Effects of clustered epitopes in multivalent ligand–receptor interactions. Biochemistry 2008, 47, 8470–8476. [Google Scholar] [CrossRef] [PubMed]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. 2012, 51, 10472–10498. [Google Scholar] [CrossRef] [PubMed]
- Sears, P.; Wong, C.H. Carbohydrate mimetics: A new strategy for tackling the problem of carbohydrate-mediated biological recognition. Angew. Chem. Int. Ed. 1999, 38, 2300–2324. [Google Scholar] [CrossRef]
- Ofek, I.; Hasty, D.L.; Sharon, N. Anti-adhesion therapy of bacterial diseases: Prospects and problems. FEMS Immunol. Med. Microbiol. 2003, 38, 181–191. [Google Scholar] [CrossRef]
- Germain, R.N. The biochemistry and cell biology of antigen presentation by MHC class I and class II molecules: Implications for development of combination vaccines. Ann. N. Y. Acad. Sci. 1995, 754, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Avci, F.Y.; Li, X.; Tsuji, M.; Kasper, D.L. Carbohydrates and T cells: A sweet twosome. Semin. Immunol. 2013, 25, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Middleton, D.R.; Wantuch, P.L.; Ozdilek, A.; Avci, F.Y. Carbohydrates as T-cell antigens with implications in health and disease. Glycobiology 2016, 26, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Guttormsen, H.-K.; Sharpe, A.H.; Chandraker, A.K.; Brigtsen, A.K.; Sayegh, M.H.; Kasper, D.L. Cognate stimulatory B-cell–T-cell interactions are critical for T-cell help recruited by glycoconjugate vaccines. Infect. Immun. 1999, 67, 6375–6384. [Google Scholar] [PubMed]
- Avci, F.Y.; Kasper, D.L. How bacterial carbohydrates influence the adaptive immune system. Annu. Rev. Immunol. 2009, 28, 107–130. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 11 February 2019).
- Walsh, C.T.; Wencewicz, T.A. Prospects for new antibiotics: A molecule-centered perspective. J. Antibiot. 2014, 67, 7–22. [Google Scholar] [CrossRef]
- Luther, A.; Bisang, C.; Obrecht, D. Advances in macrocyclic peptide-based antibiotics. Bioorg. Med. Chem. 2018, 26, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.; Hansford, K.A.; Butler, M.S.; Jia, Z.; Mark, A.E.; Cooper, M.A. Developments in glycopeptide antibiotics. ACS Infect. Dis. 2018, 4, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.G. Synthesis of Glycoproteins. Chem. Rev. 2002, 102, 579–602. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.J.; Wong, C.-H. Advances in chemical ligation strategies for the synthesis of glycopeptides and glycoproteins. Chem. Commun. 2010, 46, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Oerthel, V.; Iehl, J.; Holler, M.; Goyard, D.; Praly, J.-P.; Imberty, A.; Nierengarten, J.-F.; Vidal, S. Synthesis of Dodecavalent Fullerene-Based Glycoclusters and Evaluation of Their Binding Properties towards a Bacterial Lectin. Chem. Eur. J. 2011, 17, 3252–3261. [Google Scholar] [CrossRef] [PubMed]
- Cecioni, S.; Lalor, R.; Blanchard, B.; Praly, J.-P.; Imberty, A.; Matthews, S.E.; Vidal, S. Achieving High Affinity towards a Bacterial Lectin through Multivalent Topological Isomers of Calix[4]arene Glycoconjugates. Chem. Eur. J. 2009, 15, 13232–13240. [Google Scholar] [CrossRef]
- Ramström, O.; Yan, M. Glyconanomaterials for Combating Bacterial Infections. Chem. Eur. J. 2015, 21, 16310–16317. [Google Scholar] [CrossRef]
- Rieger, J.; Stoffelbach, F.; Cui, D.; Imberty, A.; Lameignere, E.; Putaux, J.-L.; Jérôme, R.; Jérôme, C.; Auzély-Velty, R. Mannosylated Poly(ethylene oxide)-b-Poly(ε-caprolactone) Diblock Copolymers: Synthesis, Characterization, and Interaction with a Bacterial Lectin. Biomacromolecules 2007, 8, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Ohlsen, K.; Oelschlaeger, T.; Hacker, J.; Khan, A.S. Carbohydrate receptors of bacterial adhesins: Implications and reflections. In Glycoscience and Microbial Adhesion; Lindhorst, T.K., Oscarson, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 17–65. [Google Scholar]
- Hartmann, M.; Lindhorst, T.K. The bacterial lectin FimH, a target for drug discovery–carbohydrate inhibitors of type 1 fimbriae-mediated bacterial adhesion. Eur. J. Org. Chem. 2011, 2011, 3583–3609. [Google Scholar] [CrossRef]
- Ehrmann, S.; Chu, C.-W.; Kumari, S.; Silberreis, K.; Bӧttcher, C.; Dernedde, J.; Ravoo, B.J.; Haag, R. A Toolbox Approach for Multivalent Presentation of Ligand-Receptor Recognition on a Supramolecular Scaffold. J. Mater. Chem. B 2018, 6, 4216–4222. [Google Scholar] [CrossRef]
- Sperling, O.; Fuchs, A.; Lindhorst, T.K. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: Design, synthesis and binding properties of mannoside ligands. Org. Biomol. Chem. 2006, 4, 3913–3922. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.S.; Bouckaert, J.; Hung, D.; Pinkner, J.; Widberg, C.; DeFusco, A.; Auguste, C.G.; Strouse, R.; Langermann, S.; Waksman, G.; et al. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 2002, 44, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, J.; Berglund, J.; Schembri, M.; De Genst, E.; Cools, L.; Wuhrer, M.; Hung, C.S.; Pinkner, J.; Slättegård, R.; Zavialov, A.; et al. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005, 55, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Wellens, A.; Garofalo, C.; Nguyen, H.; Van Gerven, N.; Slättegård, R.; Hernalsteens, J.-P.; Wyns, L.; Oscarson, S.; De Greve, H.; Hultgren, S.; et al. Intervening with urinary tract infections using anti-adhesives based on the crystal structure of the FimH–oligomannose-3 complex. PLoS ONE 2008, 3, e2040. [Google Scholar] [CrossRef]
- Knight, S.D.; Bouckaert, J. Structure, function, and assembly of type 1 fimbriae. In Glycoscience and Microbial Adhesion; Lindhorst, T.K., Oscarson, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 67–107. [Google Scholar]
- Han, Z.; Pinkner, J.S.; Ford, B.; Obermann, R.; Nolan, W.; Wildman, S.A.; Hobbs, D.; Ellenberger, T.; Cusumano, C.K.; Hultgren, S.J.; et al. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. [Google Scholar] [CrossRef]
- Lahmann, M. Architectures of multivalent glycomimetics for probing carbohydrate–lectin interactions. In Glycoscience and Microbial Adhesion; Lindhorst, T.K., Oscarson, S., Eds.; Springer: Berlin/Heidelberg, Germay, 2009; pp. 165–183. [Google Scholar]
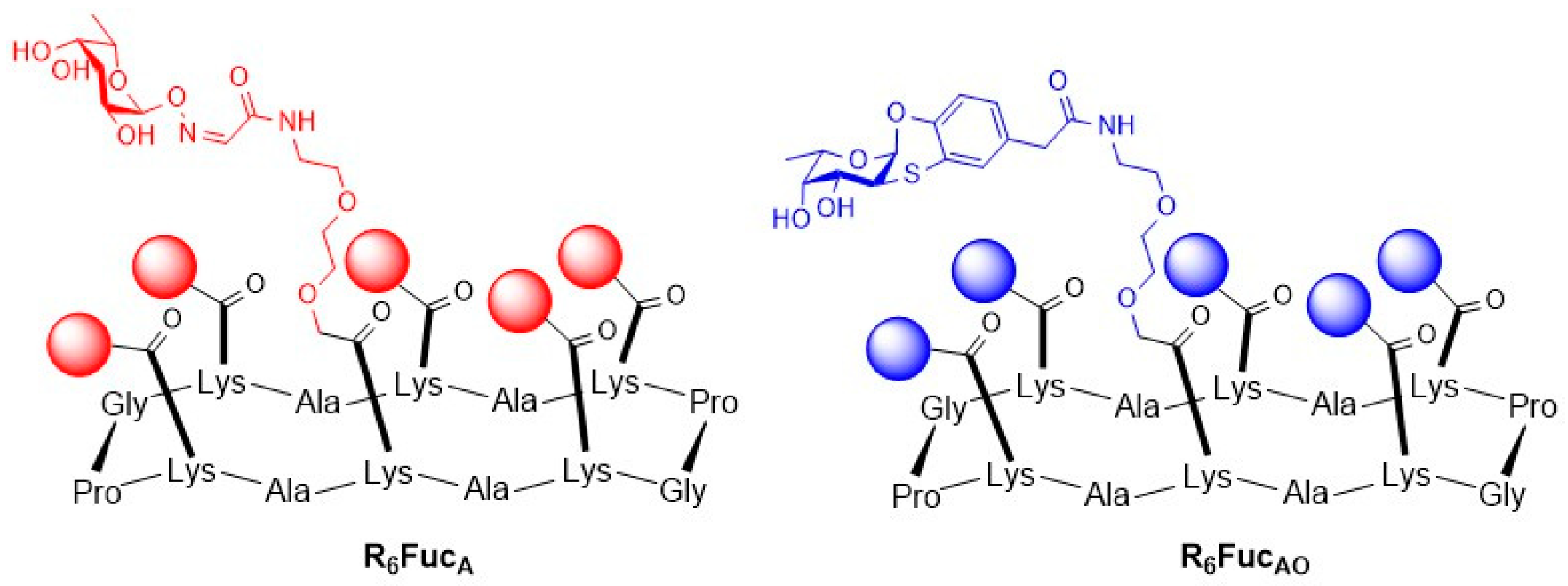
- Lindhorst, T.K.; Bruegge, K.; Fuchs, A.; Sperling, O. A bivalent glycopeptide to target two putative carbohydrate binding sites on FimH. Beilstein J. Org. Chem. 2010, 6, 801–809. [Google Scholar] [CrossRef]
- Schierholt, A.; Hartmann, M.; Schwekendiek, K.; Lindhorst, T.K. Cysteine-Based Mannoside Glycoclusters: Synthetic Routes and Antiadhesive Properties. Eur. J. Org. Chem. 2010, 2010, 3120–3128. [Google Scholar] [CrossRef]
- Wehner, J.W.; Hartmann, M.; Lindhorst, T.K. Are multivalent cluster glycosides a means of controlling ligand density of glycoarrays? Carbohydr. Res. 2013, 371, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Schierholt, A.; Hartmann, M.; Lindhorst, T.K. Bi-and trivalent glycopeptide mannopyranosides as inhibitors of type 1 fimbriae-mediated bacterial adhesion: Variation of valency, aglycon and scaffolding. Carbohydr. Res. 2011, 346, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Igde, S.; Röblitz, S.; Müller, A.; Kolbe, K.; Boden, S.; Fessele, C.; Lindhorst, T.K.; Weber, M.; Hartmann, L. Linear Precision Glycomacromolecules with Varying Interligand Spacing and Linker Functionalities Binding to Concanavalin A and the Bacterial Lectin FimH. Macromol. Biosci. 2017, 17, 1700198. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.E.; Iglewski, B.H.P. aeruginosa biofilms in CF infection. Clin. Rev. Allergy Immunol. 2008, 35, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Garber, N.; Guempel, U.; Belz, A.; Gilboa-Garber, N.; Doyle, R.J. On the specificity of the d-galactose-binding lectin (PA-I) of Pseudomonas aeruginosa and its strong binding to hydrophobic derivatives of d-galactose and thiogalactose. Biochim. Biophys. Acta 1992, 1116, 331–333. [Google Scholar] [CrossRef]
- Imberty, A.; Wimmerová, M.; Mitchell, E.P.; Gilboa-Garber, N. Structures of the lectins from Pseudomonas aeruginosa: Insights into the molecular basis for host glycan recognition. Microbes Infect. 2004, 6, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-P.; Song, S.-C.; Gilboa-Garber, N.; Chang, K.S.; Wu, A.M. Studies on the binding site of the galactose-specific agglutinin PA-IL from Pseudomonas aeruginosa. Glycobiology 1998, 8, 7–16. [Google Scholar] [CrossRef]
- Mitchell, E.; Houles, C.; Sudakevitz, D.; Wimmerova, M.; Gautier, C.; Pérez, S.; Wu, A.M.; Gilboa-Garber, N.; Imberty, A. Structural basis for oligosaccharide-mediated adhesion of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Nat. Struct. Biol. 2002, 9, 918–921. [Google Scholar] [CrossRef]
- Wittmann, V.; Pieters, R.J. Bridging lectin binding sites by multivalent carbohydrates. Chem. Soc. Rev. 2013, 42, 4492–4503. [Google Scholar] [CrossRef]
- Huang, S.F.; Lin, C.H.; Lai, Y.T.; Tsai, C.L.; Cheng, T.J.R.; Wang, S.K. Development of Pseudomonas aeruginosa Lectin LecA Inhibitor by using Bivalent Galactosides Supported on Polyproline Peptide Scaffolds. Chem. Asian J. 2018, 13, 686–700. [Google Scholar] [CrossRef]
- Kadam, R.U.; Garg, D.; Schwartz, J.; Visini, R.; Sattler, M.; Stocker, A.; Darbre, T.; Reymond, J.-L. CH–π “T-Shape” Interaction with histidine explains binding of aromatic galactosides to Pseudomonas aeruginosa lectin LecA. ACS Chem. Biol. 2013, 8, 1925–1930. [Google Scholar] [CrossRef]
- Kadam, R.U.; Bergmann, M.; Hurley, M.; Garg, D.; Cacciarini, M.; Swiderska, M.A.; Nativi, C.; Sattler, M.; Smyth, A.R.; Williams, P.; et al. A glycopeptide dendrimer inhibitor of the galactose-specific lectin LecA and of Pseudomonas aeruginosa biofilms. Angew. Chem. Int. Ed. 2011, 50, 10631–10635. [Google Scholar] [CrossRef]
- Novoa, A.; Eierhoff, T.; Topin, J.; Varrot, A.; Barluenga, S.; Imberty, A.; Römer, W.; Winssinger, N. A LecA Ligand Identified from a Galactoside-Conjugate Array Inhibits Host Cell Invasion by Pseudomonas aeruginosa. Angew. Chem. Int. Ed. 2014, 126, 9031–9035. [Google Scholar] [CrossRef]
- Zambaldo, C.; Barluenga, S.; Winssinger, N. PNA-encoded chemical libraries. Curr. Opin. Chem. Biol. 2015, 26, 8–15. [Google Scholar] [CrossRef]
- Johansson, E.M.; Crusz, S.A.; Kolomiets, E.; Buts, L.; Kadam, R.U.; Cacciarini, M.; Bartels, K.-M.; Diggle, S.P.; Cámara, M.; Williams, P.; et al. Inhibition and dispersion of Pseudomonas aeruginosa biofilms by glycopeptide dendrimers targeting the fucose-specific lectin LecB. Chem. Biol. 2008, 15, 1249–1257. [Google Scholar] [CrossRef]
- Emma, M. Inhibition of Pseudomonas aeruginosa biofilms with a glycopeptide dendrimer containing d-amino acids. Med. Chem. Commun. 2011, 2, 418–420. [Google Scholar]
- Michaud, G.; Visini, R.; Bergmann, M.; Salerno, G.; Bosco, R.; Gillon, E.; Richichi, B.; Nativi, C.; Imberty, A.; Stocker, A.; et al. Overcoming antibiotic resistance in Pseudomonas aeruginosa biofilms using glycopeptide dendrimers. Chem. Sci. 2016, 7, 166–182. [Google Scholar] [CrossRef]
- Bergmann, M.; Michaud, G.; Visini, R.; Jin, X.; Gillon, E.; Stocker, A.; Imberty, A.; Darbre, T.; Reymond, J.-L. Multivalency effects on Pseudomonas aeruginosa biofilm inhibition and dispersal by glycopeptide dendrimers targeting lectin LecA. Org. Biomol. Chem. 2016, 14, 138–148. [Google Scholar] [CrossRef]
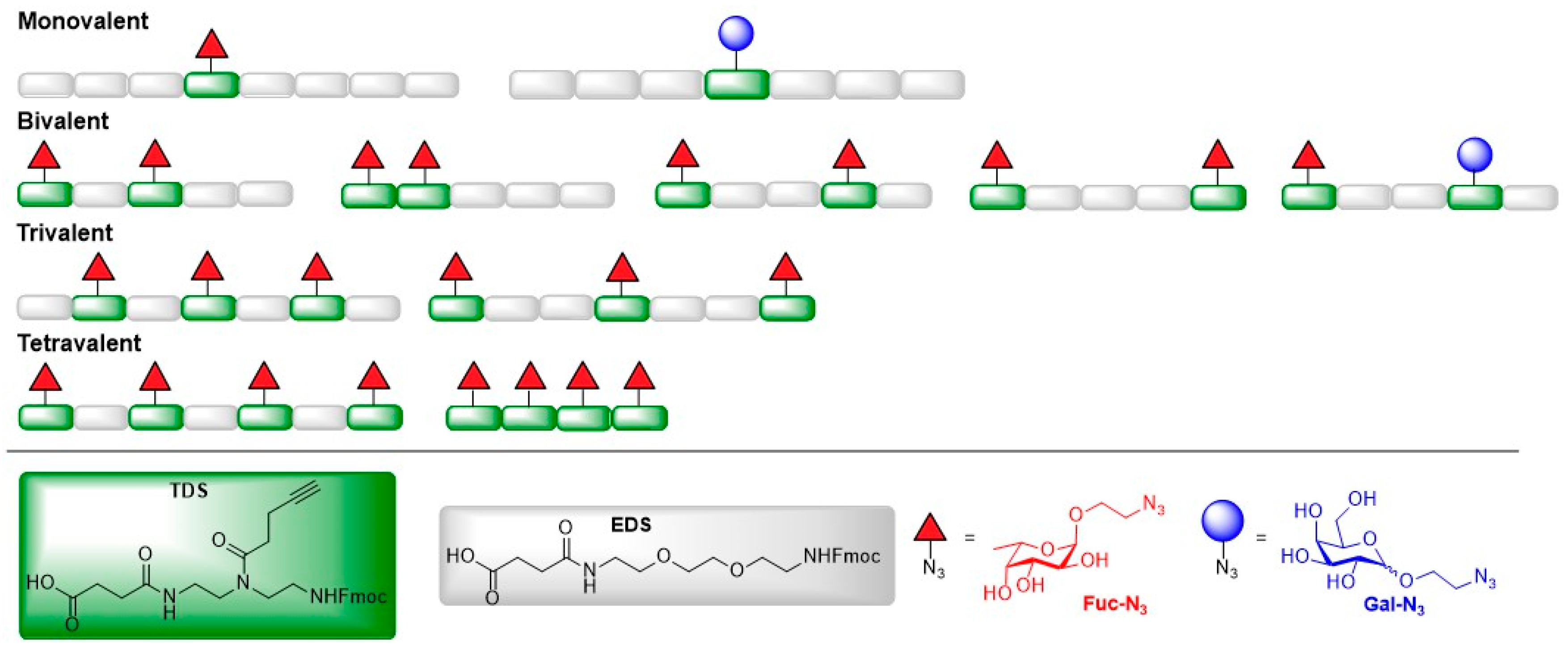
- Bücher, K.S.; Babic, N.; Freichel, T.; Kovacic, F.; Hartmann, L. Monodisperse Sequence-Controlled α-l-Fucosylated Glycooligomers and Their Multivalent Inhibitory Effects on LecB. Macromol. Biosci. 2018, 18, 1800337. [Google Scholar] [CrossRef] [PubMed]
- Bücher, K.S.; Konietzny, P.B.; Snyder, N.L.; Hartmann, L. Heteromultivalent glycooligomers as mimetics of blood group antigens. Chem. Eur. J. 2019, 25, 3301–3309. [Google Scholar] [CrossRef] [PubMed]
- Dumy, P.; Eggleston, I.; Cervigni, S.; Sila, U.; Sun, X.; Mutter, M. A convenient synthesis of cyclic peptides as regioselectively addressable functionalized templates (RAFT). Tetrahedron Lett. 1995, 36, 1255–1258. [Google Scholar] [CrossRef]
- Dumy, P.; Eggleston, I.M.; Esposito, G.; Nicula, S.; Mutter, M. Solution structure of regioselectively addressable functionalized templates: An NMR and restrained molecular dynamics investigation. Biopolymers 1996, 39, 297–308. [Google Scholar] [CrossRef]
- Grigalevicius, S.; Chierici, S.; Renaudet, O.; Lo-Man, R.; Dériaud, E.; Leclerc, C.; Dumy, P. Chemoselective assembly and immunological evaluation of multiepitopic glycoconjugates bearing clustered Tn antigen as synthetic anticancer vaccines. Bioconj. Chem. 2005, 16, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, C.; Berthet, N.; Renaudet, O. Cyclopeptide scaffolds in carbohydrate-based synthetic vaccines. Biomater. Sci. 2017, 5, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Berthet, N.; Thomas, B.; Bossu, I.; Dufour, E.; Gillon, E.; Garcia, J.; Spinelli, N.; Imberty, A.; Dumy, P.; Renaudet, O. High affinity glycodendrimers for the lectin LecB from Pseudomonas aeruginosa. Bioconj. Chem. 2013, 24, 1598–1611. [Google Scholar] [CrossRef]
- Scoffone, V.C.; Chiarelli, L.R.; Trespidi, G.; Mentasti, M.; Riccardi, G.; Buroni, S. Burkholderia cenocepacia infections in cystic fibrosis patients: Drug resistance and therapeutic approaches. Front. Microbiol. 2017, 8, 1592. [Google Scholar] [CrossRef]
- Govan, J.R.; Deretic, V. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 1996, 60, 539–574. [Google Scholar]
- Aaron, S.D.; Ferris, W.; Henry, D.A.; Speert, D.P.; MacDonald, N.E. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am. J. Respir. Crit. Care Med. 2000, 161, 1206–1212. [Google Scholar] [CrossRef]
- Lameignere, E.; Malinovská, L.; Sláviková, M.; Duchaud, E.; Mitchell, E.P.; Varrot, A.; Šedo, O.; Imberty, A.; Wimmerová, M. Structural basis for mannose recognition by a lectin from opportunistic bacteria Burkholderia cenocepacia. Biochem. J. 2008, 411, 307–318. [Google Scholar] [CrossRef]
- Lameignere, E.; Shiao, T.C.; Roy, R.; Wimmerova, M.; Dubreuil, F.; Varrot, A.; Imberty, A. Structural basis of the affinity for oligomannosides and analogs displayed by BC2L-A, a Burkholderia cenocepacia soluble lectin. Glycobiology 2009, 20, 87–98. [Google Scholar] [CrossRef]
- Marchetti, R.; Malinovska, L.; Lameignère, E.; Adamova, L.; de Castro, C.; Cioci, G.; Stanetty, C.; Kosma, P.; Molinaro, A.; Wimmerova, M. Burkholderia cenocepacia lectin A binding to heptoses from the bacterial lipopolysaccharide. Glycobiology 2012, 22, 1387–1398. [Google Scholar] [CrossRef]
- Pifferi, C.; Goyard, D.; Gillon, E.; Imberty, A.; Renaudet, O. Synthesis of Mannosylated Glycodendrimers and Evaluation against BC2L-A Lectin from Burkholderia Cenocepacia. ChemPlusChem 2017, 82, 390–398. [Google Scholar] [CrossRef]
- Goyard, D.; Baldoneschi, V.; Varrot, A.; Fiore, M.; Imberty, A.; Richichi, B.; Renaudet, O.; Nativi, C. Multivalent Glycomimetics with Affinity and Selectivity toward Fucose-Binding Receptors from Emerging Pathogens. Bioconj. Chem. 2017, 29, 83–88. [Google Scholar] [CrossRef]
- Vial, L.; Groleau, M.-C.; Lamarche, M.G.; Filion, G.; Castonguay-Vanier, J.; Dekimpe, V.; Daigle, F.; Charette, S.J.; Déziel, E. Phase variation has a role in Burkholderia ambifaria niche adaptation. ISME J. 2010, 4, 49–60. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2018. Available online: https://apps.who.int/iris/bitstream/handle/10665/274453/9789241565646-eng.pdf?ua=1 (accessed on 8 February 2019).
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Chan, J.; Fan, X.; Hunter, S.; Brennan, P.; Bloom, B. Lipoarabinomannan, a possible virulence factor involved in persistence of Mycobacterium tuberculosis within macrophages. Infect. Immun. 1991, 59, 1755–1761. [Google Scholar]
- Spencer, J.S.; Kim, H.J.; Wheat, W.H.; Chatterjee, D.; Balagon, M.V.; Cellona, R.V.; Tan, E.V.; Gelber, R.; Saunderson, P.; Duthie, M.S.J.C.; et al. Analysis of antibody responses to Mycobacterium leprae phenolic glycolipid I, lipoarabinomannan, and recombinant proteins to define disease subtype-specific antigenic profiles in leprosy. Clin. Vaccine Immunol. 2011, 18, 260–267. [Google Scholar] [CrossRef]
- Chou, Y.; Kitova, E.N.; Joe, M.; Brunton, R.; Lowary, T.L.; Klassen, J.S.; Derda, R.J.O. Genetically-encoded fragment-based discovery (GE-FBD) of glycopeptide ligands with differential selectivity for antibodies related to mycobacterial infections. Org. Biomol. Chem. 2018, 16, 223–227. [Google Scholar] [CrossRef]
- Rademacher, C.; Shoemaker, G.K.; Kim, H.-S.; Zheng, R.B.; Taha, H.; Liu, C.; Nacario, R.C.; Schriemer, D.C.; Klassen, J.S.; Peters, T. Ligand specificity of CS-35, a monoclonal antibody that recognizes mycobacterial lipoarabinomannan: A model system for oligofuranoside–protein recognition. J. Am. Chem. Soc. 2007, 129, 10489–10502. [Google Scholar] [CrossRef] [PubMed]
- Deiss, F.; Matochko, W.L.; Govindasamy, N.; Lin, E.Y.; Derda, R. Flow-through synthesis on teflon-patterned paper to produce peptide arrays for cell-based assays. Angew. Chem. Int. Ed. 2014, 53, 6374–6377. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, S.; Blixt, O.; Bergström, T.; Frank, M.; Wandall, H.H. Viral O-GalNAc peptide epitopes: A novel potential target in viral envelope glycoproteins. Rev. Med. Virol. 2016, 26, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Datema, R.; Olofsson, S.; Romero, P.A. Inhibitors of protein glycosylation and glycoprotein processing in viral systems. Pharmacol. Ther. 1987, 33, 221–286. [Google Scholar] [CrossRef]
- Olofsson, S.; Hansen, J.-E.S. Host cell glycosylation of viral glycoproteins-a battlefield for host defence and viral resistance. Scand. J. Infect. Dis. 1998, 30, 435–440. [Google Scholar] [PubMed]
- UNAIDS. Global HIV & AIDS Statistics—2018 Fact Sheet. Available online: http://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 11 February 2019).
- Burton, D.R.; Ahmed, R.; Barouch, D.H.; Butera, S.T.; Crotty, S.; Godzik, A.; Kaufmann, D.E.; McElrath, M.J.; Nussenzweig, M.C.; Pulendran, B. A blueprint for HIV vaccine discovery. Cell Host Microbe 2012, 12, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Wilson, I.A. Insights into the trimeric HIV-1 envelope glycoprotein structure. Trends Biochem. Sci. 2015, 40, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Sweet, R.W. The HIV gp120-CD4 interaction: A target for pharmacological or immunological intervention? Perspect. Drug Discov. Des. 1993, 1, 235–250. [Google Scholar] [CrossRef]
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 2001, 58, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Mizuochi, T.; Matthews, T.; Kato, M.; Hamako, J.; Titani, K.; Solomon, J.; Feizi, T. Diversity of oligosaccharide structures on the envelope glycoprotein gp 120 of human immunodeficiency virus 1 from the lymphoblastoid cell line H9. Presence of complex-type oligosaccharides with bisecting N-acetylglucosamine residues. J. Biol. Chem. 1990, 265, 8519–8524. [Google Scholar] [PubMed]
- Leonard, C.K.; Spellman, M.W.; Riddle, L.; Harris, R.J.; Thomas, J.N.; Gregory, T. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 1990, 265, 10373–10382. [Google Scholar] [PubMed]
- Reitter, J.N.; Means, R.E.; Desrosiers, R.C. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 1998, 4, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Doms, R.W.; Moss, B. Oligomeric structure of the human immunodeficiency virus type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 1990, 87, 648–652. [Google Scholar] [CrossRef]
- Wilhelm, D.; Behnken, H.N.; Meyer, B. Glycosylation assists binding of HIV protein gp120 to human CD4 receptor. ChemBioChem 2012, 13, 524–527. [Google Scholar] [CrossRef]
- Walker, L.M.; Simek, M.D.; Priddy, F.; Gach, J.S.; Wagner, D.; Zwick, M.B.; Phogat, S.K.; Poignard, P.; Burton, D.R. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV-1 infected individuals. PLoS Pathog. 2010, 6, e1001028. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.-P.; Wang, S.-K.; Ramos, A.; Chan-Hui, P.-Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, C.N.; Pantophlet, R.; Wormald, M.R.; Saphire, E.O.; Stanfield, R.; Wilson, I.A.; Katinger, H.; Dwek, R.A.; Rudd, P.M.; Burton, D.R. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1→ 2 mannose residues on the outer face of gp120. J. Virol. 2002, 76, 7306–7321. [Google Scholar] [CrossRef] [PubMed]
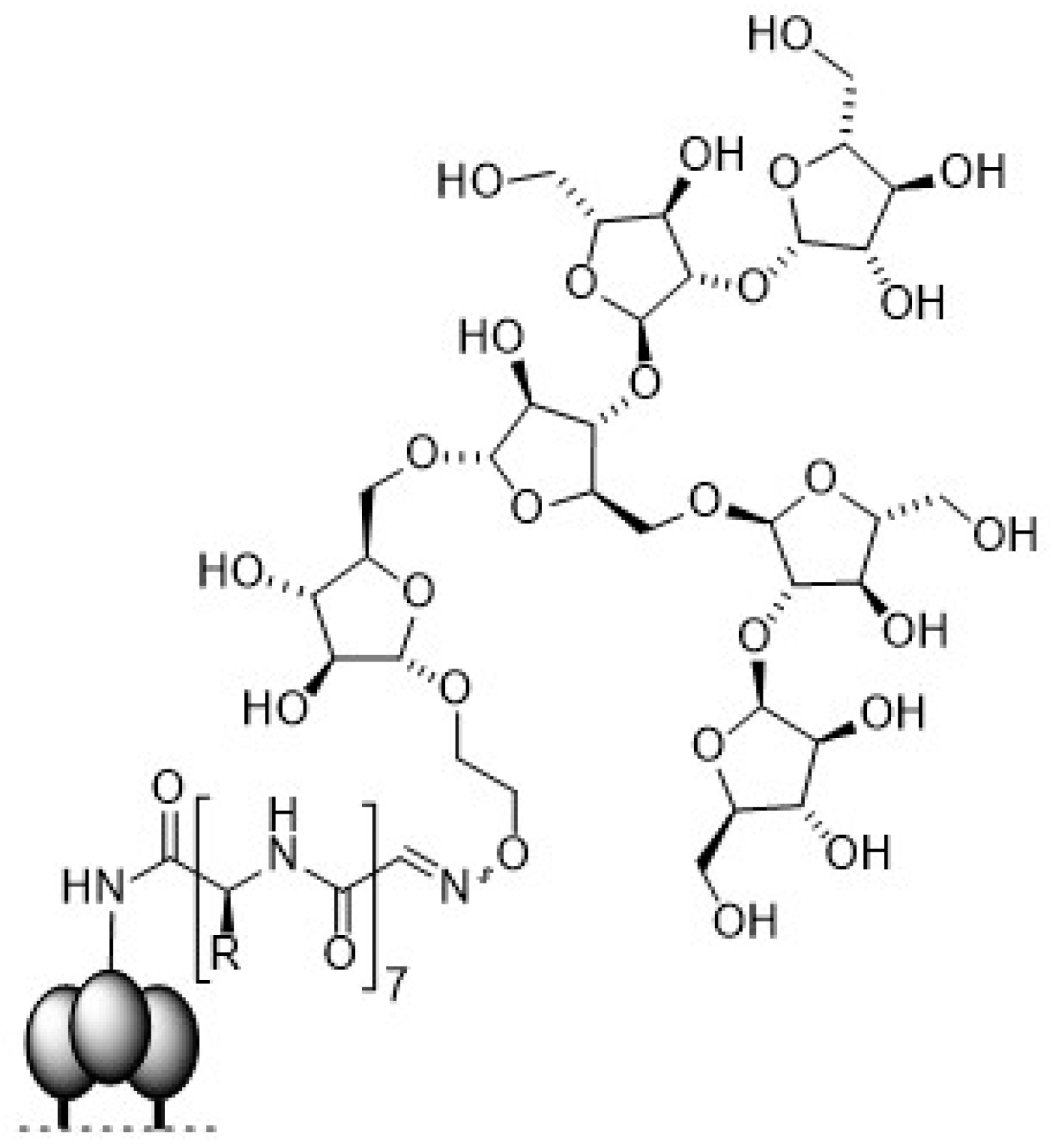
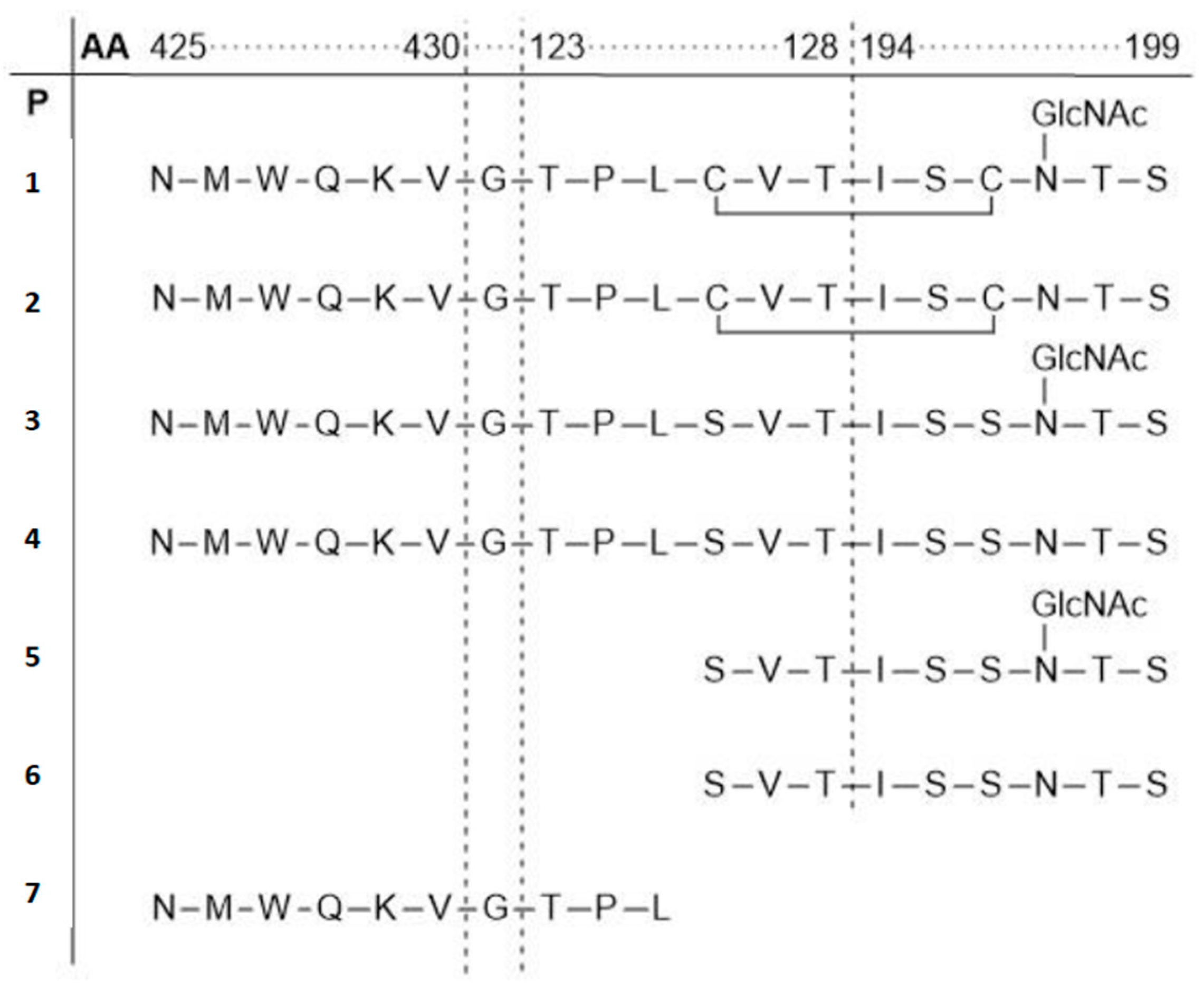
- Horiya, S.; Bailey, J.K.; Temme, J.S.; Guillen Schlippe, Y.V.; Krauss, I.J. Directed evolution of multivalent glycopeptides tightly recognized by HIV antibody 2G12. J. Am. Chem. Soc. 2014, 136, 5407–5415. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.K.; Nguyen, D.N.; Horiya, S.; Krauss, I.J. Synthesis of multivalent glycopeptide conjugates that mimic an HIV epitope. Tetrahedron 2016, 72, 6091–6098. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.-Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef] [PubMed]
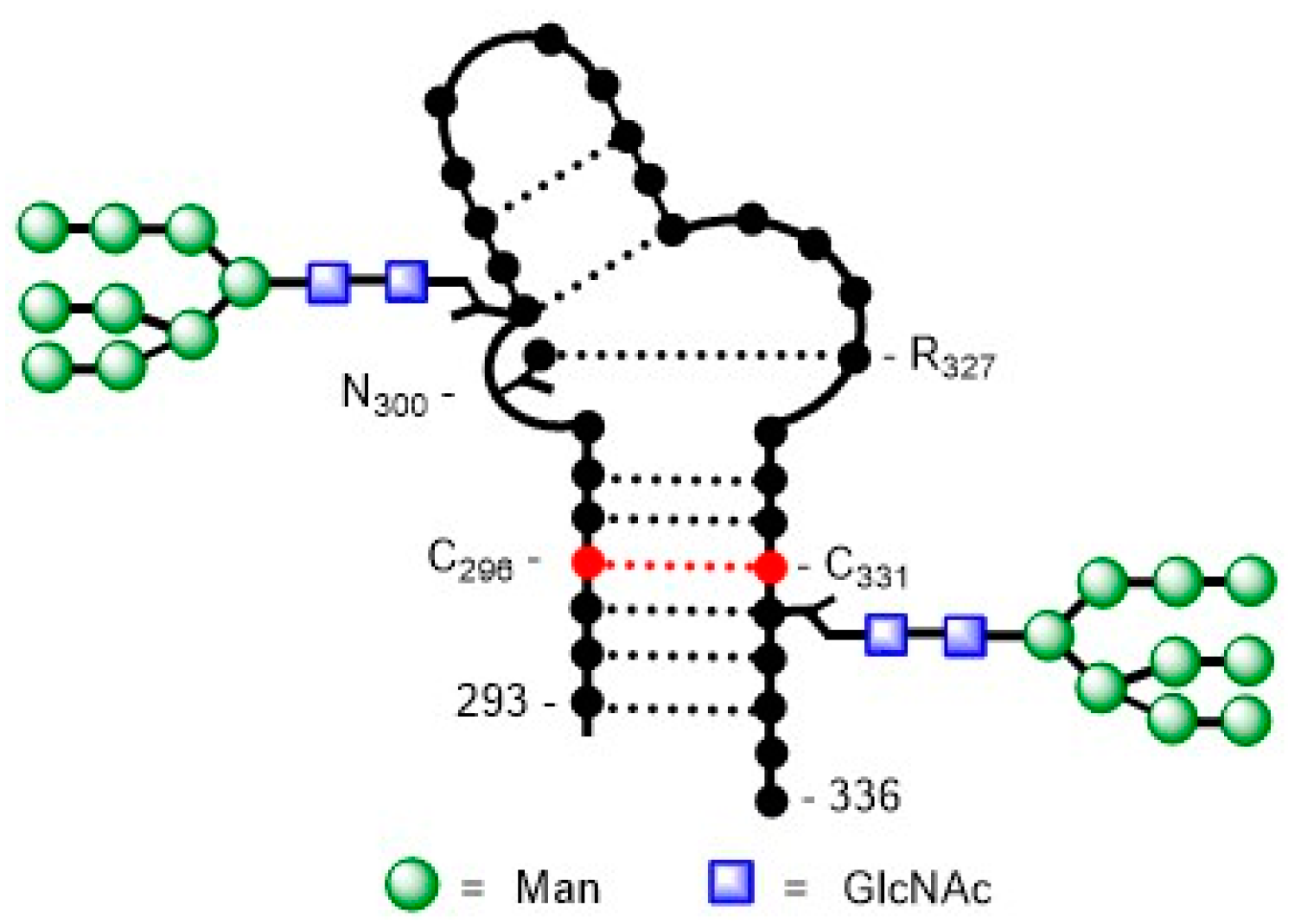
- McLellan, J.S.; Pancera, M.; Carrico, C.; Gorman, J.; Julien, J.-P.; Khayat, R.; Louder, R.; Pejchal, R.; Sastry, M.; Dai, K.; et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nat. Biotechnol. 2011, 480, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Shahzad-ul-Hussan, S.; Doria-Rose, N.A.; McLellan, J.S.; Bailer, R.T.; Dai, K.; Loesgen, S.; Louder, M.K.; Staupe, R.P.; Yang, Y.; et al. Structural basis for diverse N-glycan recognition by HIV-1–neutralizing V1–V2–directed antibody PG16. Nat. Struct. Mol. Biol. 2013, 20, 804–813. [Google Scholar] [CrossRef] [PubMed]
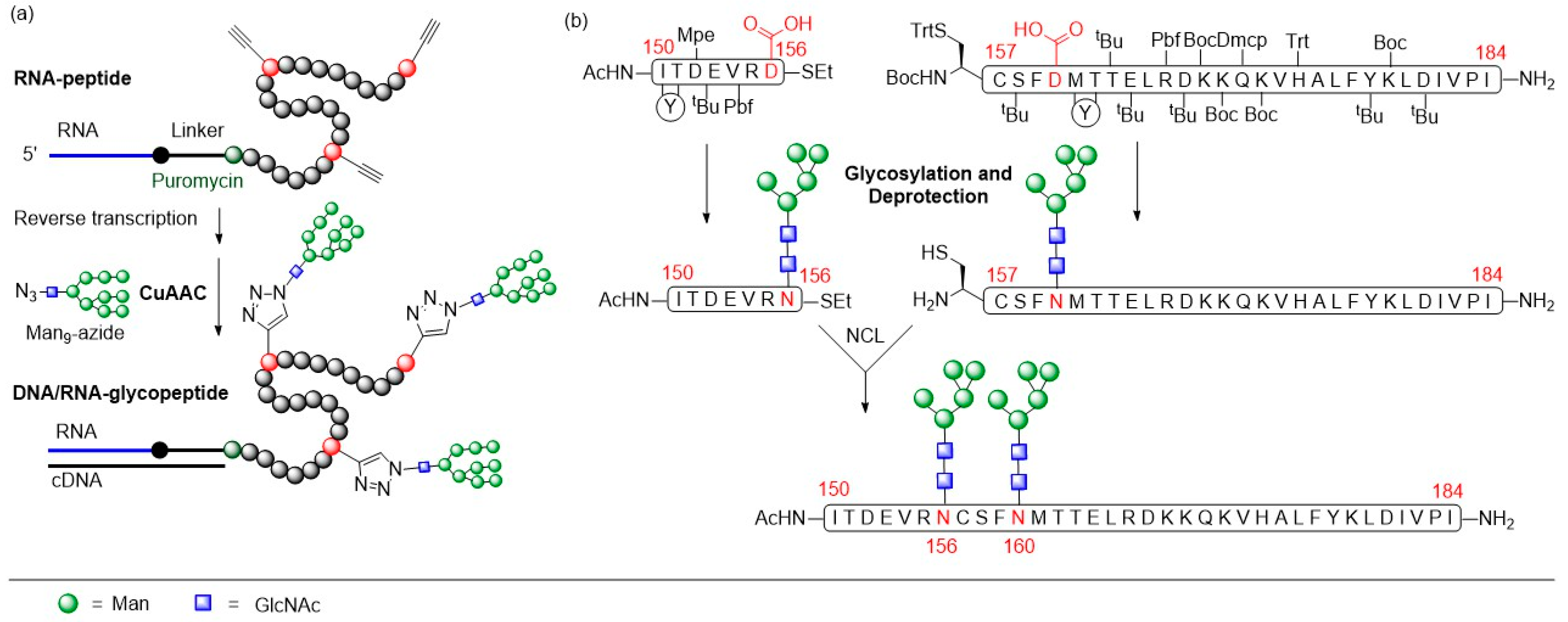
- Aussedat, B.; Vohra, Y.; Park, P.K.; Fernández-Tejada, A.; Alam, S.M.; Dennison, S.M.; Jaeger, F.H.; Anasti, K.; Stewart, S.; Blinn, J.H.; et al. Chemical Synthesis of Highly Congested gp120 V1V2 N-Glycopeptide Antigens for Potential HIV-1-Directed Vaccines. J. Am. Chem. Soc. 2013, 135, 13113–13120. [Google Scholar] [CrossRef]
- Alam, S.M.; Dennison, S.M.; Aussedat, B.; Vohra, Y.; Park, P.K.; Fernández-Tejada, A.; Stewart, S.; Jaeger, F.H.; Anasti, K.; Blinn, J.H.; et al. Recognition of synthetic glycopeptides by HIV-1 broadly neutralizing antibodies and their unmutated ancestors. Proc. Natl. Acad. Sci. USA 2013, 110, 18214–18219. [Google Scholar] [CrossRef] [PubMed]
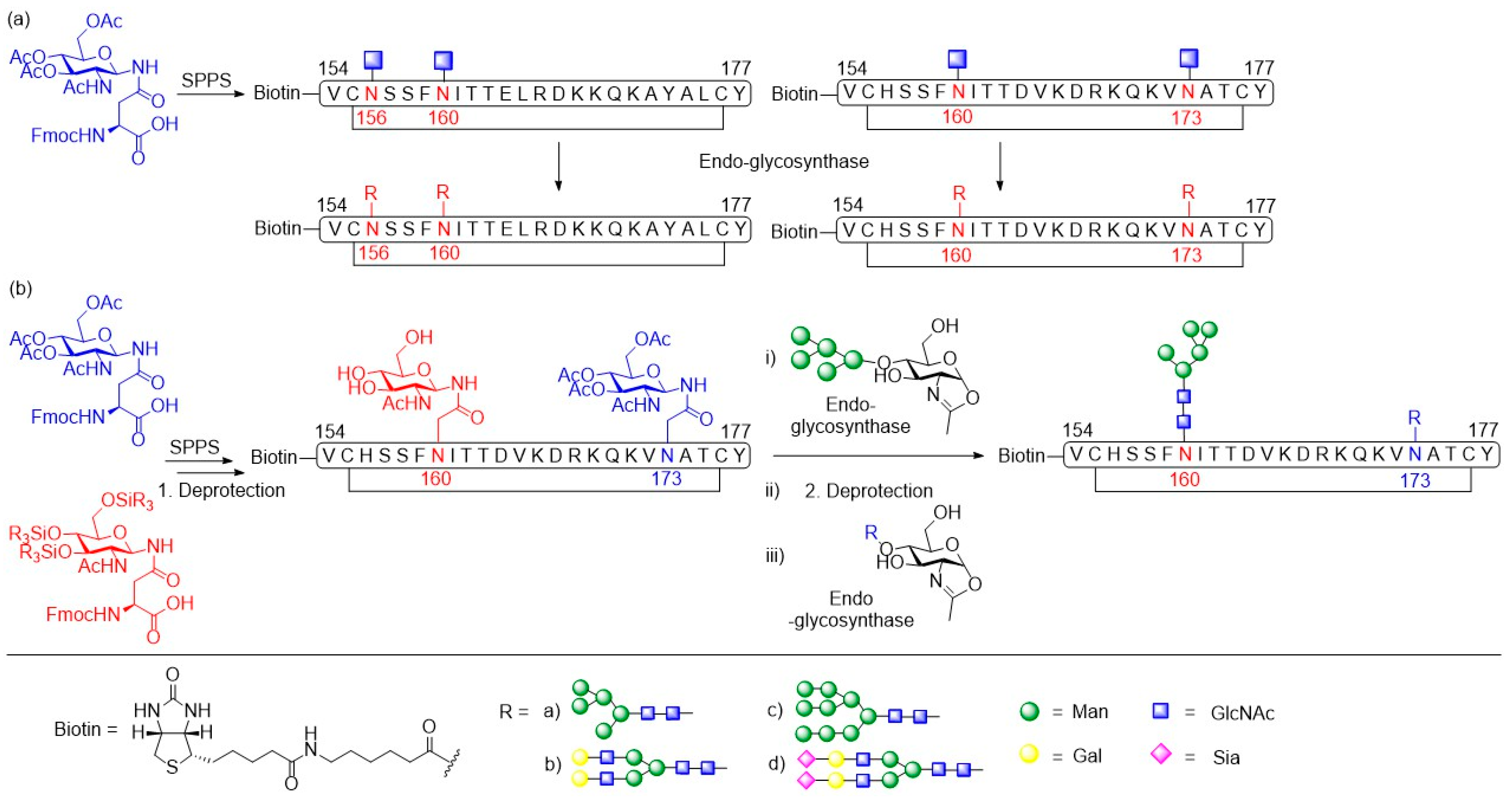
- Amin, M.N.; McLellan, J.S.; Huang, W.; Orwenyo, J.; Burton, D.R.; Koff, W.C.; Kwong, P.D.; Wang, L.-X. Synthetic glycopeptides reveal the glycan specificity of HIV-neutralizing antibodies. Nat. Chem. Biol. 2013, 9, 521–526. [Google Scholar] [CrossRef]
- Toonstra, C.; Amin, M.N.; Wang, L.-X. Site-selective chemoenzymatic glycosylation of an HIV-1 polypeptide antigen with two distinct N-glycans via an orthogonal protecting group strategy. J. Org. Chem. 2016, 81, 6176–6185. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.S.; Sutton, M.S.; Shiakolas, A.R.; Guenaga, J.; Jarosinski, M.C.; Georgiev, I.S.; McKee, K.; Bailer, R.T.; Louder, M.K.; O’Dell, S.; et al. Multiple antibody lineages in one donor target the glycan-V3 supersite of the HIV-1 envelope glycoprotein and display a preference for quaternary binding. J. Virol. 2016, 90, 10574–10586. [Google Scholar] [CrossRef]
- Orwenyo, J.; Cai, H.; Giddens, J.; Amin, M.N.; Toonstra, C.; Wang, L.-X. Systematic synthesis and binding study of HIV V3 glycopeptides reveal the fine epitopes of several broadly neutralizing antibodies. ACS Chem. Biol. 2017, 12, 1566–1575. [Google Scholar] [CrossRef]
- Kong, L.; Lee, J.H.; Doores, K.J.; Murin, C.D.; Julien, J.-P.; McBride, R.; Liu, Y.; Marozsan, A.; Cupo, A.; Klasse, P.-J.; et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat. Struct. Mol. Biol. 2013, 20, 796–803. [Google Scholar] [CrossRef]
- Gristick, H.B.; von Boehmer, L.; West, A.P., Jr.; Schamber, M.; Gazumyan, A.; Golijanin, J.; Seaman, M.S.; Fätkenheuer, G.; Klein, F.; Nussenzweig, M.C.; et al. Natively glycosylated HIV-1 Env structure reveals new mode for antibody recognition of the CD4-binding site. Nat. Struct. Mol. Biol. 2016, 23, 906–915. [Google Scholar] [CrossRef]
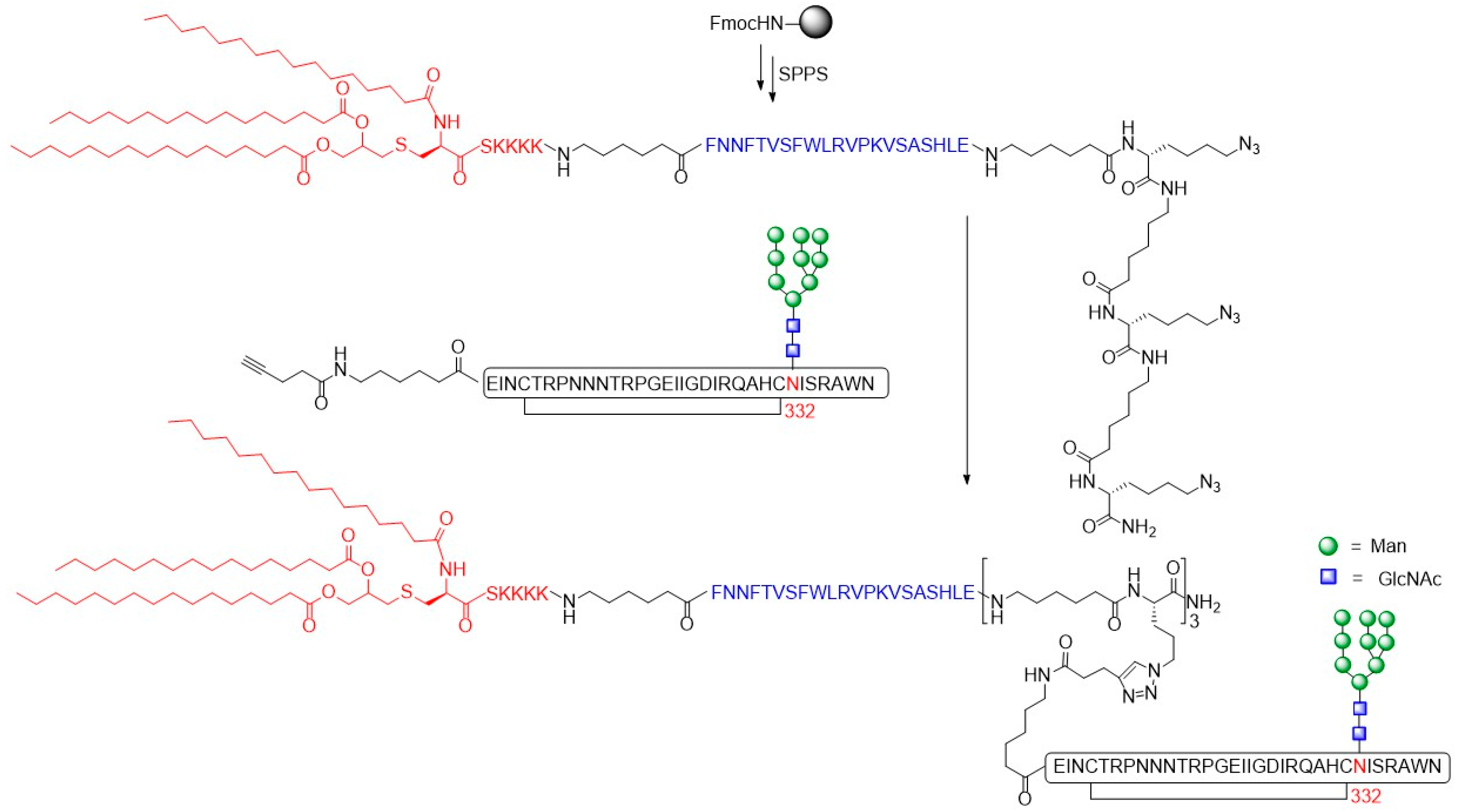
- Cai, H.; Orwenyo, J.; Guenaga, J.; Giddens, J.; Toonstra, C.; Wyatt, R.T.; Wang, L.-X. Synthetic multivalent V3 glycopeptides display enhanced recognition by glycan-dependent HIV-1 broadly neutralizing antibodies. Chem. Commun. 2017, 53, 5453–5456. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Zhang, R.; Orwenyo, J.; Giddens, J.; Yang, Q.; LaBranche, C.C.; Montefiori, D.C.; Wang, L.-X. Multivalent Antigen Presentation Enhances the Immunogenicity of a Synthetic Three-Component HIV-1 V3 Glycopeptide Vaccine. ACS Cent. Sci. 2018, 4, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Orwenyo, J.; Giddens, J.P.; Yang, Q.; Zhang, R.; LaBranche, C.C.; Montefiori, D.C.; Wang, L.-X. Synthetic Three-Component HIV-1 V3 Glycopeptide Immunogens Induce Glycan-Dependent Antibody Responses. Cell Chem. Biol. 2017, 24, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.M.; Aussedat, B.; Vohra, Y.; Meyerhoff, R.R.; Cale, E.M.; Walkowicz, W.E.; Radakovich, N.A.; Anasti, K.; Armand, L.; Parks, R.; et al. Mimicry of an HIV broadly neutralizing antibody epitope with a synthetic glycopeptide. Sci. Transl. Med. 2017, 9, eaai7521. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Zhang, R.-S.; Orwenyo, J.; Giddens, J.; Yang, Q.; LaBranche, C.C.; Montefiori, D.C.; Wang, L.-X. Synthetic HIV V3 Glycopeptide Immunogen Carrying a N334 N-Glycan Induces Glycan-Dependent Antibodies with Promiscuous Site Recognition. J. Med. Chem. 2018, 61, 10116–10125. [Google Scholar] [CrossRef] [PubMed]
- Fera, D.; Lee, M.S.; Wiehe, K.; Meyerhoff, R.R.; Piai, A.; Bonsignori, M.; Aussedat, B.; Walkowicz, W.E.; Ton, T.; Zhou, J.O.; et al. HIV envelope V3 region mimic embodies key features of a broadly neutralizing antibody lineage epitope. Nat. Commun. 2018, 9, 1111. [Google Scholar] [CrossRef]
- Cohen, J.I. Epstein–Barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef]
- Munch, M.; Riisom, K.; Christensen, T.; Møller-Larsen, A.; Haahr, S. The significance of Epstein—Barr virus seropositivity in multiple sclerosis patients? Acta Neurol. Scand. 1998, 97, 171–174. [Google Scholar] [CrossRef]
- Henle, G.; Henle, W.; Diehl, V. Relation of Burkitt’s tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc. Natl. Acad. Sci. USA 1968, 59, 94–101. [Google Scholar] [CrossRef]
- Nemerow, G.; Mold, C.; Schwend, V.K.; Tollefson, V.; Cooper, N. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: Sequence homology of gp350 and C3 complement fragment C3d. J. Virol. 1987, 61, 1416–1420. [Google Scholar]
- Ogembo, J.G.; Kannan, L.; Ghiran, I.; Nicholson-Weller, A.; Finberg, R.W.; Tsokos, G.C.; Fingeroth, J.D. Human complement receptor type 1/CD35 is an Epstein-Barr Virus receptor. Cell Rep. 2013, 3, 371–385. [Google Scholar] [CrossRef]
- D’Arrigo, I.; Cló, E.; Bergström, T.; Olofsson, S.; Blixt, O. Diverse IgG serum response to novel glycopeptide epitopes detected within immunodominant stretches of Epstein-Barr virus glycoprotein 350/220: Diagnostic potential of O-glycopeptide microarrays. Glycoconj. J. 2013, 30, 633–640. [Google Scholar] [CrossRef]
- Patel, A.; Patel, R. Recent insights into HSV infection and disease: Results of wider genome analysis. Curr. Opin. Infect. Dis. 2019, 32, 51–55. [Google Scholar] [CrossRef]
- Whitley, R.J.; Kimberlin, D.W.; Roizman, B. Herpes simplex viruses. Clin. Infect. Dis. 1998, 26, 541–553. [Google Scholar] [CrossRef]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef]
- Cló, E.; Kračun, S.K.; Nudelman, A.S.; Jensen, K.J.; Liljeqvist, J.-Å.; Olofsson, S.; Bergström, T.; Blixt, O. Characterization of the viral O-glycopeptidome: A novel tool of relevance for vaccine design and serodiagnosis. J. Virol. 2012, 86, 6268–6278. [Google Scholar] [CrossRef]
- Risinger, C.; Sørensen, K.K.; Jensen, K.J.; Olofsson, S.; Bergström, T.; Blixt, O. Linear Multiepitope (Glyco) peptides for Type-Specific Serology of Herpes Simplex Virus (HSV) Infections. ACS Infect. Dis. 2017, 3, 360–367. [Google Scholar] [CrossRef]
- Hay, A.; Wolstenholme, A.; Skehel, J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [CrossRef]
- Wang, C.; Takeuchi, K.; Pinto, L.; Lamb, R. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [Google Scholar]
- Edmond, J.; Johnston, R.; Kidd, D.; Rylance, H.; Sommerville, R. The inhibition of neuraminidase and antiviral action. Br. J. Pharmacol. Chemother. 1966, 27, 415–426. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Kyo, Y.; Kato, K.; Park, Y.S.; Gajhate, S.; Umehara, T.; Lillehoj, E.P.; Suzaki, H.; Kim, K.C. Antiinflammatory Role of MUC1 Mucin during Infection with Nontypeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol. 2012, 46, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lindén, S.K.; Sheng, Y.H.; Every, A.L.; Miles, K.M.; Skoog, E.C.; Florin, T.H.; Sutton, P.; McGuckin, M.A. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 2009, 5, e1000617. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.; Corcilius, L.; Tan, H.; Payne, R.; McGuckin, M.; Brown, L. The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol. 2017, 10, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Foundation, C. Global Burden of Norovirus and Prospects for Vaccine Development. Available online: https://www.cdc.gov/norovirus/downloads/global-burden-report.pdf (accessed on 6 Februrary 2019).
- Tan, M.; Jiang, X. The p domain of norovirus capsid protein forms a subviral particle that binds to histo-blood group antigen receptors. J. Virol. 2005, 79, 14017–14030. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, S.; Ruvoën, N.; Le Moullac–Vaidye, B.; Clement, M.; Cailleau–Thomas, A.; Ruiz–Palacois, G.; Huang, P.; Jiang, X.; Le Pendu, J. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 2002, 122, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Rydell, G.E.; Kindberg, E.; Larson, G.; Svensson, L. Susceptibility to winter vomiting disease: A sweet matter. Rev. Med. Virol. 2011, 21, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Koromyslova, A.D.; Leuthold, M.M.; Bowler, M.W.; Hansman, G.S. The sweet quartet: Binding of fucose to the norovirus capsid. Virology 2015, 483, 203–208. [Google Scholar] [CrossRef]
- Bücher, K.S.; Yan, H.; Creutznacher, R.; Ruoff, K.; Mallagaray, A.; Grafmüller, A.; Dirks, J.S.; Kilic, T.; Weickert, S.; Rubailo, A.; et al. Fucose-functionalized precision glycomacromolecules targeting human norovirus capsid protein. Biomacromolecules 2018, 19, 3714–3724. [Google Scholar] [CrossRef] [PubMed]
- Celine, A.; Claudio, S. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar]
- Brik, A. New Strategies for Glycopeptide, Neoglycopeptide, and Glycoprotein Synthesis. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 55–89. [Google Scholar]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2014, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [PubMed]
- Pudelko, M.; Bull, J.; Kunz, H. Chemical and Chemoenzymatic Synthesis of Glycopeptide Selectin Ligands Containing Sialyl Lewis X Structures. ChemBioChem 2010, 11, 904–930. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Patton, J.T.; Sarkar, A.; Ernst, B.; Magnani, J.L.; Frenette, P.S. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010, 116, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized phase 2 study of GMI-1070 in SCD: Reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015, 125, 2656–2664. [Google Scholar] [CrossRef]
- Taylor-Papadimitriou, J.; Burchell, J.M.; Graham, R.; Beatson, R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018, 46, 659–668. [Google Scholar] [CrossRef]
- Rivalland, G.; Loveland, B.; Mitchell, P. Update on Mucin-1 immunotherapy in cancer: A clinical perspective. Expert Opin. Biol. Ther. 2015, 15, 1773–1787. [Google Scholar] [CrossRef]

















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behren, S.; Westerlind, U. Glycopeptides and -Mimetics to Detect, Monitor and Inhibit Bacterial and Viral Infections: Recent Advances and Perspectives. Molecules 2019, 24, 1004. https://doi.org/10.3390/molecules24061004
Behren S, Westerlind U. Glycopeptides and -Mimetics to Detect, Monitor and Inhibit Bacterial and Viral Infections: Recent Advances and Perspectives. Molecules. 2019; 24(6):1004. https://doi.org/10.3390/molecules24061004
Chicago/Turabian StyleBehren, Sandra, and Ulrika Westerlind. 2019. "Glycopeptides and -Mimetics to Detect, Monitor and Inhibit Bacterial and Viral Infections: Recent Advances and Perspectives" Molecules 24, no. 6: 1004. https://doi.org/10.3390/molecules24061004
APA StyleBehren, S., & Westerlind, U. (2019). Glycopeptides and -Mimetics to Detect, Monitor and Inhibit Bacterial and Viral Infections: Recent Advances and Perspectives. Molecules, 24(6), 1004. https://doi.org/10.3390/molecules24061004