An Improved Method for the Quaternization of Nicotinamide and Antifungal Activities of Its Derivatives



 ,
, 
Abstract
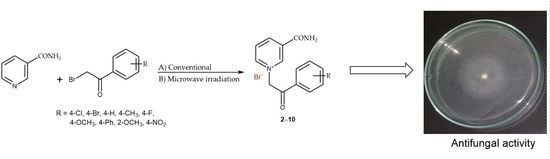
1. Introduction
2. Results
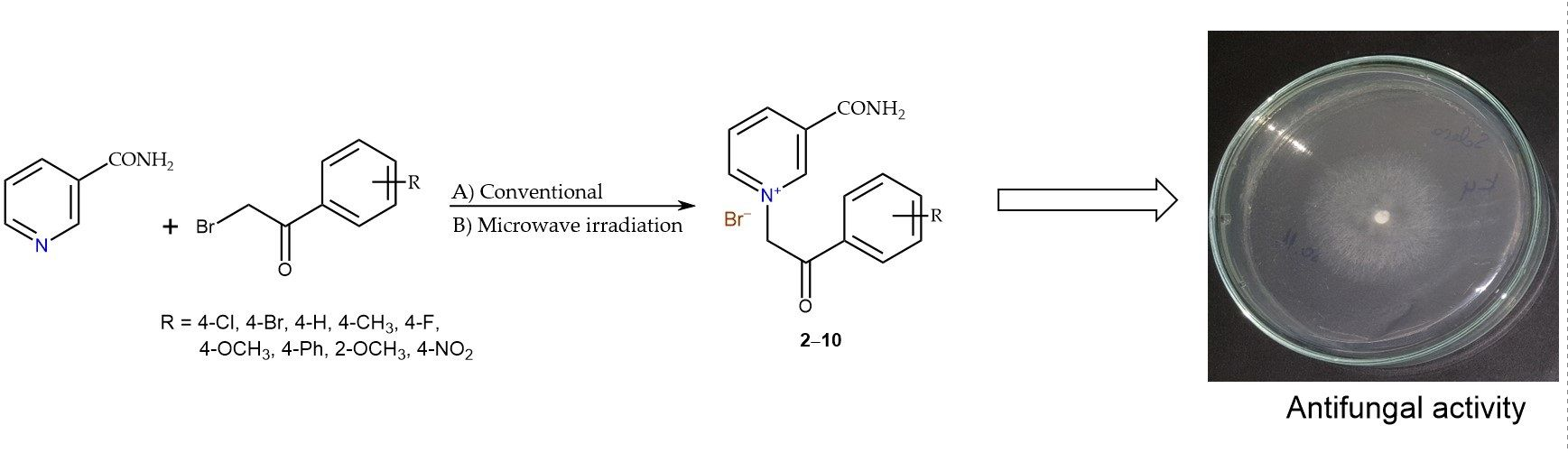
2.1. Chemistry
2.2. Antifungal Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Synthesis of 3-Carbamoyl-1-methylpyridinium Iodide (1)
3.3. Synthesis of Nicotinamide Derivatives with Substituted 2-Bromoacetophenones (2–10)
3.4. Antifungal Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sauve, A.A. NAD+ and Vitamin B3: From Metabolism to Therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Asif, M. Antimicrobial Potential of Nicotinic Acid Derivatives Against Various Pathogenic Microbes. Eur. Rev. Chem. Res. 2014, 1, 10–21. [Google Scholar] [CrossRef]
- Ammar, Y.A.; Mohamed, Y.A.; El-Sharief, A.M.S.; El-Gaby, M.S.A.; Abbas, S.Y. Reactivity of 2,3-Pyridine Dicarboxylic Anhydride Towards some Nitrogen Nucleophilic Reagents: Synthesis and Antimicrobial Evaluation of some Pyridine Carboxamide and Pyrrolo[3,4-B]Pyridine-5,7-Dione Derivatives. Chem. Sci. 2011, CSJ-16. [Google Scholar] [CrossRef]
- Thomas, A.B.; Nanda, R.K.; Kothapalli, L.P.; Deshpande, A.D. Synthesis and antimicrobial activity of N-[2-(aryl/substituted aryl)-4-oxo-1,3-thiazolidin- 3-yl]pyridine-4-carboxamide. J. Kor. Chem. Soc. 2011, 55, 960–968. [Google Scholar] [CrossRef]
- Vijay, V.; Soni, N.; Khanna, V. Synthesis and antimicrobial activity of 5-arylidine derivatives of N-(2-(4-chlorophenyl)-4-oxo-3-thiazolidinyl)isonicotinamide. Inter. J. Res. Pharm. Sci. 2011, 1, 34–43. [Google Scholar] [CrossRef]
- Wu, J.; Kang, S.H.; Song, B.A.; Hu, D.Y.; He, M.; Jin, L.H.; Song, Y. Synthesis and antibacterial activity against Ralstonia solanacearum for novel hydrazone derivatives containing a pyridine moiety. Chem. Cent. J. 2012, 6, 28. [Google Scholar] [CrossRef]
- Dhore, J.W.; Pethe, G.B.; Wagh, S.P.; Thorat, G.D. Synthesis, characterization and biological studies of some triazolyl isonicotinamide. Arch. Appl. Sci. Res. 2011, 3, 407–414. [Google Scholar]
- Wu, J.; Kang, S.; Luo, L.; Shi, Q.; Ma, J.; Yin, J.; Song, B.; Hu, D.; Yang, S. Synthesis and antifungal activities of novel nicotinamide derivatives containing 1,3,4-oxadiazole. Chem. Central J. 2013, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hu, D.; Kuang, J.; Cai, H.; Wu, S.; Xue, W. Synthesis and antifungal activity of N-(substituted pyridinyl)-1-methyl(phenyl)-3-(trifluoromethyl)-1H-pyrazole-4-carboxa-mide derivatives. Molecules 2012, 17, 14205–14218. [Google Scholar] [CrossRef]
- Kozlevčar, B.; Leban, I.; Turel, I.; Segedin, P.; Petrič, M.; Pohleven, F.; White Andrew, J.P.; Williams, D.J.; Sieler, J. Complexes of copper(II) acetate with nicotinamide: Preparation, characterization and fungicidal activity; crystal structures of [Cu2(O2CCH3)4(nia)] and [Cu2(O2CCH3)4(nia)2]. Polyhedron 1999, 18, 755–762. [Google Scholar] [CrossRef]
- Komori, T.; Ujita, S.; Inoue, T. Preparation of N-(3-phenoxybenzyl) nicotinamide derivatives as plant fungicides. Chem. Abstr. 2010, 154, 64642. [Google Scholar]
- Wen, F.; Zhang, H.; Yu, Z.; Jin, H.; Yang, Q.; Houb, T. Design, synthesis and antifungal/insecticidal evaluation of novel nicotinamide derivatives. Pestic. Biochem. Physiol. 2010, 98, 2–248. [Google Scholar] [CrossRef]
- Frackenpohl, J.; Hense, A.; Arnold, C.; Franken, E.M.; Malsam, O.; Sanwald, E.; Goergens, U. Preparation of Nicotinamides as Agrochemical Insecticides. Chem. Abstr. 2008, 152, 287405. [Google Scholar]
- Zhu, Y.C.; Yuan, L.P.; Cao, J.; Shen, X.; Lu, D.H.; Ni, C.C.; Shen, Z.; Chen, L.; Zhang, Y.B. Synthesis and bioactivity of 2-benzylsulfanyl nicotinamide derivatives. Agrichemicals 2008, 10, 287–291. [Google Scholar]
- Gesing, E.R.F.; Mueller, K.H.; Kysela, E.; Drewes, M.W.; Dahmen, P.; Feucht, D.; Pontzen, R. Preparation of N-phenylnicotinamides for use as herbicides. Chem. Abstr. 2001, 134, 266207. [Google Scholar]
- Jeschke, P.; Lindner, W.; Bonse, G.; Santel, H.J.; Luerssen, K.; Schmidt, R.R.; Hartwig, J. Herbicidal and plant-mematocidal compositions based on mercaptonicotinic acid derivatives. Chem. Abstr. 1993, 119, 160137. [Google Scholar]
- Chung, I.M.; Kim, J.J.; Chun, S.C.; Ahmad, A. Potential herbicidals and growth regulators constituents in rice hulls of Oryza sativa. J. Chem. 2008, 20, 820–822. [Google Scholar]
- Girgis, A.S.; Hosni, H.M.; Barsoum, F.F. Novel synthesis of nicotinamide derivatives of cytotoxic properties. Bioorg. Med. Chem. 2006, 14, 4466–4476. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Shi, L.; Ke, S. Nicotinamide-based diamides derivatives as potential cytotoxic agents: Synthesis and biological evaluation. Chem. Cent. J. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.E.; Yoo, M.S.; Choi, J.H.; Lee, J.Y.; Kim, J.H.; Kim, J.H.; Lee, J.K.; Kim, G.I.; Park, Y.; Chi, Y.H.; et al. BRN-103, a novel nicotinamide derivative, inhibits VEGF-induced angiogenesis and proliferation in human umbilical vein endothelial cells. Bioorg. Med. Chem. Lett. 2011, 1, 6236–6241. [Google Scholar] [CrossRef]
- Bušić, V.; Pavlović, H.; Roca, S.; Vikić-Topić, D.; Gašo-Sokač, D. Microwave-assisted Quaternization of Various Pyridine Derivatives and their Antibacterial Activity. Croat. Chem. Acta 2017, 90, 425–433. [Google Scholar] [CrossRef]
- Gašo-Sokač, D.; Bušić, V.; Cetina, M.; Jukić, M. An Efficient Synthesis of Pyridoxal Oxime Derivatives under Microwave Irradiation. Molecules 2014, 19, 7610–7620. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, O.E.; Verolainen, N.V.; Voronchikhina, L.I. Effect of structure of nucleophile and substrate on the quaternization of heterocyclic amines. Russian J. General Chem. 2010, 80, 1025–1028. [Google Scholar] [CrossRef]
- Pirrung, M. Handbook of Synthetic Organic Chemistry; Elsevier Science Publishing Co Inc.: San Diego, CA, USA, 2016; pp. 57–60. [Google Scholar]
- Gullino, M.L.; Minuto, A.; Gilardi, G.; Garibaldi, A. Efficacy of azoxystrobin and other strobilurins against Fusarium wilts of carnation, cyclamen and Paris daisy. Crop Protection 2002, 21, 57–61. [Google Scholar] [CrossRef]
- Haidukowski, M.; Pascale, M.; Perrone, G.; Pancaldi, D.; Campagna, C.; Visconti, A. Effect of fungicides on the development of Fusarium head blight, yield and deoxynivalenol accumulation in wheat inoculated under field conditions with Fusarium graminearum and Fusarium culmorum. J. Sci. Food Agric. 2005, 85, 191–198. [Google Scholar] [CrossRef]
- Xu, C.; Liang, X.; Hou, Y.; Zhou, M. Effects of the Novel Fungicide Benzothiostrobin on Sclerotinia sclerotiorum in the Laboratory and on Sclerotinia Stem Rot in Rape Fields. Plant Dis. 2015, 99, 969–975. [Google Scholar] [CrossRef]
- Parmar, H.V.; Kapadiya, H.J.; Bhaliya, C.M. Efficacy of different fungicides against Macrophomina phaseolina (Tassi) Goid causing castor root rot. IJCS 2017, 5, 1807–1809. [Google Scholar]
- Ye, Y.H.; Ma, L.; Dai, Z.-C.; Xiao, Y.; Zhang, Y.-Y.; Li, D.-D.; Wang, J.-X.; Zhu, H.-L. Synthesis and Antifungal Activity of Nicotinamide Derivatives as Succinate Dehydrogenase Inhibitors. J. Agric. Food Chem. 2014, 62, 4063–4071. [Google Scholar] [CrossRef]
- SAS/STAT. User’s Guide, version 9.2; SAS Institute Inc.: Cary, NC, USA, 2008. [Google Scholar]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 |
|---|---|---|---|---|---|---|---|---|
| Yield (%) | 22 | 28 | 55 | 71 | 93 | 85 | 89 | 90 |
| Compound | Conventional Method (A) | Microwave-Assisted Method (B) | ||||
|---|---|---|---|---|---|---|
| Solvent | Time (h) | Yield (%) | Time (min) | EtOH | Acetone | |
| Yield (%) | Yield (%) | |||||
| (1) | EtOH | 17 (rt) | 78 | 20 | 95 | 92 |
| (2) | Acetone | 1 (60 °C) + 24 (rt) | 32 | 10 | 67 | 65 |
| (3) | 51 | 64 | 56 | |||
| (4) | 25 | 93 | 73 | |||
| (5) | 18 | 88 | 76 | |||
| (6) | 37 | 71 | 46 | |||
| (7) | 11 | 90 | 75 | |||
| (8) | 31 | 44 | 80 | |||
| (9) | 36 | 41 | 36 | |||
| (10) | 39 | 43 | 42 | |||
| Mycelial Growth (mm) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | M. phaseolina | F. culmorum | F. oxysporum | S. sclerotiorum | ||||
| 10 µg/mL | 100 µg/mL | 10 µg/mL | 100 µg/mL | 10 µg/mL | 100 µg/mL | 10 µg/mL | 100 µg/mL | |
| (1) | 21.5 ± 5.8 | 16.75 ± 3.2 | 25.25 ± 3 | 19.25 ± 0.9 | 15 ± 1.4 | 12.75 ± 1.2 | 4 ± 0 | 4 ± 0 |
| (2) | 18.75 ± 4.6 | 18.75 ± 4.3 | 23.75 ± 1.5 | 20.25 ± 1.7 | 15 ± 2.8 | 14 ± 1.6 | 4.5 ± 1 | 4 ± 0 |
| (3) | 16.25 ± 4.5 | 15 ± 4.8 | 24 ± 0 | 19.5 ± 0.6 | 15 ± 1.4 | 12.25 ± 0.9 | 4.25 ± 0.5 | 4 ± 0 |
| (4) | 20.5 ± 5.2 | 1.9 ± 4.5 | 22.5 ± 1.9 | 18.5 ± 0.6 | 15.5 ± 0.6 | 13.5 ± 2.4 | 4.75 ± 0.9 | 4 ± 0 |
| (5) | 23.75 ± 3.9 | 23 ± 3.4 | 25.75 ± 1.5 | 26 ± 1.8 | 16.5 ± 1 | 12.5 ± 1.3 | 4 ± 0 | 4 ± 0 |
| (6) | 21 ± 6.2 | 21 ± 5.2 | 26.5 ± 2.6 | 26 ± 2.4 | 15 ± 1.6 | 12.5 ± 0.6 | 4 ± 0 | 4 ± 0 |
| (7) | 20.5 ± 4.2 | 20 ± 8.1 | 26.5 ± 3.1 | 25 ± 2.4 | 13.75 ± 1.2 | 12.25 ± 0.9 | 4 ± 0 | 4 ± 0 |
| (8) | 16.5 ± 6.4 | 24.2 ± 3.9 | 26.5 ± 2.2 | 31.75 ± 1.2 | 14.5 ± 1 | 16.25 ± 0.9 | 4.25 ± 0.5 | 4 ± 0 |
| (9) | 20.5 ± 5.8 | 22 ± 2.9 | 29.5 ± 1.3 | 29.75 ± 0.9 | 16 ± 0.8 | 14.25 ± 0.9 | 4.5 ± 1 | 4 ± 0 |
| (10) | 22.25 ± 2.2 | 19 ± 3.6 | 29 ± 1.1 | 31.75 ± 0.5 | 16.25 ± 0.5 | 15 ± 0.8 | 4 ± 0 | 4 ± 0 |
| Strobilurin a | 4 ± 0 | 4 ± 0 | 4 ± 0 | 4 ± 0 | 4 ± 0 | 4 ± 0 | ||
| Dithio-carbamates a | 4 ± 0 | 4 ± 0 | ||||||
| LSD test | 6.91 | 6.34 | 2.79 | 2.06 | 1.91 | 1.74 | 0.80 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siber, T.; Bušić, V.; Zobundžija, D.; Roca, S.; Vikić-Topić, D.; Vrandečić, K.; Gašo-Sokač, D. An Improved Method for the Quaternization of Nicotinamide and Antifungal Activities of Its Derivatives. Molecules 2019, 24, 1001. https://doi.org/10.3390/molecules24061001
Siber T, Bušić V, Zobundžija D, Roca S, Vikić-Topić D, Vrandečić K, Gašo-Sokač D. An Improved Method for the Quaternization of Nicotinamide and Antifungal Activities of Its Derivatives. Molecules. 2019; 24(6):1001. https://doi.org/10.3390/molecules24061001
Chicago/Turabian StyleSiber, Tamara, Valentina Bušić, Dora Zobundžija, Sunčica Roca, Dražen Vikić-Topić, Karolina Vrandečić, and Dajana Gašo-Sokač. 2019. "An Improved Method for the Quaternization of Nicotinamide and Antifungal Activities of Its Derivatives" Molecules 24, no. 6: 1001. https://doi.org/10.3390/molecules24061001
APA StyleSiber, T., Bušić, V., Zobundžija, D., Roca, S., Vikić-Topić, D., Vrandečić, K., & Gašo-Sokač, D. (2019). An Improved Method for the Quaternization of Nicotinamide and Antifungal Activities of Its Derivatives. Molecules, 24(6), 1001. https://doi.org/10.3390/molecules24061001