Abstract
A simple method has been developed for the synthesis of cyclopropa[c]coumarins, which belong to the donor-acceptor cyclopropane family and, therefore, are promising substrates for the preparation of chromene-based fine chemicals. The method, based on the acetic acid-induced intramolecular transesterification of 2-arylcyclopropane-1,1-dicarboxylates, was found to be efficient for substrates containing hydroxy group directly attached to the aromatic ring.
1. Introduction
The longstanding interest to coumarin derivatives stems from their wide distribution in Nature and a broad spectrum of bioactivity, including anticoagulant, anticonvulsant, antidepressant, anti-HIV, antiinflammatory, antimicrobial, antioxidative, antituberculosis, antitumor activities, etc. [1,2,3,4,5,6,7,8,9,10,11]. Coumarin derivatives are used in medicinal practice for the treatment of dementia [12], varicose veins [13], hemorrhoids [13], etc. [14]; they are applied as rodenticides [15] and as fluorophores in cell biology [16]. The high value of these compounds has stimulated their intensive investigation, i.e. the synthesis of novel coumarin derivatives, the study of their transformations and screening of their bioactivity, 3- and 4-substituted coumarins as well as their 3,4-dihydro derivatives being the most interesting substrates for the medicinal applications.
Cyclopropa[c]coumarins, being the members of the donor-acceptor cyclopropanes subclass, are highly potent substrates for the preparation of a large diversity of functionalized coumarin derivatives due to a rich reactivity of the three-membered ring. Even though the first cyclopropanation of 3-acylcoumarins and coumarin-3-carboxylates with α-haloketones was described by Widman exactly 100 years ago (Scheme 1a) [17,18], cyclopropacoumarins remain poorly investigated due to the absence of efficient methods for their synthesis. Since Widman’s study, a number of cyclopropa[c]coumarins bearing diverse acceptor substituents at both C(1) and C(1a) atoms have been obtained by the related cyclopropanations under phase transfer catalysis (Scheme 1a) [19] or using diazoketones (Scheme 1b) [20,21,22]. Nevertheless, the application of diazoketones usually resulted in the formation of complex mixtures and low yields of target products. Moreover, 1,1,1a-substituted cyclopropa[c]coumarins were synthesized in reasonable yields by the treatment of coumarin-3-carboxylates and -carboxamides with α,α-dibromoketones and zinc (Scheme 1c) [23,24,25], trichloroacetic acid [26], phenyliodonium (Scheme 1d) [27] and diphenylsulfonium ylides (Scheme 1e) [28].
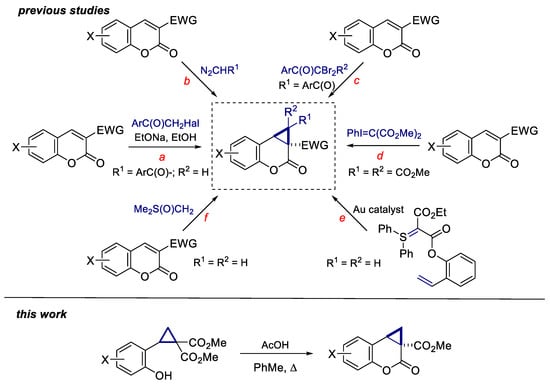
Scheme 1.
Syntheses of cyclopropa[c]coumarins.
Oppositely, the reaction of much more reactive dimethylsulfoxonium methylide [29,30] with 3-acylcoumarins, coumarin-3-carboxylates, the corresponding nitriles as well as sulfones under the typical conditions of Corey-Chaykovsky cyclopropanation was found to afford cyclopenta[b]-benzofuran derivatives as a result of the fast secondary reaction of the initially formed cyclopropa[c]coumarins with a second equivalent of the Corey ylide [31,32,33]. The target cyclopropa[c]coumarins were obtained in low-to-moderate yields when the reaction was performed at 0 °C (for 3-ethoxycarbonyl-, 3-pivaloyl- and 3-cyanocoumarins) or at −40 °C (for 3-acetyl and 3-benzoyl derivatives) (Scheme 1f) [31]. Very recently it was proposed to perform this cyclopropanation using very slow addition of the Corey ylide solution (5 mL for 6 h) to the corresponding coumarin derivative, however, product yields were not given [34,35]. Therefore, the development of efficient procedure for the synthesis of 1-unsubstituted cyclopropa[c]coumarins remains a challenging task for organic chemists. Herein, we describe a new general approach to cyclopropa[c]coumarins based on the use of 2-(2-hydroxyphenyl)cyclopropane-1,1-dicarboxylates (Scheme 1) which can be easily obtained from the corresponding salicylic aldehydes [36,37].
2. Results and Discussion
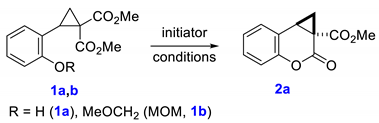
We started our research with a search for the optimal conditions for the transesterification of 2-hydroxy- and 2-(methoxymethoxy)phenyl-substituted cyclopropane-1,1-dicarboxylates 1a,b as model substrates using their treatment with base (K2CO3 or N,N-diisopropylethylamine, DIPEA) or Brønsted acid (Table 1). We have found that the highest yield of the target methyl cyclopropa[c]coumarin-3-carboxylate 2a was obtained when toluene solution of cyclopropane 1a was heated under reflux with 2 equiv of acetic acid (Table 1, entry 8). The use of stronger acids such as trifluoroacetic acid (TFA) or p-toluenesulfonic acid (TsOH) resulted in the formation of complex mixture of products; any attempts to isolate 2a were unsuccessful. Base-induced reaction produced the desired product 2a in low yield only.

Table 1.
Optimization of reaction conditions for the intramolecular transesterification of model cyclopropanes 1a,b.
With the optimized reaction conditions in hand, we studied the scope of this transesterification and found that diverse substituents in the benzene ring (halogens, alkyl or nitro group) are tolerant to the reaction conditions (Scheme 2). Oppositely, the reaction of 2-hydroxy-3-methoxyphenyl-substituted cyclopropane 1g produced only trace amounts of the corresponding cyclopropa[c]coumarin 2g.
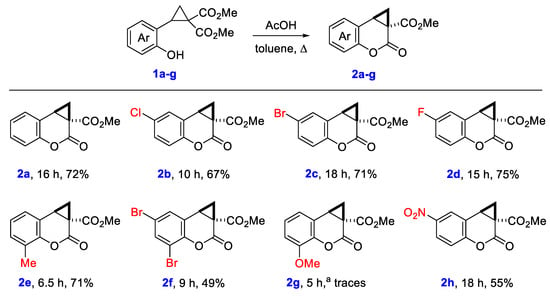
Scheme 2.
Synthesis of cyclopropa[c]coumarins 2a–h. a In chlorobenzene.
To extend the scope of the reaction, we have studied reactions of cyclopropane 3 with a 2-(hydroxymethyl)phenyl group as a donor and its analogue 8 bearing a 3-hydroxymethyl-4-indolyl substituent. However, in both cases we failed to obtain the corresponding products of intramolecular esterification. Thus, the heating of 3 with 2 equiv of acetic acid in both toluene and chlorobenzene did not produce the desired oxepanone 4 at all. When a large excess of acetic acid was applied, benzyl acetate 5 was obtained as a single low-molecular-weight product (Scheme 3). The use of a stronger acid led to the formation of complex mixtures containing predominantly products of the three-membered ring opening.
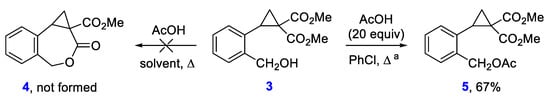
Scheme 3.
The attempt to synthesize benzoxepane 4. a Substrate concentration: 0.04 M.
Meanwhile, indolyl-substituted cyclopropane 8, which was obtained in two steps from the reported cyclopropane 6 [38], under heating with acetic acid afforded bis(indolyl)methane 9 (Scheme 4). Similar AcOH-induced transformations of 3-indolylmethanols to bis(indolyl)methanes was previously described [39,40,41]. Meanwhile, the presence of highly reactive donor-acceptor cyclopropane functionality in the starting alcohol 8 and two such moieties in the product 9 provides non-trivial nature this process.
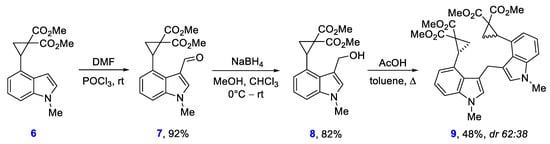
Scheme 4.
Synthesis and acetic acid-induced dimerization of indolylmethanol 8.
The obtained cyclopropa[c]coumarins 2 are potent substrates for the synthesis of diverse coumarin derivatives (Scheme 5). Thus, it was recently shown that such cyclopropacoumarins undergo Ni(ClO4)2-catalyzed nucleophilic ring opening under treatment with indole affording 4-(indolylmethyl)chroman-2-one-3-carboxylates [34].
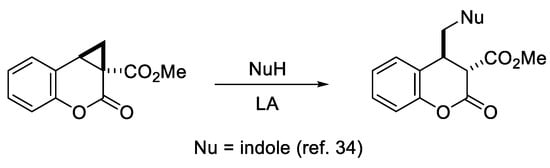
Scheme 5.
Nucleophilic ring opening of cyclopropa[c]coumarins.
Nevertheless, our attempts to transform compounds 2 to the corresponding benzoxepan derivatives via their reduction with Zn/AcOH system [42] or Lewis acid-induced isomerization [43] were unsuccessful. Unexpectedly, the treatment of cyclopropacoumarin 2d with zinc and acetic acid in methanol did not afford the products of three-membered ring reduction. Instead, it led to the methanolysis of the lactone moiety in 2d producing cyclopropane 1d and a small amount of acyclic product 10 formed by the reduction of 1d (Scheme 6). Moreover, this cyclopropacoumarin remains intact under heating with both tin(II) triflate in dichloromethane and trimethylsilyl triflate in chlorobenzene for several hours. The absence of benzoxepane derivative in the reaction mixtures together with the aforementioned literature data on the nucleophilic attack of cyclopa[c]coumarins at the CH2 atom [34] demonstrate the decelerating effect of annulation on the reactivity of the C-C bond between atoms connected to donor and acceptor substituents. The effect of the annulated ring nature on the reactivity of donor-acceptor cyclopropanes deserve, evidently, a careful study.
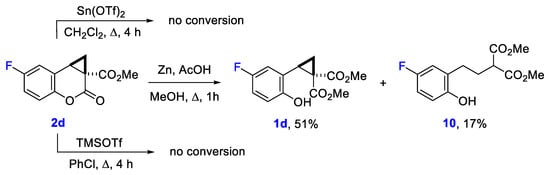
Scheme 6.
Attempts of a ring enlargement of cyclopropa[c]coumarin 2d.
3. Experimental
3.1. General Information
The structures of synthesized compounds were elucidated with the aid of 1D NMR (1H, 13C) and 2D NMR (HSQC and HMBC 1H-13C) spectroscopy. NMR spectra were acquired on Avance 500 (Bruker, Billerica, MA, USA) and 400-MR (Agilent, Santa Clara, CA, USA) spectrometers at room temperature; the chemical shifts δ were measured in ppm with respect to solvent (1H: CDCl3, δ = 7.27 ppm; 13C: CDCl3, δ = 77.0). Splitting patterns are designated as s, singlet; d, doublet; m, multiplet; dd, double doublet; br., broad. Coupling constants (J) are in Hertz. Infrared spectra were recorded on an Infralum FT-801 spectrometer (Simex, Novosibirsk, Russia). High resolution and accurate mass measurements were carried out using a micrOTOF-QTM ESI-TOF (Electrospray Ionization/Time of Flight, Bruker, Billerica, MA, USA), LTQ Orbitrap (Thermo Fischer Scientific, Waltham, MA, USA) mass spectrometers and a Triple TOF 5600+ instrument (AB Sciex, Darmstadt, Germany) using ESI modes. Elemental analyses were performed with an EA-1108 CHNS elemental analyser instrument (Fisons, Ipswich, UK). Melting points (mp) are uncorrected and were measured on a 9100 capillary melting point apparatus (Electrothermal, Stone, UK). Analytical thin layer chromatography (TLC) was carried out with silica gel plates (silica gel 60, F254, supported on aluminium); visualization was done using a UV lamp (365 nm). Column chromatography was performed on silica gel 60 (230–400 mesh, Merck, Darmstadt, Germany). All reactions were carried out using freshly distilled and dry solvents. Dimethyl 2-(2-hydroxyaryl)cyclopropane-1,1-dicarboxylate was synthesized by the published procedure [36,37]. Commercial reagents employed in the synthesis were analytical grade, obtained from Aldrich (St. Louis, MI, USA) or Alfa Aesar (Ward Hill, MO, USA). Cyclopropanes 3 and 6 were obtained by the reported procedures [36,38]. The 1H NMR, 13C NMR for synthesized compounds as well as 2D (HSQC and HMBC) NMR spectra for selected compounds are available in the Supplementary Material.
3.2. General Procedure for the Synthesis of Cyclopropa[c]coumarins
A toluene solution of cyclopropane 1 (0.03 or 0.04 M, 1 equiv) and glacial acetic acid (2 equiv) was heated for the specified time under reflux or using backflow condenser without cooling with water providing slow removal of the formed methanol. When 1 was completely converted (TLC control), the reaction mixture was cooled, diluted with ether (10 mL), washed with saturated NaHCO3 solution (3 × 15 mL), dried with Na2SO4 and concentrated in vacuo. The resulting residue was purified by flash chromatography on silica gel (eluent: 10–50% ethyl acetate in petroleum ether) to afford the target cyclopropacoumarin 2 (Figure 1).
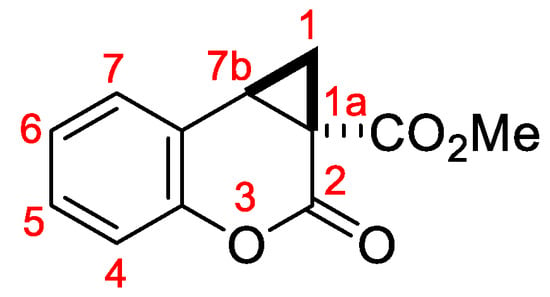
Figure 1.
Atom numbering in compounds 2.
Methyl (1aRS,7bRS)-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2a). Dimethyl 2- (2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1a, 120 mg, 0.48 mmol), AcOH (58 mg, 55 µL, 0.96 mmol), toluene (12 mL), reflux, 16 h. Rf = 0.55 (ethyl acetate:petroleum ether 1:2). Yield 75 mg (72%); colorless solid; mp = 98–100 °C (lit. 100–102 °C [34,35]; oil [37]). Spectral data are consistent with the reported ones [34,35].
Methyl (1aRS,7bRS)-6-chloro-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2b). Dimethyl 2-(5-chloro-2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1b) (120 mg, 0.42 mmol), AcOH (51 mg, 48 µL, 0.84 mmol), toluene (12 mL), reflux, 10 h. Rf = 0.31 (ethyl acetate:petroleum ether 1:4). Yield 71 mg (67%); colorless solid; mp = 111–112 °C (lit. 119–121 °C [34]). 1H-NMR (CDCl3, 500 MHz) δ = 1.41 (dd, 2J = 5.2 Hz, 3J = 6.4 Hz, 1H, CH2), 2.50 (dd, 2J = 5.2 Hz, 3J =9.2 Hz, 1H, CH2), 2.89 (dd, 3J = 9.2 Hz, 3J = 6.4 Hz, 1H, CH), 3.86 (s, 3H, CH3O), 7.00 (d, 3J = 8.7 Hz, 1H, Ar), 7.24 (dd, 3J = 8.7 Hz, 4J = 2.3 Hz, 1H, Ar), 7.37 (d, 4J = 2.3 Hz, 1H, Ar). 13C-NMR (CDCl3, 125 MHz) δ = 21.1 (CH2), 28.7 (CH), 29.7 (C), 53.4 (CH3O), 118.6 (CH, Ar), 121.7 (C, Ar), 127.6 (CH, Ar), 128.6 (CH, Ar), 129.8 (C, Ar), 148.0 (C, Ar), 161.7 (CO), 167.7 (CO2Me). IR (сm−1) 3110, 3066, 2950, 2955, 2922, 2853, 1763, 1723, 1484, 1435, 1369, 1319, 1283, 1259, 1107, 1085, 1052, 946. HRMS ESI-TOF: m/z = 253.0265 [M + H]+ (253.0262 сalcd for C12H10ClO4).
Methyl (1aRS,7bRS)-6-bromo-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2c). Dimethyl 2-(5-bromo-2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1c, 200 mg, 0.61 mmol), AcOH (73 mg, 70 µL, 1.2 mmol), toluene (15 mL), reflux, 18 h. Rf = 0.70 (ethyl acetate:petroleum ether 1:4). Yield 126 mg (71%); colorless solid; mp = 114–115 °C (lit. 105–107 °C [34]). 1H-NMR (CDCl3, 500 MHz) δ = 1.40 (dd, 2J = 5.1 Hz, 3J = 6.4 Hz, 1H, CH2), 2.21 (dd, 2J = 5.1 Hz, 3J = 9.2 Hz, 1H, CH2), 2.89 (dd, 3J = 9.2 Hz, 3J = 6.4 Hz, 1H, CH), 3.85 (s, 3H, CH3O), 6.92 (d, 3J = 8.7 Hz, 1H, Ar), 7.38 (dd, 3J = 8.7 Hz, 4J = 2.3 Hz, 1H, Ar), 7.50 (d, 4J = 2.3 Hz, 1H, Ar). 13C-NMR (CDCl3, 125 MHz) δ = 21.1 (CH2), 28.3 (C), 28.5 (CH), 53.3 (CH3O), 117.0 (C, Ar), 118.8 (CH, Ar), 122.1 (C, Ar), 130.4 (CH, Ar), 131.5 (CH, Ar), 148.5 (C, Ar), 161.5 (CO), 167.6 (CO2Me). IR (сm−1) 3256, 2956, 1762, 1673, 1498, 1440, 1415, 1352, 1276, 1196, 1154, 1108, 1074. HRMS ESI-TOF: m/z = 296.9757 [M + H]+ (296.9757 сalcd for C12H10BrO4).
Methyl (1aRS,7bRS)-6-fluoro-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2d). Dimethyl 2-(5-fluoro-2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1d, 200 mg, 0.75 mmol), AcOH (90 mg, 85 µL, 1.49 mmol), toluene (11 mL), boiling with backflow condenser, 8 h; then an extra portion of AcOH (90 mg, 85 µL, 1.49 mmol), reflux, 7 h. Rf = 0.69 (ethyl acetate:petroleum ether 1:2). Yield 150 mg (75%); colorless solid; mp = 90–91 °C (lit. 93–95 °C [34]). 1H-NMR (CDCl3, 400 MHz) δ = 1.40 (dd, 2J = 5.1 Hz, 3J = 6.3 Hz, 1H, CH2), 2.59 (dd, 2J = 5.1 Hz, 3J =9.0 Hz, 1H, CH2), 2.88 (dd, 3J = 9.0 Hz, 3J = 6.3 Hz, 1H, CH), 3.84 (s, 3H, CH3O), 6.93–7.02 (m, 2H, Ar), 7.07 (dd, 3J = 8.2 Hz, 4JHF = 3.0 Hz, 1H, Ar). 13C- NMR (CDCl3, 100 MHz) δ = 21.2 (CH2), 28.3 (C), 29.0 (CH), 53.5 (CH3O), 114.5 (d, 2JCF = 24 Hz, CH, Ar), 115.5 (d, 2JCF = 24 Hz, CH, Ar), 118.7 (d, 3JCF = 9 Hz, CH, Ar), 121.7 (d, 3JCF = 8 Hz, C, Ar), 145.6 (C, Ar), 158.5 (d, 1JCF = 243 Hz, C, Ar), 162.1 (CO), 167.9 (CO2Me). IR (сm−1) 1759, 1724, 1579, 1496, 1441, 1375, 1325, 1288, 1250, 1197, 1149, 1107, 1057, 987, 966, 931. HRMS ESI-TOF: m/z = 237.0555 [M + H]+ (237.0558 сalcd for C12H10 FO4).
Methyl (1aRS,7bRS)-4-methyl-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2e). Dimethyl 2-(3-methyl-2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1e, 180 mg, 0.64 mmol), AcOH (158 mg, 160 µL, 2.6 mmol), toluene (12 mL), reflux, 10 h. Rf = 0.75 (ethyl acetate:petroleum ether 1:2). Yield 113 mg (76%); colorless solid; mp = 82–83 °C. 1H-NMR (CDCl3, 500 MHz) δ = 1.37 (dd, 2J = 4.9 Hz, 3J = 6.7 Hz, 1H, CH2), 2.30 (s, 3H, CH3), 2.46 (dd, 2J = 4.9 Hz, 3J =9.2 Hz, 1H, CH2), 2.90 (dd, 3J = 9.2 Hz, 3J = 6.7 Hz, 1H, CH), 3.84 (s, 3H, CH3O), 7.03 (dd, 3J = 7.6 Hz, 3J = 7.4 Hz, 1H, Ar), 7.11 (br. d, 3J = 7.6 Hz, 1H, Ar), 7.18 (br. d, 3J = 7.4 Hz, 1H, Ar). 13C-NMR (CDCl3, 125 MHz) δ = 15.6 (CH3), 20.9 (CH2), 28.6 (C), 29.3 (CH), 53.1 (CH3O), 119.6 (C, Ar), 124.0 (CH, Ar), 125.2 (CH, Ar), 126.5 (C, Ar), 130.0 (CH, Ar), 147.7 (C, Ar), 162.4 (CO), 168.1 (CO2Me). IR (сm−1) 3106, 3040, 2957, 2924, 2853, 1746, 1730, 1471, 1434, 1395, 1327, 1292, 1230, 1192, 1116, 1098, 1057, 1039, 946, 925. HRMS ESI-TOF: m/z = 233.0810 [M + H]+ (233.0808 сalcd for C13H13O4).
Methyl (1aRS,7bRS)-4,6-dibromo-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2f). Dimethyl 2-(3,5-dibromo-2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1f, 200 mg, 0.49 mmol), AcOH (30 mg, 28 µL, 0.49 mmol), toluene (11 mL), boiling with backflow condenser, 5 h, then an extra portion of AcOH (60 mg, 56 µL, 0.98 mmol), reflux, 4 h. Rf = 0.52 (ethyl acetate:petroleum ether 1:4). Yield 90 mg (49%); colorless solid; mp = 77–78 °C. 1H-NMR (CDCl3, 500 MHz) δ = 1.45 (dd, 2J = 5.2 Hz, 3J = 6.4 Hz, 1H, CH2), 2.51 (dd, 2J = 5.2 Hz, 3J =9.2 Hz, 1H, CH2), 2.90 (dd, 3J = 9.2 Hz, 3J = 6.4 Hz, 1H, CH), 3.86 (s, 3H, CH3O), 7.47 (d, 4J = 2.3 Hz, 1H, Ar), 7.67 (d, 4J = 2.3 Hz, 1H, Ar). 13C-NMR (CDCl3, 125 MHz) δ = 21.1 (CH2), 28.8 (C), 29.0 (CH), 53.7 (CH3O), 112.0 (C, Ar), 117.2 (C, Ar), 123.5 (C, Ar), 129.8 (CH, Ar), 134.9 (CH, Ar), 146.0 (C, Ar), 160.6 (CO), 167.5 (CO2Me). IR (film, сm−1) 3447, 3078, 3005, 2954, 2924, 2851, 1777, 1725, 1453, 1439, 1368, 1324, 1239, 1207, 1071, 1045, 991, 985. HRMS ESI-TOF: m/z = 374.8856 [M + H]+ (374.8862 сalcd for C12H9Br2O4).
Methyl (1aRS,7bRS)-6-nitro-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2h). Dimethyl 2-(5-nitro-2-hydroxyphenyl)cyclopropane-1,1-dicarboxylate (1h, 200 mg, 0.68 mmol), AcOH (81 mg, 78 µL, 1.35 mmol), toluene (17 mL), boiling with backflow condenser, 7 h, then an extra portion of AcOH (81 mg, 78 µL, 1.35 mmol), reflux, 10 h. Rf = 0.63 (ethyl acetate:petroleum ether 1:4). Yield 97 mg (55%); colorless solid; mp = 146–147 °C. 1H-NMR (CDCl3, 500 MHz) δ = 1.49 (dd, 2J = 5.3 Hz, 3J = 6.4 Hz, 1H, CH2), 2.59 (dd, 2J = 5.3 Hz, 3J =9.2 Hz, 1H, CH2), 3.06 (dd, 3J = 9.2 Hz, 3J = 6.4 Hz, 1H, CH), 3.88 (s, 3H, CH3O), 7.19 (d, 3J = 9.0 Hz, 1H, Ar), 8.18 (dd, 3J = 9.0 Hz, 4J = 2.8 Hz, 1H, Ar), 8.33 (br. d, 4J = 2.8 Hz, 1H, Ar). 13C-NMR (CDCl3, 125 MHz) δ = 21.2 (CH2), 28.3 (C), 28.4 (CH), 53.5 (CH3O), 118.2 (CH, Ar), 121.3 (C, Ar), 123.8 (CH, Ar), 124.3 (CH, Ar), 144.2 (C, Ar), 153.7 (C, Ar), 160.6 (CO), 167.2 (CO2Me). IR (film, сm−1) 3117, 2957, 2923, 2854, 1778, 1730, 1591, 1527, 1492, 1443, 1337, 1316, 1252, 1128, 1107, 1048, 979. HRMS ESI-TOF: m/z = 264.0502 [M + H]+ (264.0503 сalcd for C12H10NO6).
Dimethyl 2-[2-(acetoxymethyl)phenyl]cyclopropane-1,1-dicarboxylate (5). A solution of cyclopropane 3 (190 mg, 0.72 mmol) and glacial acetic acid (1.6 mL, 28 mmol, 40 equiv.) in chlorobenzene (14 mL) was refluxed for 16 h, cooled, diluted with CH2Cl2 (10 mL), washed with saturated NaHCO3 solution (3 × 15 mL), dried with Na2SO4 and concentrated in vacuo. The resulting residue was purified by flash chromatography on silica gel (eluent: 10–30% ethyl acetate in petroleum ether) to afford acetate 4 as colorless oil. Rf = 0.57 (ethyl acetate:petroleum ether 1:3). Yield 148 mg (67%). 1H-NMR (CDCl3, 400 MHz) δ = 1.74 (dd, 2J = 5.2 Hz, 3J = 9.2 Hz, 1H, CH2), 2.07 (s, 3H, CH3CO), 2.28 (dd, 2J = 5.2 Hz, 3J = 8.2 Hz, 1H, CH2), 3.28 (s, 3H, CH3O), 3.30 (dd, 3J = 9.2 Hz, 3J = 8.2 Hz, 1H, CH), 3.78 (s, 3H, CH3O), 5.12 (AB system, 2J = 12.7 Hz, 1H, CH2O), 5.27 (AB system, 2J = 12.7 Hz, 1H, CH2O), 7.07–7.10 (m, 1H, Ar), 7.23–7.27 (m, 2H, Ar), 7.30–7.33 (m, 1H, Ar). 13C-NMR (CDCl3, 100 MHz) δ = 18.2 (CH2), 20.8 (CH3), 29.9 (CH), 36.6 (C), 52.1 (CH3O), 52.8 (CH3O), 64.2 (CH2O), 127.5 (CH, Ar), 127.7 (CH, Ar), 128.1 (CH, Ar), 129.3 (CH, Ar), 133.1 (C, Ar), 136.2 (C, Ar), 166.7 (MeCO2), 169.8 (CO2Me), 170.7 (CO2Me). IR (сm−1) 3066, 3027, 3005, 2954, 2904, 2848, 2256, 1741, 1735, 1496, 1438, 1378, 1332, 1282, 1230, 1207, 1134, 1026, 968, 920. HRMS ESI-TOF: m/z = 307.1176 [M + Na]+ (307.1176 сalcd for C16H19O6).
Dimethyl 2-(3-formyl-1-methyl-1H-indol-4-yl)cyclopropane-1,1-dicarboxylate (7). DMF (1 mL) and POCl3 (0.3 mL, 3.2 mmol) were mixed at 10 °C under argon atmosphere. After 20 min to the resulting mixture cyclopropane 6 (840 mg, 2.9 mmol) in DMF (1 mL) was added. The obtained mixture was stirred at room temperature for 2 h and poured into ice-cold water (15 mL). The formed clear solution was treated with aq. NaOH (620 mg in 3 mL). Product was extracted with ethyl acetate. Combined organic fractions were washed with brine (4 × 10 mL) and dried with Na2SO4. Solvent was evaporated. Product was obtained as brown crystals (850 mg, 92%). Analytical sample was obtained by column chromatography on silica gel as yellow crystals. Rf = 0.54 (ethyl acetate); mp 192–193 °C. 1H-NMR (CDCl3, 500 MHz) δ = 1.95 (dd, 2J = 5.0 Hz, 3J = 9.0 Hz, 1H, CH2), 2.40 (dd, 2J = 5.0 Hz, 3J = 8.1 Hz, 1H, CH2), 3.18 (s, 3H, CH3O), 3.87 (s, 6H, CH3N, CH3O), 4.01 (dd, 3J = 9.0 Hz, 3J = 8.1 Hz, 1H, CH), 7.07 (br.d, 3J = 7.3 Hz, 1H, Ind), 7.24 (dd, 3J = 8.2 Hz, 3J = 7.3 Hz, 1H, Ind), 7.31 (br.d, 3J = 8.2 Hz, 1H, Ind), 7.83 (s, 1H, Ind), 10.13 (s, 1H, CHO). 13C-NMR (CDCl3, 125 MHz) δ = 19.2 (CH2), 33.2 (CH), 33.8 (CH3N), 37.5 (C), 51.8 (CH3O), 52.7 (CH3O), 109.8 (CH, Ind), 119.1 (C, Ind), 122.1 (CH, Ind), 123.1 (CH, Ind), 125.9 (C, Ind), 128.3 (C, Ind), 138.1 (C, Ind), 138.5 (CH, Ind), 167.2 (CO2Me), 170.1 (CO2Me), 184.3 (CHO). IR (Nujol, сm−1): 2938, 2873, 1726, 1657, 1530, 1462, 1447, 1356, 1296, 1214, 1081, 1042. Anal. calcd for C17H17NO5: C, 64.75; H, 5.43; N, 4.44. Found: C, 64.59; H, 5.21; N, 4.35.
Dimethyl 2-(3-hydroxymethyl-1-methyl-1H-indol-4-yl)cyclopropane-1,1-dicarboxylate (8). To a stirred solution of cyclopropane 7 (381 mg, 1.2 mmol) in MeOH (1.2 mL) and CHCl3 (5 mL), NaBH4 (51 mg, 1.3 mmol) was added at 0 °C and stirred for 10 min. Then the mixture was allowed to warm to room temperature and stirred for 5 h until full conversion (TLC control). The solvent was removed in vacuo and the slurry was transferred to a separatory funnel with water and CH2Cl2. The product solution was extracted with CH2Cl2 (3 × 5 mL), the organic fractions were washed with water (3 × 5 mL) and dried with Na2SO4 affording the title compound. Yield 314 mg (82%). 1H-NMR (CDCl3, 500 MHz) δ = 1.84 (dd, 2J = 5.0 Hz, 3J = 8.9 Hz, 1H, CH2), 2.32 (dd, 2J = 5.0 Hz, 3J = 7.9 Hz, 1H, CH2), 3.05 (br. s, 1H, OH), 3.20 (s, 3H, CH3), 3.76 (s, 3H, CH3), 3.87 (s, 3H, CH3), 3.92 (dd, 3J = 8.9 Hz, 3J = 7.9 Hz, 1H, CH), 4.73 (d, 2J = 12.5 Hz, 1H, CH2), 4.97 (d, 2J = 12.5 Hz, 1H, CH2), 6.85 (br. d, 3J = 7.3 Hz, 1H, Ar), 7.13 (s, 1H, Ar), 7.14 (dd, 3J = 8.2 Hz, 3J = 7.3 Hz, 1H, Ar), 7.22 (br. d, 3J = 8.2 Hz, 1H, Ar). 13C-NMR (CDCl3, 125 MHz) δ = 18.3 (CH2), 31.4 (CH3N), 32.8 (CH), 37.4 (C), 52.1 (CH3O), 53.0 (CH3O), 57.1 (CH2O), 109.3 (CH, Ar), 115.0 (C, Ar), 118.6 (CH, Ar), 121.2 (CH, Ar), 126.1 (C, Ar), 126.9 (C, Ar), 129.9 (CH, Ar), 137.5 (C, Ar), 167.8 (CO2Me), 170.6 (CO2Me). IR (сm−1) 3476, 3022, 3003, 2951, 2925, 2855, 1726, 1674, 1455, 1437, 1334, 1281, 1210, 1132. HRMS ESI-TOF: m/z = 340.1159 [M + Na]+ (340.1155 сalcd for C17H19NNaO5).
Dimethyl 2-[3-({4-[2,2-bis(methoxycarbonyl)cyclopropyl]-1-methyl-1H-indol-3-yl}methyl)-1-methyl-1H-indol-4-yl]cyclopropane-1,1-dicarboxylate (9). Cyclopropane 8 (170 mg, 0.53 mmol), AcOH (32 mg, 30 L, 0.53 mmol), toluene (13 mL), boiling with backflow condenser, 6 h. Rf = 0.63 (ethyl acetate:petroleum ether 1:2). Yield 75 mg (48%); colorless solid; mp = 79–80 °C. Mixture of diastereoisomers in the A:B ratio of 62:38; 1H-NMR (CDCl3, 500 MHz) for mixture of isomers: δ = 1.74 (dd, 2J = 5.0 Hz, 3J = 9.0 Hz, 2H, CH2, A), 1.83 (dd, 2J = 5.2 Hz, 3J = 9.4 Hz, 2H, CH2, B), 2.40 (dd, 2J = 5.2 Hz, 3J = 8.2 Hz, 2H, CH2, B), 2.45 (dd, 2J = 5.0 Hz, 3J = 8.4 Hz, 2H, CH2, A), 3.23 (s, 6H, CH3O, B), 3.29 (s, 6H, CH3O, A), 3.37 (s, 6H, CH3O, B), 3.56 3.29 (s, 6H, CH3O, A), 3.68 (s, 6H+6H, CH3N, A, B), 3.82 (dd, 3J = 9.0 Hz, 3J = 8.4 Hz, 2H, CH, A), 3.85 (dd, 3J = 9.4 Hz, 3J = 8.2 Hz, 2H, CH, B), 6.66 (broad signal, ν1/2 = 34 Hz, 2H, Ind, A), 6.69 (br. d, 3J = 7.3 Hz, 2H, Ind, B), 6.76 (br. s, 2H, Ind, B), 6.79 (br. d, 3J = 7.3 Hz, 2H, Ar, A), 7.07–7.13 (m, 2H + 2H, Ind, A, B), 7.18–7.21 (m, 2H + 2H, Ind, A, B). 13C-NMR (CDCl3, 125 MHz) for mixture of isomers: δ = 18.3 (2 × CH2, A), 19.7 (2 × CH2, B), 24.6 (CH2, A), 25.7 (CH2, B), 31.2 (2 × CH, A), 31.5 (2 × CH, B), 32.7 (2 × CH3N, B), 32.8 (2 × CH3N, A), 38.4 (2 × C, A), 38.7 (2 × C, B), 52.1 (2 × CH3O + 2 × CH3O, A, B), 52.2 (CH3O + CH3O, A, B), 52.4 (CH3O + CH3O, A, B), 108.9 (2 × CH, Ind, B), 109.0 (2 × CH, Ind, A), 114.9 (2 × C + 2 × C, Ind, A, B), 116.4 (2 × CH, Ind, A), 117.7 (2 × CH, A, B), 120.87 (2 × CH, Ar, B), 120.94 (2 × CH, Ind, A), 127.0 (2 × C, Ind, A), 127.4 (2 × C, Ind, B), 127.6 (2 × C, Ind, B), 127.7 (2 × C, Ind, A), 128.5 (2 × CH, Ind, A), 129.2 (2 × CH, Ind, B), 137.9 (2 × C + 2 × C, Ind, A, B), 167.27 (2 × CO2Me, B), 167.33 (2 × CO2Me, A), 169.8 (2 × CO2Me, B), 170.0 (2 × CO2Me, A). IR (сm−1) 3618, 3442, 3055, 3022, 2950, 2924, 2852, 2254, 1728, 1608, 1570, 1550, 1456, 1437, 1371, 1330, 1281, 1209, 1128, 1103, 910. HRMS ESI-TOF: m/z = 587.2390 [M + H]+ (587.2388 сalcd for C33H35N2O8).
Dimethyl 2-[2-(5-fluoro-2-hydroxyphenyl)ethyl]malonate (10). To a solution of cyclopropane 1d (236 mg, 1 mmol) in MeOH (10 mL) Zn dust (0.69 g, 0.01 mol) and glacial acetic acid (0.29 mL, 5 mmol) were added. The reaction mixture was refluxed for 1 h, cooled, remaining Zn was filtered off and washed with ethyl acetate (5 mL). Filtrates were combined and diluted with water (8 mL), MeOH was evaporated under reduced pressure and aqueous phase was extracted with ethyl acetate (3 × 10 mL). Combined organic fractions were washed with saturated NaHCO3 solution (2 × 5 mL), dried with Na2SO4 and concentrated in vacuo. The resulting residue was purified by flash chromatography on silica gel (eluent: 10–50% ethyl acetate in petroleum ether) to afford product 10 as a colorless liquid; Rf = 0.63 (ethyl acetate:petroleum ether 1:2). 1H-NMR (CDCl3, 400 MHz) δ = 2.11–2.17 (m, 2H, CH2), 2.61–2.65 (m, 2H, CH2), 3.44 (dd, 3J = 7.6 Hz, 3J = 6.5 Hz, 1H, CH), 3.77 (s, 6H, 2 × CH3O), 6.10 (br. s, 1H, OH), 6.76–6.81 (m, 3H, Ar). 13C-NMR (CDCl3, 100 MHz) δ = 28.0 (CH2), 28.8 (CH2), 50.6 (CH), 53.0 (2 × CH3O), 114.1 (d, 2JCF = 23 Hz, CH, Ar), 116.5 (d, 2JCF = 23 Hz, CH, Ar), 116.9 (d, 3JCF = 8 Hz, CH, Ar), 127.9 (d, 3JCF = 7 Hz, C, Ar), 150.4 (C, Ar), 157.4 (d, 1JCF = 238 Hz, C, Ar), 170.3 (2 × CO2Me). IR (сm−1) 3442, 3394, 2954, 1730, 1620, 1510, 1437, 1342, 1273, 1230, 1186, 1101, 1079, 1045. HRMS ESI-TOF: m/z = 271.0968 [M + H]+ (271.0976 сalcd for C13H16 FO5).
4. Conclusions
The simple method for the synthesis of cyclopropa[c]coumarins based on the acid-induced intramolecular transesterification of 2-(2-hydroxyaryl)cyclopropane-1,1-dicarboxylates has been developed. Various functional groups in the aromatic ring including alkyl, halogen and nitro functionalities were shown to be tolerant to the optimized reaction conditions. The proposed method was, however, found to be inefficient for the preparation of the corresponding lactones with a larger ring size. The investigation of the reactivity of the synthesized compounds is in progress.
Supplementary Materials
Copies of 1H NMR and 13C NMR spectra for synthesized compounds as well as 2D (HSQC and HMBC) NMR spectra for selected compounds are available online.
Author Contributions
Conceptualization, O.A.I. and I.V.T.; methodology, O.A.I.; investigation, O.A.I., V.A.A., A.O.C., and I.I.L.; resources, I.V.T. and L.G.V.; writing, O.A.I., A.O.C., and I.V.T.; supervision, O.A.I. and I.V.T.; project administration, O.A.I.; funding acquisition, A.O.C., I.V.T., and L.G.V.
Funding
This research was funded by the Russia Foundation for Basic Research (grant 18-03-00954) and the Ministry of Education and Science of the Russia Federation (Project 4.5386.2017/8.9 and grant MK-1567.2018.3).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef]
- Pereira, T.M.; Franco, D.P.; Vitorio, F.; Kummerle, A.E. Coumarin Compounds in Medicinal Chemistry: Some Important Examples from the Last Years. Curr. Top. Med. Chem. 2018, 18, 124–148. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, D.; Godugu, C.; Dubey, P.K. A Review of Pharmacological Properties of Coumarins. Mini-Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar] [CrossRef] [PubMed]
- Detsi, A.; Kontogiorgis, C.; Hajipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Exp. Opin. Ther. Patents 2017, 27, 1201–1226. [Google Scholar] [CrossRef] [PubMed]
- Tejada, S.; Martorell, M.; Capo, X.; Tur, J.A.; Pons, A.; Sureda, A. Coumarin and Derivates as Lipid Lowering Agents. Curr. Top. Med. Chem. 2017, 17, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.-W.; Lv, Z.-S.; Feng, L.-S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Dandriyal, J.; Singla, R.; Kumar, M.; Jaitak, V. Recent developments of C-4 substituted coumarin derivatives as anticancer agents. Eur. J. Med. Chem. 2016, 119, 141–168. [Google Scholar] [CrossRef]
- Penta, S. (Ed.) Advances in Structure and Activity Relationship of Coumarin Derivatives; Academic Press-Elsevier: Amsterdam, The Netherlands, 2015; ISBN 978-0-12-803797-3. [Google Scholar]
- Medina, F.G.; Marrero, J.G.; Macias-Alonso, M.; Gonzalez, M.C.; Cordova-Guerrero, I.; Teissier Garcia, A.G.; Osegueda-Robles, S. Coumarin heterocyclic derivatives: Chemical synthesis and biological activity. Nat. Prod. Rep. 2015, 32, 1472–1507. [Google Scholar] [CrossRef]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef]
- Keri, R.S.; Sasidhar, B.S.; Nagaraja, B.M.; Santos, M.A. Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem. 2015, 100, 257–269. [Google Scholar] [CrossRef]
- Hoerr, R.; Noeldner, M. Ensaculin (KA-672.HCl): A Multitransmitter Approach to Dementia Treatment. CNS Drug Rev. 2002, 8, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Steglich, W.; Fugmann, B.; Lang-Fugmann, S. RÖMPP Encyclopedia Natural Products, 1st ed.; Georg Thieme Verlag: Stuttgart, Germany, 2000; pp. 187–188. ISBN 3-13-117711-X. [Google Scholar]
- Enejoh, O.S.; Suleiman, M.M. Anthelmintics and Their Application in Veterinary Medicine. Res. Med. Eng. Sci. 2017, 2. [Google Scholar] [CrossRef]
- Ravina, E. The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs; Wiley-VCH: Weinheim, Germany, 2011; p. 148. ISBN 978-3-527-32669-3. [Google Scholar]
- Lee, M.M.; Peterson, B.R. Quantification of Small Molecule–Protein Interactions Using FRET between Tryptophan and the Pacific Blue Fluorophore. ACS Omega 2016, 1, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Widman, O. Über eine neue Gruppe von Cyclopropan-Derivaten. I: Die Einwirkung von Phenyl-acylhalogeniden auf 3-Acidyl-cumarine bei Gegenwart von Natrium-alkoholat. Ber. Dtsch. Chem. Ges. 1918, 51, 533–541. [Google Scholar] [CrossRef]
- Widman, O. Über eine neue Gruppe von Cyclopropan-Derivaten. II: Die Einwirkung einiger Analoga der Phenacylhalogenide auf 3-Acidyl-cumarine. Ber. Dtsch. Chem. Ges. 1918, 51, 907–911. [Google Scholar] [CrossRef]
- Bojilova, A.; Trendafilova, A.; Ivanov, C.; Rodios, N.A. Cyclopropanation reaction of 3-acyl-2H-1-benzopyran-2-ones with phenacylbromide in phase transfer systems. Tetrahedron 1993, 49, 2275–2286. [Google Scholar] [CrossRef]
- Bojilova, A.; Videnova, I.; Ivanov, C.; Rodios, N.A.; Terzis, A.; Raptopoulou, C.P. Regio- and stereo-Selective 1,3-dipolar cycloaddition reactions of ethyl diazoacetate to 3-substituted 2H-1-benzopyran-2-ones. Tetrahedron 1994, 50, 13023–13036. [Google Scholar] [CrossRef]
- Wawzonek, S.; Morreal, C.E. The Action of Alkali on 3,4-Phenacylidene-3-acetylcoumarin. J. Am. Chem. Soc. 1960, 82, 439–441. [Google Scholar] [CrossRef]
- Abdallah, H.; Gree, R.; Carrie, R. Reactions du dimethylacetal du diazoacetaldehyde avec des coumarines at une chromone electrophiles. Bull. Soc. Chim. Fr. 1984, 2, 338–344. [Google Scholar]
- Shchepin, V.V.; Silaichev, P.S.; Kodess, M.I. Reaction of Zinc Enolates Prepared from 2,2-Dibromoindan-1-one or 2,2-Dibromo-1-tetralone and Zinc with 2-Oxochromen-3-carboxylic Acid Derivatives. Russ. J. Org. Chem. 2007, 43, 1441–1445. [Google Scholar] [CrossRef]
- Shchepin, V.V.; Silaichev, P.S.; Stepanyan, Y.G.; Kalyuzhnyi, M.M.; Russkikh, N.Y.; Kodess, M.I. Cyclopropanation of N-Substituted 3-Aryl-2-cyanoprop-2-enamides and Derivatives of 5,5-Dimethyl-2-oxo-2,5-dihydrofuran-3-carboxylic Acid and 2-Oxochromene-3-carboxylic Acid with Bromine-Containing Zinc Enolates. Russ. J. Org. Chem. 2006, 42, 973–980. [Google Scholar] [CrossRef]
- Shchepin, V.V.; Silaichev, P.S.; Vakhrin, M.I.; Russkikh, N.Y. Cyclopropanation of N-Substituted 2-Oxochromene-3-carboxamides and 3-Oxobenzo[f]chromene-2-carboxamides with Bromine-containing Zinc Enolate Prepared from α,α-Dibromopinacolin and Zinc. Russ. J. Org. Chem. 2005, 41, 1219–1221. [Google Scholar] [CrossRef]
- Bojilova, A. Interaction of Trichloroacetic Acid with Some 3-Substituted 2H-1-Benzopyran-2-ones. Synth. Commun. 1990, 20, 1967–1976. [Google Scholar] [CrossRef]
- Guo, J.; Liu, Y.; Li, X.; Liu, X.; Lin, L.; Feng, X. Nickel(II)-catalyzed enantioselective cyclopropanation of 3-alkenyl-oxindoles with phenyliodonium ylide via free carbene. Chem. Sci. 2016, 7, 2717–2721. [Google Scholar] [CrossRef]
- Huang, X.; Klimczyk, S.; Veiros, L.F.; Maulide, N. Stereoselective intramolecular cyclopropanation through catalytic olefin activation. Chem. Sci. 2013, 4, 1105–1110. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Gololobov, Y.G.; Nesmeyanov, A.N.; Lysenko, V.P.; Bordeskul, I.E. Twenty-five years of dimethylsulfoxonium methylide (Corey’s reagent). Tetrahedron 1987, 43, 2609–2651. [Google Scholar] [CrossRef]
- Yamashita, M.; Okuyama, K.; Kawajiri, T.; Takada, A.; Inagaki, Y.; Nakano, H.; Tomiyama, M.; Ohnaka, A.; Terayama, I.; Kawasaki, I.; et al. A novel tandem reaction of 3-substituted coumarins with two equivalents of dimethylsulfoxonium ylide to 2-substituted cyclopenta[b]benzofuran-3-ol derivatives. Tetrahedron 2002, 58, 1497–1505. [Google Scholar] [CrossRef]
- Arimitsu, K.; Nomura, S.; Iwasaki, H.; Ozeki, M.; Yamashita, M. First total synthesis of (±)-adunctin B. Tetrahedron Lett. 2011, 52, 7046–7048. [Google Scholar] [CrossRef]
- Yamashita, M.; Okuyama, K.; Kawasaki, I.; Ohta, S. One-Step Synthesis of 2-Substituted Cyclopenta[b]benzofuran-3-ol Derivatives from 3-Substituted Coumarins. Tetrahedron Lett. 1995, 36, 5603–5605. [Google Scholar] [CrossRef]
- Lee, J.; Ko, K.M.; Kim, S.-G. Ni(ClO4)2-Catalyzed Friedel-Crafts Reaction of Coumarin-Fused Donor-Acceptor Cyclopropanes with Indoles: Stereoselective Synthesis of trans-3,4-Disubstituted-3,4-dihydrocoumarins. Eur. J. Org. Chem. 2018, 4166–4170. [Google Scholar] [CrossRef]
- Choi, S.; Kim, S.-G. (±)-Methyl 1,1a,2,7b-Tetrahydro-2-oxocyclopropa[c]chromene-1a-carboxylate. Molbank 2017, 2017, M966. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Andronov, V.A.; Vasin, V.S.; Shumsky, A.N.; Rybakov, V.B.; Voskressensky, L.G.; Trushkov, I.V. Expanding the Reactivity of Donor-Acceptor Cyclopropanes: Synthesis of Benzannulated Five-Membered Heterocycles via Intramolecular Attack of a Pendant Nucleophilic Group. Org. Lett. 2018, 20, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Bezzubov, S.I.; Melnikov, M.Y.; Bludynina, E.M. Donor-acceptor cyclopropanes as ortho-quinone methide equivalents in formal (4 + 2)-cycloaddition to alkenes. Org. Biomol. Chem. 2018, 16, 3897–3909. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V.; Melnikov, M.Y. Ring Opening of Donor-Acceptor Cyclopropanes with the Azide Ion: A Tool for Construction of N-Heterocycles. Chem. Eur. J. 2015, 21, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Borpatra, P.J.; Deka, B.; Rajbongshi, B.K.; Deb, M.L.; Baruah, P.K. One-pot sequential multi-component reaction: Synthesis of 3-substituted indoles. Synth. Commun. 2018, 48, 2074–2082. [Google Scholar] [CrossRef]
- Misztal, S.; Mokrosz, J.L.; Bielecka, Z. Oxidation of 2,3-Bis(hydroxymethyl)indole. J. Prakt. Chem. 1989, 331, 751–756. [Google Scholar] [CrossRef]
- Fusco, R.; Sannicolo, F. Rearrangement of Arylhydrazones of α,β-Unsaturated Carbonyl Compounds in Polyphosphoric Acid. 6. J. Org. Chem. 1984, 49, 4374–4378. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Villemson, E.V.; Latyshev, G.V.; Bezzubov, S.I.; Majouga, A.G.; Melnikov, M.Y.; Bludynina, E.M. Regioselective Hydrogenolysis of Donor-Acceptor Cyclopropanes with Zn-AcOH Reductive System. J. Org. Chem. 2017, 82, 9537–9549. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanova, O.A.; Rakhmankulov, E.R.; Budynina, E.M.; Trushkov, I.V.; Melnikov, M.Y. Lewis Acid-Catalyzed Isomerization of 2-Arylcyclopropane-1,1-dicarboxylates: A New Efficient Route to 2-Styrylmalonates. Adv. Synth. Catal. 2010, 352, 3179–3184. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).