
Sulfonylimino Group Transfer Reaction Using Imino-λ3-iodanes with I2 as Catalyst Under Metal-free Conditions

 , , , and
, , , and
Abstract

1. Introduction
2. Results
2.1. Optimization of Reaction Conditions
2.2. Scope of Reactions
3. Discussion
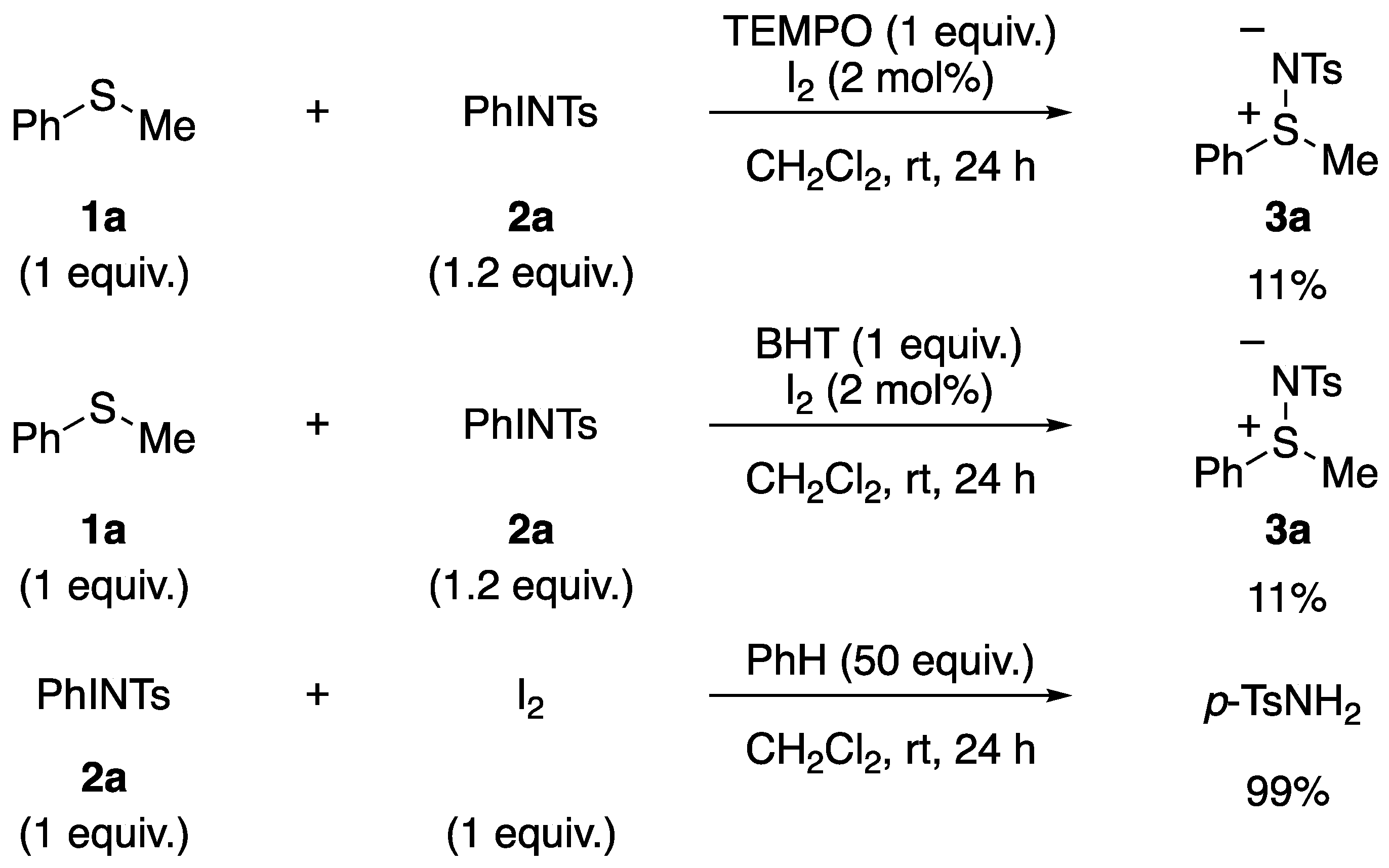
3.1. Mechanistic Study
3.2. Proposed Reaction Mechanism
4. Materials and Methods
4.1. General Experimental Remarks
4.2. General Procedure for Imination of Sulfides 1 with Imino-phenyl-λ3-iodane 2 in the Presence of I2
4.3. Large Scale Reaction of 1a
4.4. Reaction of Triphenylphosphine 4
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aggarwal, V.K.; Winn, C.L. Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis. Acc. Chem. Res. 2004, 37, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K. Catalytic asymmetric epoxidation and aziridination mediated by sulfur ylides. Evolution of a project. Synlett 1998, 329–336. [Google Scholar] [CrossRef]
- Lu, L.-Q.; Li, T.-R.; Wang, Q.; Xiao, W.-J. Beyond sulfide-centric catalysis: Recent advances in the catalytic cyclization reactions of sulfur ylides. Chem. Soc. Rev. 2017, 46, 4135–4149. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-L.; Tang, Y. Ylide-Initiated Michael Addition-Cyclization Reactions beyond Cyclopropanes. Acc. Chem. Res. 2008, 41, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Richardson, J. Product class 1: Sulfur ylides. Sci. Synth. 2004, 27, 21–104. [Google Scholar] [CrossRef]
- Bizet, V.; Hendriks, C.M.M.; Bolm, C. Sulfur imidations: Access to sulfimides and sulfoximines. Chem. Soc. Rev. 2015, 44, 3378–3390. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, T.L.; Moody, C.J. The chemistry of sulfilimines. Chem. Rev. 1977, 77, 409–435. [Google Scholar] [CrossRef]
- Taylor, P.C. Sulfimides (sulfilimines): Applications in stereoselective synthesis. Sulfur Rep. 1999, 21, 241–280. [Google Scholar] [CrossRef]
- Yoshimura, T.; Akasaka, T.; Furukawa, N.; Oae, S. Free sulfilimines. 9. One step synthesis of optically active aziridine with optically active o-methoxyphenyl phenyl sulfilimine and olefin. Heterocycles 1977, 7, 287–291. [Google Scholar] [CrossRef]
- Baird, C.P.; Taylor, P.C. Asymmetric synthesis of epoxides using chiral sulfimides. J. Chem. Soc. Chem. Commun. 1995, 893–894. [Google Scholar] [CrossRef]
- Thakur, V.V.; Kumar, N.S.C.R.; Sudalai, A. Sulfilimine palladacycles: Stable and efficient catalysts for carbon-carbon coupling reactions. Tetrahedron Lett. 2004, 45, 2915–2918. [Google Scholar] [CrossRef]
- Johnson, C.R. Utilization of sulfoximines and derivatives as reagents for organic synthesis. Acc. Chem. Res. 1973, 6, 341–347. [Google Scholar] [CrossRef]
- Lüecking, U. Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem. Int. Ed. 2013, 52, 9399–9408. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.G. Applications of chiral sulfoximines to diastereoselective and catalytic asymmetric synthesis. Sulfur Rep. 1999, 21, 281–334. [Google Scholar] [CrossRef]
- Reggelin, M.; Zur, C. Sulfoximines. Structures, properties, and synthetic applications. Synthesis 2000, 1–64. [Google Scholar] [CrossRef]
- Garcia Mancheno, O.; Bolm, C. Synthesis of N-(1H)-Tetrazole Sulfoximines. Org. Lett. 2007, 9, 2951–2954. [Google Scholar] [CrossRef] [PubMed]
- Garcia Mancheno, O.; Bistri, O.; Bolm, C. Iodinane- and Metal-Free Synthesis of N-Cyano Sulfilimines: Novel and Easy Access of NH-Sulfoximines. Org. Lett. 2007, 9, 3809–3811. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.B.; Li, S.N.; Zhou, Z.H.; Shen, H.S.; Li, X.; Sun, Q.; He, L.; Xue, Y. Sulfur–Nitrogen and Carbon–Nitrogen Bond Formation by Intermolecular Imination and Amidation without Catalyst. Eur. J. Org. Chem. 2012, 1554–1562. [Google Scholar] [CrossRef]
- Ochiai, M.; Naito, M.; Miyamoto, K.; Hayashi, S.; Nakanishi, W. Imination of Sulfides and Sulfoxides with Sulfonylimino-λ3-Bromane under Mild, Metal-Free Conditions. Chem. Eur. J. 2010, 16, 8713–8718. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Saito, A.; Zhdankin, V.V. Iodonium Salts as Benzyne Precursors. Chem. Eur. J. 2018, 24, 15156–15166. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Kita, Y. Metal-Free Oxidative Biaryl Coupling by Hypervalent Iodine Reagents. Curr. Org. Chem. 2016, 20, 580–615. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent Iodine Chemistry. In Topics in Current Chemistry; Springer: Cham, Switzerland, 2016; Volume 373, pp. 1–310. ISBN 978-3-319-33733-3. [Google Scholar]
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds. In Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2014; ISBN 9781118341155. [Google Scholar]
- Dauban, P.; Dodd, R.H. Iminoiodanes and C-N bond formation in organic synthesis. Synlett 2003, 1571–1586. [Google Scholar] [CrossRef]
- Karila, D.; Dodd, R.H. Recent progress in iminoiodane-mediated aziridination of olefins. Curr. Org. Chem. 2011, 15, 1507–1538. [Google Scholar] [CrossRef]
- Chang, J.W.W.; Ton, T.M.U.; Chan, P.W.H. Transition-metal-catalyzed aminations and aziridinations of C-H and C-C bonds with iminoiodinanes. Chem. Rec. 2011, 11, 331–357. [Google Scholar] [CrossRef] [PubMed]
- Takada, H.; Nishibayashi, Y.; Ohe, K.; Uemura, S. Novel asymmetric catalytic synthesis of sulfimides. Chem. Commun. (Cambridge) 1996, 931–932. [Google Scholar] [CrossRef]
- Takada, H.; Nishibayashi, Y.; Ohe, K.; Uemura, S.; Baird, C.P.; Sparey, T.J.; Taylor, P.C. Catalytic Asymmetric Sulfimidation. J. Org. Chem. 1997, 62, 6512–6518. [Google Scholar] [CrossRef]
- Nishikori, H.; Ohta, C.; Oberlin, E.; Irie, R.; Katsuki, T. Mn-salen catalyzed nitrene transfer reaction: Enantioselective imidation of alkyl aryl sulfides. Tetrahedron 1999, 55, 13937–13946. [Google Scholar] [CrossRef]
- Miyake, Y.; Takada, H.; Ohe, K.; Uemura, S. Catalytic asymmetric sulfimidation of 1,3-dithianes. J. Chem. Soc. Perkin Trans. 1 1998, 2373–2376. [Google Scholar] [CrossRef]
- Kantam, M.L.; Kavita, B.; Neeraja, V.; Haritha, Y.; Chaudhuri, M.K.; Dehury, S.K. Heterogeneous catalytic sulfimidation using immobilized Cu(acac)2. Adv. Synth. Catal. 2005, 347, 641–645. [Google Scholar] [CrossRef]
- Wang, J.; Frings, M.; Bolm, C. Enantioselective Nitrene Transfer to Sulfides Catalyzed by a Chiral Iron Complex. Angew. Chem. Int. Ed. 2013, 52, 8661–8665. [Google Scholar] [CrossRef]
- Lamar, A.A.; Nicholas, K.M. Iodine-Catalyzed Aminosulfonation of Hydrocarbons by Imidoiodinanes. a Synthetic and Mechanistic Investigation. J. Org. Chem. 2010, 75, 7644–7650. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.D.; Scott, K.A.; DeMier, B.C.; Morgan, H.R.; Macgruder, J.A.; Lamar, A.A. Formation of N-sulfonyl imines from iminoiodinanes by iodine-promoted, N-centered radical sulfonamidation of aldehydes. Org. Biomol. Chem. 2017, 15, 9209–9216. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, K.; Kosaka, T.; Minakata, S. Metal-Free Aziridination of Styrene Derivatives with Iminoiodinane Catalyzed by a Combination of Iodine and Ammonium Iodide. Org. Lett. 2013, 15, 4858–4861. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.F.; Hair, N.J.; Morris, D.G. Structural investigations of ylides. IV. Crystal and molecular structure of N-(p-toluenesulfonyl)iminotriphenylphosphorane. Acta Crystallogr. Sect. B 1974, 30, 221–225. [Google Scholar] [CrossRef]
- Yamada, Y.; Yamamoto, T.; Okawara, M. Synthesis and reaction of new type iodine-nitrogen ylide, N-tosyliminoiodinane. Chem. Lett. 1975, 361–362. [Google Scholar] [CrossRef]
- Yoshimura, A.; Nemykin, V.N.; Zhdankin, V.V. o-Alkoxyphenyliminoiodanes: Highly Efficient Reagents for the Catalytic Aziridination of Alkenes and the Metal-Free Amination of Organic Substrates. Chem. Eur. J. 2011, 17, 10538–10541. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, A.C.; Hancock, E.N.; Anders, E.J.; Tierney, M.M.; Morgan, H.R.; Scott, K.A.; Lamar, A.A. Visible-light-mediated, nitrogen-centered radical amination of tertiary alkyl halides under metal-free conditions to form α-tertiary amines. Org. Biomol. Chem. 2016, 14, 4387–4392. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Luedtke, M.W.; Zhdankin, V.V. (Tosylimino)phenyl-λ3-iodane as a Reagent for the Synthesis of Methyl Carbamates via Hofmann Rearrangement of Aromatic and Aliphatic Carboxamides. J. Org. Chem. 2012, 77, 2087–2091. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R.H. Copper(I) Catalyzed Regioselective Asymmetric Alkoxyamination of Aryl Enamide Derivatives. Org. Lett. 2011, 13, 5792–5795. [Google Scholar] [CrossRef] [PubMed]
- Besenyei, G.; Nemeth, S.; Simandi, L.I. A new method for the preparation of (arylsulfonyliminoiodo)benzenes. Tetrahedron Lett. 1993, 34, 6105–6106. [Google Scholar] [CrossRef]
- Combee, L.A.; Raya, B.; Wang, D.; Hilinski, M.K. Organocatalytic nitrenoid transfer: Metal-free selective intermolecular C(sp3)-H amination catalyzed by an iminium salt. Chem. Sci. 2018, 9, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Sutherlin, K.D.; Vardhaman, A.K.; Yan, J.J.; Park, S.; Lee, Y.-M.; Jang, S.; Lu, X.; Ohta, T.; Ogura, T.; et al. A Mononuclear Nonheme Iron(V)-Imido Complex. J. Am. Chem. Soc. 2017, 139, 8800–8803. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Tatami, A.; Maeda, T.; Ju Kim, B.; Kawashima, W.; Yoshimura, T.; Abe, H.; Akasaka, T. Generation of Nitrene by the Photolysis of N-Substituted Iminodibenzothiophene. J. Org. Chem. 2008, 73, 7159–7163. [Google Scholar] [CrossRef] [PubMed]
- Kapovits, I.; Ruff, F.; Kucsman, A. Basicity of sulfur(IV)-nitrogen(sp2)-sulfur(VI) group in N-sulfonyl sulfilimines. Tetrahedron 1972, 28, 4413–4417. [Google Scholar] [CrossRef]
- Aida, T.; Furukawa, N.; Oae, S. Mechanism of the reaction of N-arylsulfonylsulfimides with trivalent phosphorus compounds. J. Chem. Soc. Perkin Trans. 2 1976, 1438–1444. [Google Scholar] [CrossRef]
- Giribabu, L.; Singh, S.P.; Patil, N.M.; Kantam, M.L.; Gupte, S.P.; Chaudhari, R.V. Highly Efficient Sulfimidation of 1,3-Dithianes by Cu(I) Complexes. Synth. Commun. 2008, 38, 619–625. [Google Scholar] [CrossRef]
- Marzinzik, A.L.; Sharpless, K.B. A simple method for the preparation of N-sulfonylsulfilimines from sulfides. J. Org. Chem. 2001, 66, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Chen, Z.-C. Hypervalent iodine in synthesis XXXII: A novel way for the synthesis of N-sulfonylsulfilimines from sulfides and sulfonamides using iodosobenzene diacetate. Synth. Commun. 1999, 29, 4443–4449. [Google Scholar] [CrossRef]
- Ruff, F.; Komoto, K.; Furukawa, N.; Oae, S. Steric effects in the reaction of alkylphenyl and dialkylsulfides with chloramine-T. Tetrahedron 1976, 32, 2763–2767. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yoshida, D.; Hojyo, J.; Terauchi, H. Preparation of tetraalkylammonium N-chloro-p-toluenesulfonamides and their application to imination of phosphorus compounds and sulfides. Bull. Chem. Soc. Jpn. 1984, 57, 3341–3342. [Google Scholar] [CrossRef]
- Hopper, R.J. N-Sulfonyl-sulfilimines as premature vulcanization inhibitors. Rubber Chem. Technol. 1980, 53, 1106–1116. [Google Scholar] [CrossRef]
- Okamura, H.; Bolm, C. Rhodium-Catalyzed Imination of Sulfoxides and Sulfides: Efficient Preparation of N-Unsubstituted Sulfoximines and Sulfilimines. Org. Lett. 2004, 6, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Akutagawa, K.; Furukawa, N.; Oae, S. Preparation of N-(arylsulfonyl)sulfoximines by oxidation of N-(arylsulfonyl)sulfilimines with sodium hypochlorite in a two-phase system. J. Org. Chem. 1984, 49, 2282–2284. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2a and 3a are available from the authors. |





{kind=link}
{kind=link}
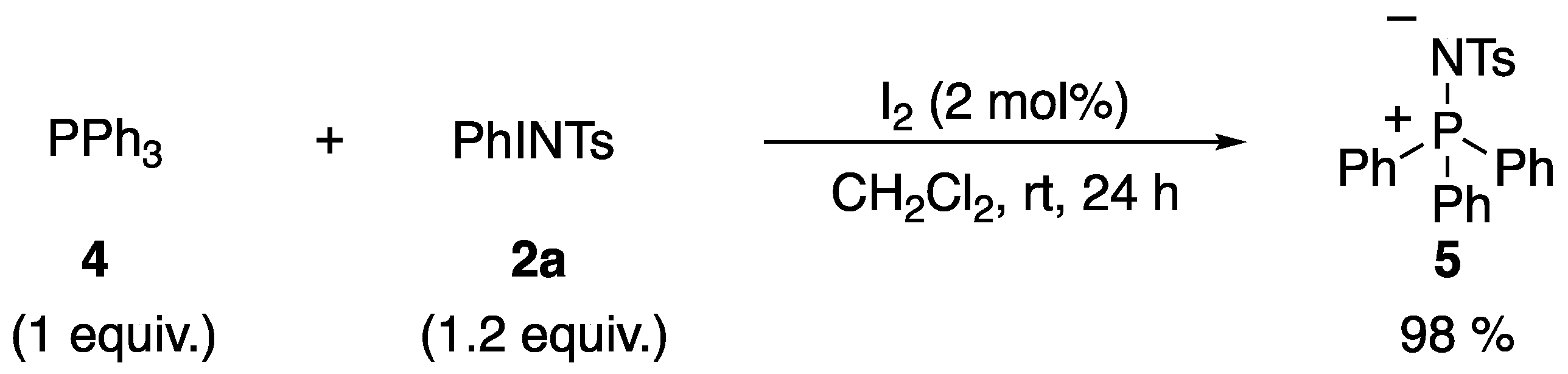{kind=link}
{kind=link}
{kind=link}

| Entry | Time (h) | Solvent | I2 (mol%) | TBAI (mol%) | 3a Yield (%) 2 |
|---|---|---|---|---|---|
| 1 | 3 | MeCN | 20 | 10 | 81 (80) |
| 2 | 3 | Hexane | 20 | 10 | 15 |
| 3 | 3 | AcOEt | 20 | 10 | 49 |
| 4 | 3 | MeOH | 20 | 10 | 77 |
| 5 | 3 | THF | 20 | 10 | 4 |
| 6 | 3 | Et2O | 20 | 10 | Trace |
| 7 | 3 | PhH | 20 | 10 | 47 |
| 8 | 3 | CCl4 | 20 | 10 | 7 |
| 9 | 3 | CH2Cl2 | 20 | 10 | 83 (83) |
| 10 | 3 | CH2Cl2 | 20 | none | 84 |
| 11 | 3 | CH2Cl2 | none | 10 | 7 |
| 12 | 3 | CH2Cl2 | none | none | 5 |
| 13 | 3 | CH2Cl2 | 10 | none | 72 |
| 14 | 24 | CH2Cl2 | 10 | none | 87 |
| 15 | 24 | CH2Cl2 | 5 | none | 91 |
| 16 | 24 | CH2Cl2 | 2 | none | 92 (88) |
| 17 | 24 | CH2Cl2 | 1 | none | 68 |
| 18 | 48 | CH2Cl2 | 1 | none | 66 |
| 19 3 | 24 | CH2Cl2 | 2 | none | 77 |

| Entry | 1 R1, R2 | 2 R3 | 3 Yield(%) 2 |
|---|---|---|---|
| 1 | 1a Ph, Me | 2ap-Tol | 3a 88% (76%) 3 |
| 2 | 1bp-tol, Me | 2ap-Tol | 3b 72% |
| 3 | 1cp-MeOC6H4, Me | 2ap-Tol | 3c 78% |
| 4 | 1dp-ClC6H4, Me | 2ap-Tol | 3d 79% |
| 5 | 1em-ClC6H4, Me | 2ap-Tol | 3e 80% |
| 6 | 1fo-ClC6H4, Me | 2ap-Tol | 3f 68% |
| 7 | 1gp-BrC6H4, Me | 2ap-Tol | 3g 70% |
| 8 | 1hp-NCC6H4, Me | 2ap-Tol | 3h 86% |
| 9 | 1ip-NO2C6H4, Me | 2ap-Tol | 3i 59% |
| 10 | 1j Ph, Ph | 2ap-Tol | 3j 56% |
| 11 | 1k Ph, Bn | 2ap-Tol | 3k 65% |
| 12 | 1l Bn, Bn | 2ap-Tol | 3l 66% |
| 13 | 1mnBu, nBu | 2ap-Tol | 3m 79% |
| 14 | 1nnOctyl, nOctyl | 2ap-Tol | 3n 90% |
| 15 | 1o –(CH2)3– | 2ap-Tol | 3o 97% |
| 16 | 1p –(CH2)4– | 2ap-Tol | 3p 76% |
| 17 | 1qtBu, tBu | 2ap-Tol | 3q 8% |
| 18 | 1a Ph, Me | 2bp-NO2C6H4 | 3r 92% |
| 19 | 1a Ph, Me | 2co-NO2C6H4 | 3s 51% |
| 20 | 1a Ph, Me | 2d Ph | 3t 67% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshimura, A.; Makitalo, C.L.; Jarvi, M.E.; Shea, M.T.; Postnikov, P.S.; Rohde, G.T.; Zhdankin, V.V.; Saito, A.; Yusubov, M.S. Sulfonylimino Group Transfer Reaction Using Imino-λ3-iodanes with I2 as Catalyst Under Metal-free Conditions. Molecules 2019, 24, 979. https://doi.org/10.3390/molecules24050979
Yoshimura A, Makitalo CL, Jarvi ME, Shea MT, Postnikov PS, Rohde GT, Zhdankin VV, Saito A, Yusubov MS. Sulfonylimino Group Transfer Reaction Using Imino-λ3-iodanes with I2 as Catalyst Under Metal-free Conditions. Molecules. 2019; 24(5):979. https://doi.org/10.3390/molecules24050979
Chicago/Turabian StyleYoshimura, Akira, Cody L. Makitalo, Melissa E. Jarvi, Michael T. Shea, Pavel S. Postnikov, Gregory T. Rohde, Viktor V. Zhdankin, Akio Saito, and Mekhman S. Yusubov. 2019. "Sulfonylimino Group Transfer Reaction Using Imino-λ3-iodanes with I2 as Catalyst Under Metal-free Conditions" Molecules 24, no. 5: 979. https://doi.org/10.3390/molecules24050979
APA StyleYoshimura, A., Makitalo, C. L., Jarvi, M. E., Shea, M. T., Postnikov, P. S., Rohde, G. T., Zhdankin, V. V., Saito, A., & Yusubov, M. S. (2019). Sulfonylimino Group Transfer Reaction Using Imino-λ3-iodanes with I2 as Catalyst Under Metal-free Conditions. Molecules, 24(5), 979. https://doi.org/10.3390/molecules24050979