
APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
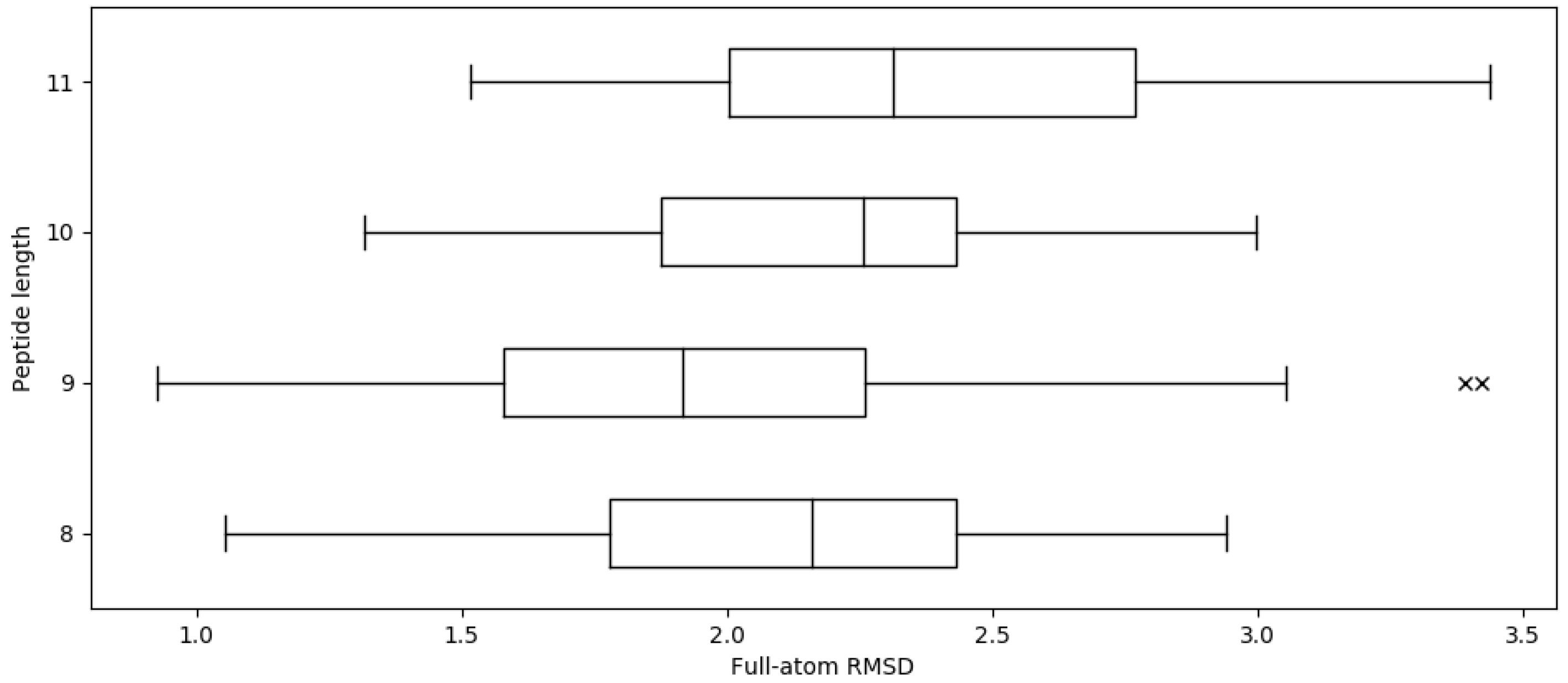
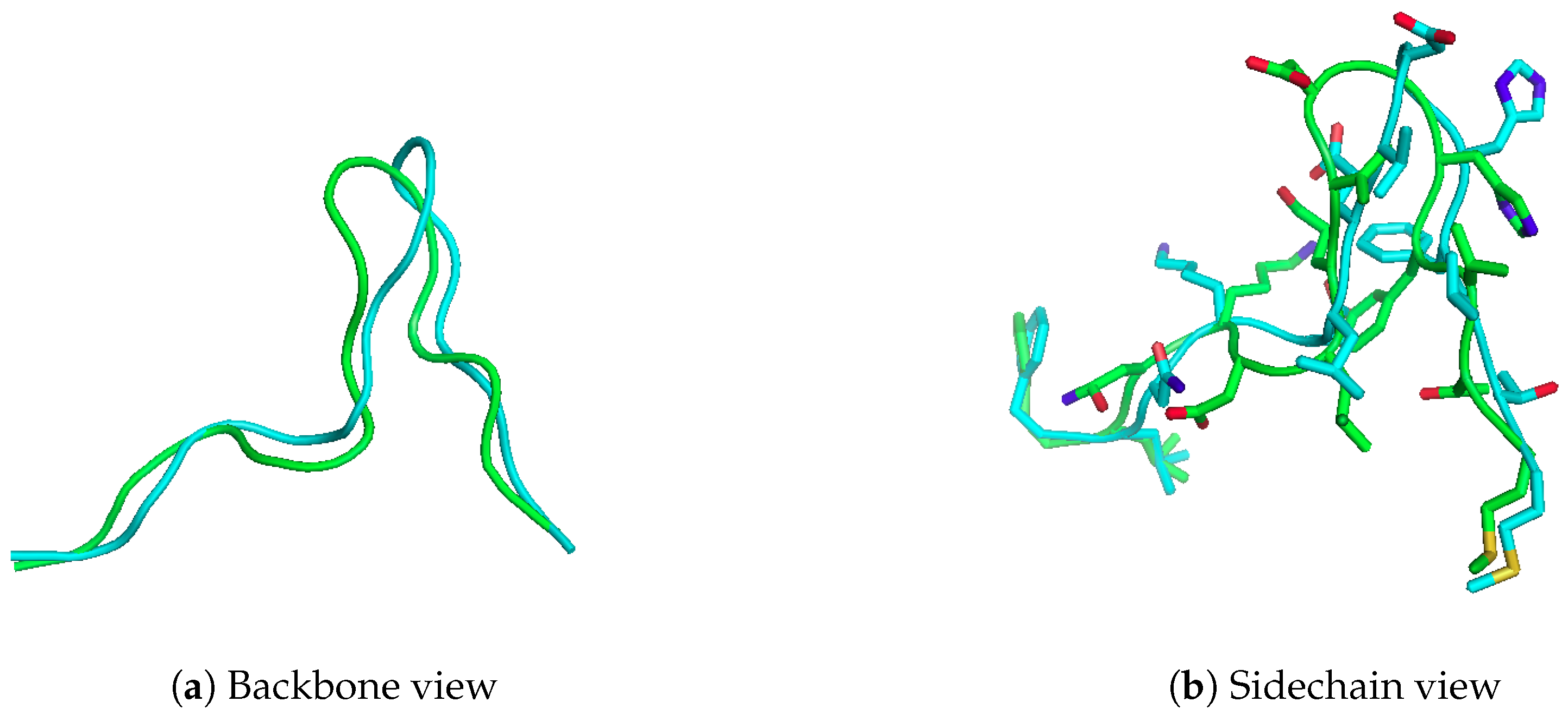
2.1. Reproducing Crystal Structures
2.2. Using Only Sequence Information
2.3. Application: Modelling Thousands of pMHCs
2.4. Application: Modelling a 15-mer Peptide
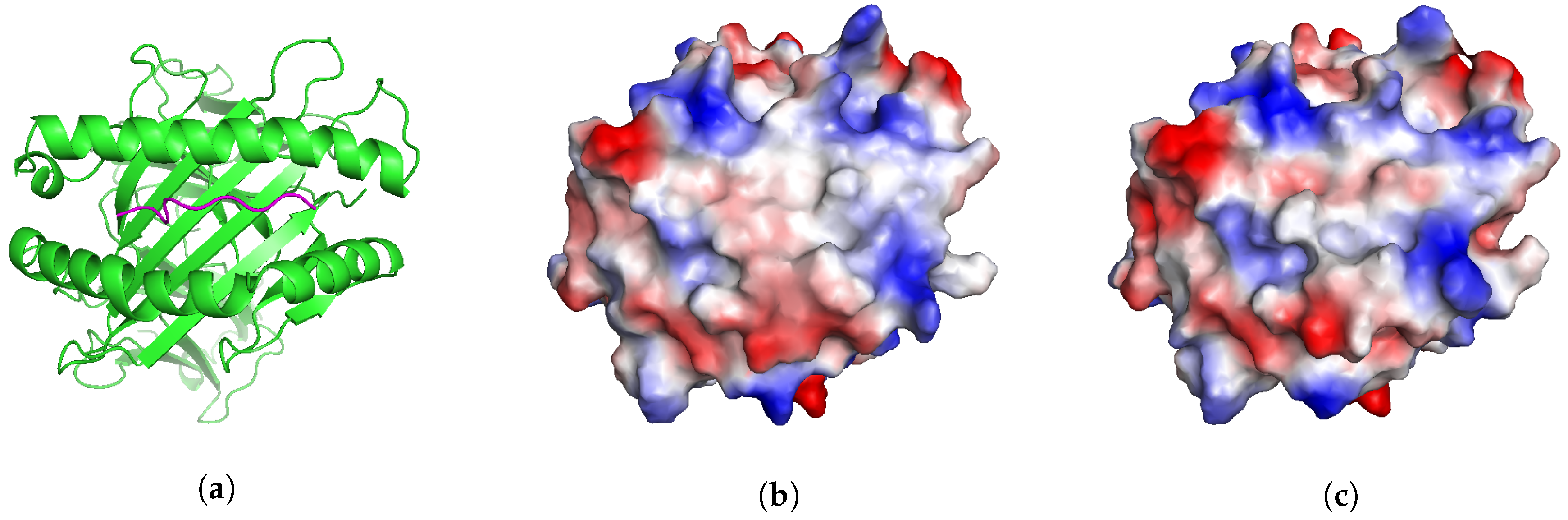
2.5. Application: Studying Cross-Reactivity
3. Materials and Methods
3.1. Input Preparation
3.2. Anchor Alignment
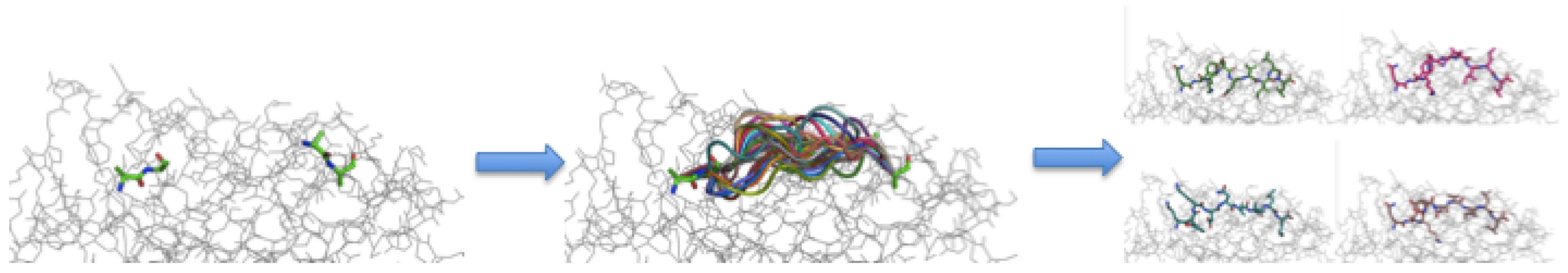
3.3. Peptide Backbone Sampling
3.4. Sidechain Sampling and Energy Minimization
3.5. Running APE-Gen for Multiple Rounds
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MHC | Major Histocompatibility Complex |
| pMHC | peptide-MHC |
| PDB | Protein data bank |
| RMSD | Root mean square deviation |
| C | alpha-carbon |
References
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M. Bioinformatics tools for the prediction of T-cell epitopes. Methods Mol. Biol. 2018, 1785, 269–281. [Google Scholar] [PubMed]
- Luo, H.; Ye, H.; Ng, H.W.; Shi, L.; Tong, W.; Mendrick, D.L.; Hong, H. Machine Learning Methods for Predicting HLA-Peptide Binding Activity. Bioinform. Biol. Insights 2015, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Abella, J.R.; Devaurs, D.; Rigo, M.M.; Kavraki, L.E. Structure-based methods for binding mode and binding affinity prediction for peptide-MHC complexes. Curr. Top. Med. Chem. 2018, 18, 2239–2255. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Devaurs, D.; Kavraki, L.E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discov. 2015, 10, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, R. Classification of current scoring functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Moll, M.; Devaurs, D.; Jackson, K.R.; Lizee, G.; Kavraki, L.E. DINC 2.0: A New Protein-Peptide Docking Webserver Using an Incremental Approach. Cancer Res. 2017, 77, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.; Zheng, Q.; Vajda, S.; DeLisi, C. Computing the Structure of Bound Peptides: Application to Antigen Recognition by Class I Major Histocompatibility Complex Receptors. J. Mol. Biol. 1993, 234, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Rognan, D.; Lauemøller, S.L.; Holm, A.; Buus, S.; Tschinke, V. Predicting Binding Affinities of Protein Ligands from Three-Dimensional Models: Application to Peptide Binding to Class I Major Histocompatibility Proteins. J. Med. Chem. 1999, 42, 4650–4658. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.C.; Tan, T.W.; Ranganathan, S. Modeling the structure of bound peptide ligands to major histocompatibility complex. Protein Sci. 2004, 13, 2523–2532. [Google Scholar] [CrossRef] [PubMed]
- Kyeong, H.H.; Choi, Y.; Kim, H.S. GradDock: rapid simulation and tailored ranking functions for peptide-MHC Class I docking. Bioinformatics 2018, 34, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Fodor, J.; Riley, B.T.; Borg, N.A.; Buckle, A.M. Previously Hidden Dynamics at the TCR-Peptide-MHC Interface Revealed. J. Immunol. 2018, 200, 4134–4145. [Google Scholar] [CrossRef] [PubMed]
- Knapp, B.; Demharter, S.; Esmaielbeiki, R.; Deane, C.M. Current status and future challenges in T-cell receptor/peptide/MHC molecular dynamics simulations. Brief. Bioinform. 2015, 16, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.; Campos-Rodriguez, R.; Rojas-Hernandez, S.; Contis-Montes de Oca, A.; Correa-Basurto, J. Predicting peptide vaccine candidates against H1N1 influenza virus through theoretical approaches. Immunol. Res. 2015, 62, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.; Correa-Basurto, J. Energetic and flexibility properties captured by long molecular dynamics simulations of a membrane-embedded pMHCII-TCR complex. Mol. Biosyst. 2016, 12, 1350–1366. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Wehmeyer, C.; Abualrous, E.T.; Wu, H.; Crabtree, M.D.; Schoneberg, J.; Clarke, J.; Freund, C.; Weikl, T.R.; Noe, F. Protein-peptide association kinetics beyond the seconds timescale from atomistic simulations. Nat. Commun. 2017, 8, 1095. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.; Kavraki, L.E.; Clementi, C. On the characterization of protein native state ensembles. Biophys. J. 2007, 92, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Canutescu, A.A.; Dunbrack, R.L. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003, 12, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A.; Kavraki, L.E. Modeling Structures and Motions of Loops in Protein Molecules. Entropy 2012, 14, 252–290. [Google Scholar] [CrossRef]
- Chys, P.; Chacon, P. Random Coordinate Descent with Spinor-matrices and Geometric Filters for Efficient Loop Closure. J. Chem. Theory Comput. 2013, 9, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Rigo, M.M.; Antunes, D.A.; Vaz de Freitas, M.; Fabiano de Almeida Mendes, M.; Meira, L.; Sinigaglia, M.; Vieira, G.F. DockTope: A Web-based tool for automated pMHC-I modelling. Sci. Rep. 2015, 5, 18413. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Ehrenmann, F.; Kaas, Q.; Lefranc, M.P. IMGT/3Dstructure-DB and IMGT/DomainGapAlign: A database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res. 2010, 38, D301–D307. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [PubMed]
- Hassan, C.; Chabrol, E.; Jahn, L.; Kester, M.G.; de Ru, A.H.; Drijfhout, J.W.; Rossjohn, J.; Falkenburg, J.H.; Heemskerk, M.H.; Gras, S. Naturally processed non-canonical HLA-A*02:01 presented peptides. J. Biol. Chem. 2015, 290, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.C.; Rizkallah, P.J.; Simmons, R.; Donnellan, Z.; Dukes, J.; Bossi, G.; Le Provost, G.S.; Todorov, P.; Baston, E.; Hickman, E.; et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci. Rep. 2016, 6, 18851. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, J.H.; Gomez-Eerland, R.; van de Wiel, B.; Hulshoff, L.; van den Broek, D.; Bins, A.; Tan, H.L.; Harper, J.V.; Hassan, N.J.; Jakobsen, B.K.; et al. Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol. Ther. 2015, 23, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Degauque, N.; Brouard, S.; Soulillou, J.P. Cross-reactivity of TCR repertoire: Current concepts, challenges, and implication for allotransplantation. Front. Immunol. 2016, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Rigo, M.M.; Freitas, M.V.; Mendes, M.F.A.; Sinigaglia, M.; Lizee, G.; Kavraki, L.E.; Selin, L.K.; Cornberg, M.; Vieira, G.F. Interpreting T-Cell Cross-reactivity through Structure: Implications for TCR-Based Cancer Immunotherapy. Front Immunol. 2017, 8, 1210. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Rigo, M.M.; Silva, J.P.; Cibulski, S.P.; Sinigaglia, M.; Chies, J.A.; Vieira, G.F. Structural in silico analysis of cross-genotype-reactivity among naturally occurring HCV NS3-1073-variants in the context of HLA-A*02:01 allele. Mol. Immunol. 2011, 48, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, S.; Nabney, I.T.; Flower, D.R. Scrutinizing human MHC polymorphism: Supertype analysis using Poisson-Boltzmann electrostatics and clustering. J. Mol. Graph. Model. 2017, 77, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, P.; Picaud, S.; Baumgaertner, P.; Montandon, N.; Schmidt, J.; Speiser, D.E.; Coukos, G.; Bassani-Sternberg, M.; Filippakopoulos, P.; Gfeller, D. The C-terminal extension landscape of naturally presented HLA-I ligands. Proc. Natl. Acad. Sci. USA 2018, 115, 5083–5088. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.R.; Ensign, D.L.; Pande, V.S. Enhanced modeling via network theory: Adaptive sampling of Markov state models. J. Chem. Theory Comput. 2010, 6, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Doerr, S.; De Fabritiis, G. On-the-fly learning and sampling of ligand binding by high-throughput molecular simulations. J. Chem. Theory Comput. 2014, 10, 2064–2069. [Google Scholar] [CrossRef] [PubMed]
- Preto, J.; Clementi, C. Fast recovery of free energy landscapes via diffusion-map-directed molecular dynamics. Phys. Chem. Chem. Phys. 2014, 16, 19181–19191. [Google Scholar] [CrossRef] [PubMed]
- Hruska, E.; Abella, J.R.; Nuske, F.; Kavraki, L.E.; Clementi, C. Quantitative comparison of adaptive sampling methods for protein dynamics. J. Chem. Phys. 2018, 149, 244119. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abella, J.R.; Antunes, D.A.; Clementi, C.; Kavraki, L.E. APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations. Molecules 2019, 24, 881. https://doi.org/10.3390/molecules24050881
Abella JR, Antunes DA, Clementi C, Kavraki LE. APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations. Molecules. 2019; 24(5):881. https://doi.org/10.3390/molecules24050881
Chicago/Turabian StyleAbella, Jayvee R., Dinler A. Antunes, Cecilia Clementi, and Lydia E. Kavraki. 2019. "APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations" Molecules 24, no. 5: 881. https://doi.org/10.3390/molecules24050881
APA StyleAbella, J. R., Antunes, D. A., Clementi, C., & Kavraki, L. E. (2019). APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations. Molecules, 24(5), 881. https://doi.org/10.3390/molecules24050881