New Caffeoylquinic Acid Derivatives and Flavanone Glycoside from the Flowers of Chrysanthemum morifolium and Their Bioactivities

Abstract
:
1. Introduction
2. Results and Discussion
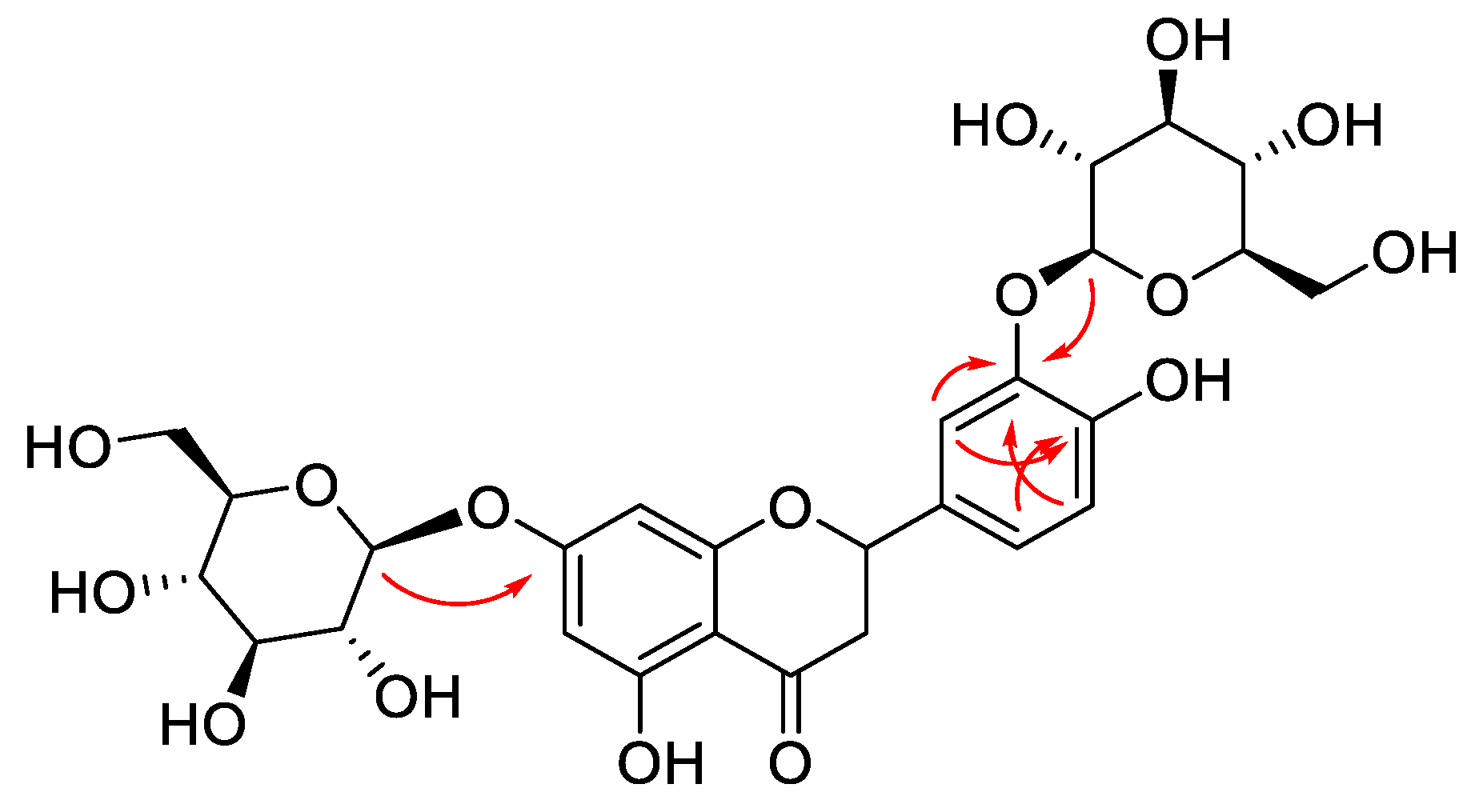
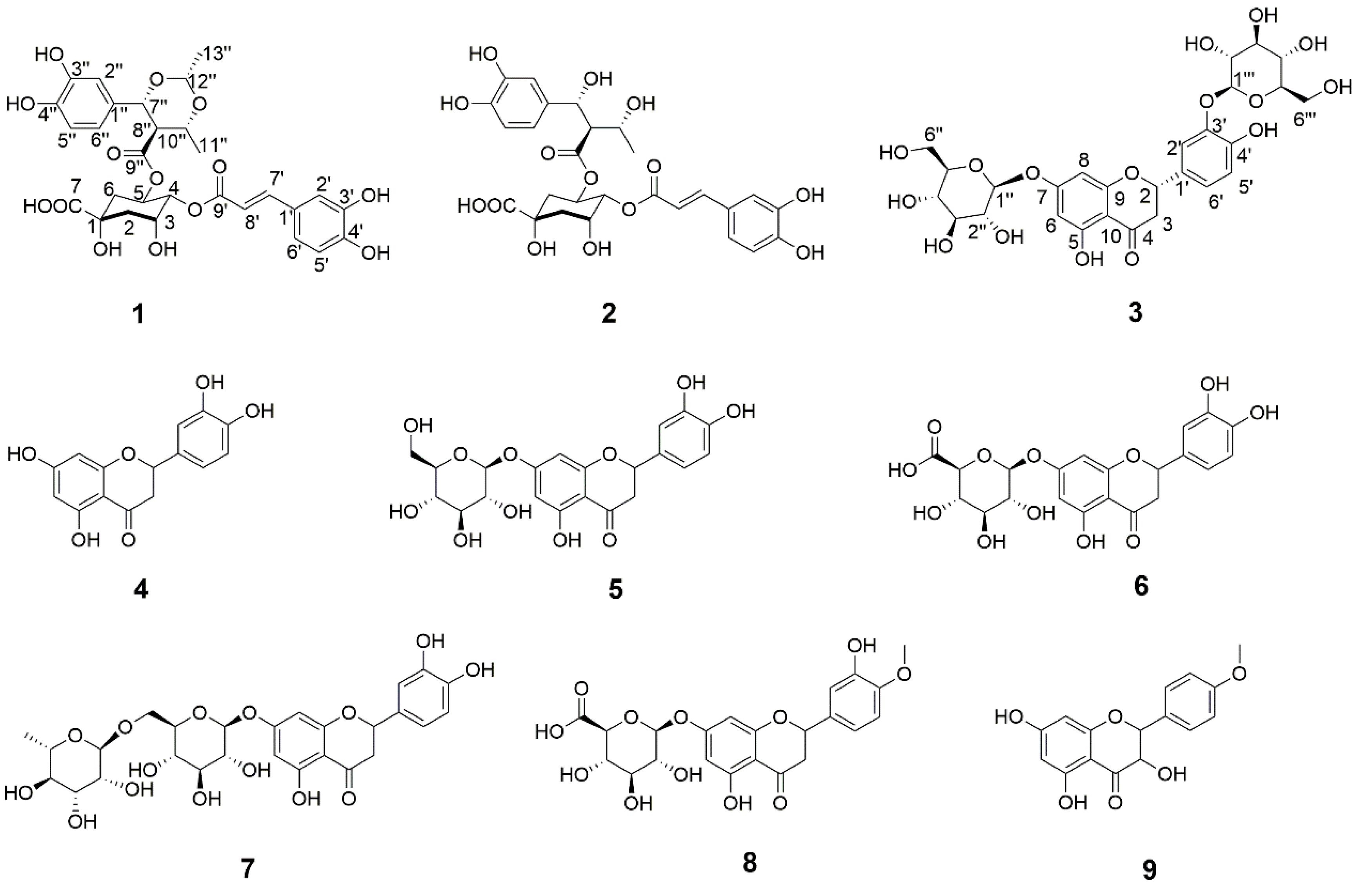
2.1. Structural Elucidation
2.2. Biological Activities
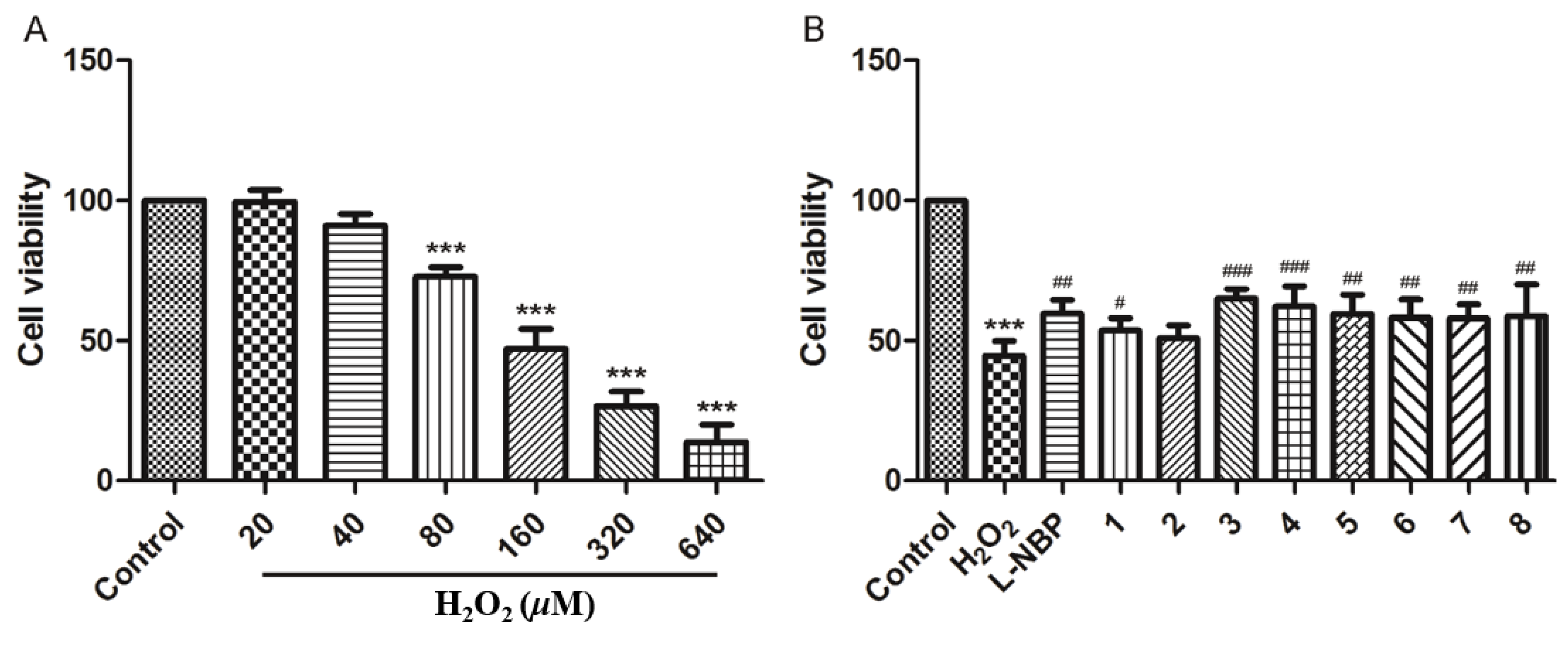
2.2.1. Hepatoprotective Activity
2.2.2. Neuroprotective Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Materials
3.3. Extraction and Isolation
3.4. Characterization
3.5. Hydrolysis of Compound 3 and GC Analysis
3.6. Hepatoprotective Activity Assay
3.7. Neuroprotective Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chinese Pharmacopeia Commission. Pharmacopeia of the People′s Republic of China; China Medical Science Publisher: Beijing, China, 2010; Part 1; p. 292. [Google Scholar]
- Ma, R.L.; Huang, C.L.; Zhang, X.H.; Wu, Z.Y. Advancement on the study of tea chrysanthemum. North. Hortic. 2009, 8, 151–154. [Google Scholar]
- Peng, Y.R.; Shi, L.; Luo, Y.H.; Ding, Y.F. Protective effect of total flavones from chrysanthemum on isoprenaline-induced myocardial ischemia in rats. Lishizhen Med. Mater. Med. Res. 2006, 17, 1131–1132. [Google Scholar]
- Kim, H.J.; Lee, Y.S. Identification of new dicaffeoylquinic acids from Chrysanthemum morifolium and their antioxidant activities. Planta Med. 2005, 71, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Duh, P.D.; Yen, G.C. Antioxidative activity of three herbal water extracts. Food Chem. 1997, 60, 639–645. [Google Scholar] [CrossRef]
- He, D.X.; Ru, X.C.; Wen, L.; Wen, Y.C.; Jiang, H.D.; Bruce, I.C.; Jin, J.; Ma, X.; Xia, Q. Total flavonoids of Flos Chrysanthemi protectarterial endothelial cells against oxidative stress. J. Ethnopharmacol. 2012, 139, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, H.J.; Lee, Y.S. A new anti-HIV flavonoid glucuronide from Chrysanthemum morifolium. Planta Med. 2003, 69, 859–861. [Google Scholar] [PubMed]
- Hu, C.Q.; Chen, K.; Shi, Q.; Kilkuskie, R.E.; Cheng, Y.C.; Lee, K.H. Anti-AIDS agents, 10. Acacetin-7-O-β-D-galactopyranoside, an anti HIV principle from Chrysanthemum morifolium and a structure- activity correlation with some related flavonoids. J. Nat. Prod. 1994, 57, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Ukiya, M.; Akihisa, T.; Yasukawa, K.; Kasahara, Y.; Kimura, Y.; Koike, K.; Nikaido, T.; Takido, M. Constituents of compositae plants. 2. Triterpene diol, triols, and their 3-O-fatty acid esters from edible chrysanthemum flower extract and their anti-inflammatory effects. J. Agric. Food Chem. 2001, 49, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.D.; Wang, L.F.; Zhou, X.M.; Xia, Q. Vasorelaxant effects and underlying mechanism of EtOAc extract from Chrysanthemum morifolium in rat thoracic aorta. Chin. J. Pathophysiol. 2005, 21, 334–338. [Google Scholar]
- Kim, I.S.; Koppula, S.; Park, P.J.; Kim, E.H.; Kim, C.J.; Choi, W.S.; Lee, K.H.; Choi, D.K. Chrysanthemum morifolium Ramat (CM) extract protects human neuroblastoma SH-SY5Y cells against MPP+-induced cytotoxicity. J. Ethnopharmacol. 2009, 126, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Ukiya, M.; Akihisa, T.; Tokuda, H.; Suzuki, H.; Mukainaka, T.; Ichiishi, E.; Yasukawa, K.; Kasahara, Y.; Nishino, H. Constituents of Compositae plants III. Anti-tumor promoting effects and cytotoxic activity against human cancer cell lines of triterpene diols and triols from edible chrysanthemum flowers. Cancer Lett. 2002, 177, 7–12. [Google Scholar] [CrossRef]
- Xie, Y.Y.; Yuan, D.; Yang, J.Y.; Wang, L.H.; Wu, C.F. Cytotoxic activity of flavonoids from Flos Chrysanthemum on human colon cancer Colon 205 cells. J. Asian Nat. Prod. Res. 2009, 11, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.Y.; Huang, X.; Lian, T.T.; Xu, Q.T. Protective Effect of Dendranthema morifolium on CCl4- induced Liver Injury in Mice. Nat. Prod. Res. Dev. 2012, 24, 1634–1636. [Google Scholar]
- Wang, P.; Pan, X.; Chen, G. Increased exposure of vitamin A by Chrysanthemum morifolium Ramat extract in rat was not via induction of CYP1A1, CYP1A2 and CYP2B1. J. Food Sci. 2012, 77, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Morikawa, T.; Toguchida, I.; Yoshikawa, M. Structural requirements of flavonoids and related compounds for aldose reductase inhibitory activity. Chem. Pharm. Bull. 2002, 50, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Terashima, S.; Shimizu, M.; Horie, S.; Morita, N. Studies on aldose reductase inhibitors from natural products. IV. Constituents and aldose reductase inhibitory effect of Chrysanthemum morifolium, Bixa orellana and Ipomoea batatus. Chem. Pharm. Bull. 1991, 39, 3346–3347. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Hisama, M. Antimutagenic activity of flavonoids from Chrysanthemum morifolium. Biosci. Biotechnol. Biochem. 2003, 67, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.H.; Chen, Z.L. Sesquiterpenoid alcohols from Chrysanthemum morifolium. Phytochemistry 1997, 44, 1287–1290. [Google Scholar]
- Tsao, R.; Attygalle, A.B.; Schroeder, F.C.; Marvin, C.H.; McGarvey, B.D. Isobutylamides of unsaturated fatty acids from Chrysanthemum morifolium associated with host-plant resistance against the western Flower Thrips. J. Nat. Prod. 2003, 66, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Shen, L.T.; Zeng, Y.Y.; Tian, Y.Q.; Xu, H.H. Flavonoids from Ficus sarmentosa var. henryi. Chin. Tradit. Herb. Drugs 2010, 41, 526–529. [Google Scholar]
- Sun, Y.; Ma, X.B.; Liu, J.X. Compounds from fraction with cardiovascular activity of Chrysanthemum indicum. China J. Chin. Mater. Med. 2012, 37, 61–65. [Google Scholar]
- Zhou, D.N.; Ruan, J.L.; Cai, Y.L. Flavonoids from Aerial Parts of Arachniodes exilis. Chin. Pharm. J. 2008, 43, 1218–1220. [Google Scholar]
- Shang, H.Q.; Qin, M.J.; Wu, J.R. Constituents of Rhizomes of Iris tectorum. Chin. J. Nat. Med. 2007, 5, 312–314. [Google Scholar]
- Abate, A.; Brenna, E.; Costantini, A.; Fuganti, C.; Gatti, F.G.; Malpezzi, L.; Serra, S. Enzymatic approach to enantiomerically pure 5-alken-2,4-diols and 4-hydrocy-5-alken-2-ones: Application to the synthesis of chiral synthons. J. Org. Chem. 2006, 71, 5228–5240. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Chiou, Y.N.; Lin, Y.L. Phenolic Glycosides from Viscum angulatum. J. Nat. Prod. 2002, 65, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Kato, M.; Iinuma, M.; Tanaka, T.; Kimura, A.; Ohashi, H.; Sakai, H. Acylated luteolin glucosides from Salix gilgiana. Phytochemistry 1987, 26, 2418–2420. [Google Scholar] [CrossRef]
- Yang, Y.N.; Huang, X.Y.; Feng, Z.M.; Jiang, J.S.; Zhang, P.C. New butyrolactone type lignans from Arctii Fructus and their anti-inflammatory activities. J. Agric. Food Chem. 2015, 63, 7958–7966. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–3 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 a | 2 b | |||
|---|---|---|---|---|
| Position | δH | δC | δH | δC |
| 1 | 75.2 | 75.1 | ||
| 2 | 2.06, m | 38.2 | 2.01, m 2.19, m | 38.4 |
| 3 | 4.28, m | 70.3 | 4.24, m | 70.1 |
| 4 | 4.90, m | 76.3 | 5.01, dd (3.0, 10.0) | 75.7 |
| 5 | 5.44, m | 69.4 | 5.29, m | 69.8 |
| 6 | 1.70, m | 40.2 | 1.89, m | 39.9 |
| 7 | 175.9 | 175.2 | ||
| 1′ | 127.7 | 127.7 | ||
| 2′ | 7.03, d (2.0) | 115.1 | 7.07, d (2.0) | 115.2 |
| 3′ | 146.8 | 146.8 | ||
| 4′ | 149.8 | 149.7 | ||
| 5′ | 6.78, d (8.0) | 116.5 | 6.78, d (8.0) | 115.9 |
| 6′ | 6.94, dd (2.0, 8.0) | 123.1 | 6.97, dd (2.0, 8.0) | 123.2 |
| 7′ | 7.58, d (16.0) | 147.8 | 7.63, d (16.0) | 147.6 |
| 8′ | 6.24, d (16.0) | 114.9 | 6.27, d (16.0) | 114.9 |
| 9′ | 168.2 | 168.4 | ||
| 1′′ | 131.7 | 134.3 | ||
| 2′′ | 6.76, d (2.0) | 115.2 | 6.66, d (2.0) | 115.4 |
| 3′′ | 146.6 | 146.2 | ||
| 4′′ | 146.7 | 146.5 | ||
| 5′′ | 6.67, d (8.0) | 116.0 | 6.66, d (8.0) | 116.5 |
| 6′′ | 6.58, dd (2.0, 8.0) | 119.8 | 6.81, dd (2.0, 8.0) | 120.2 |
| 7′′ | 4.56, d (10.0) | 81.8 | 4.77, d (10.0) | 76.7 |
| 8′′ | 2.38, dd (10.0, 10.0) | 57.2 | 2.72, dd (8.5, 10.0) | 62.4 |
| 9′′ | 171.8 | 171.2 | ||
| 10′′ | 3.93, dq (6.0, 10.0) | 75.4 | 4.14, dq (6.0, 8.5) | 70.3 |
| 11′′ | 1.10, d (6.0) | 20.3 | 1.14, d (6.0) | 21.7 |
| 12′′ | 4.90, m | 100.2 | ||
| 13′′ | 1.29, d (5.0) | 21.2 | ||
| 3 | 3 | ||||
|---|---|---|---|---|---|
| Position | δH | δC | Position | δH | δC |
| 2 | 5.50, m | 78.8 | 6′ | 7.03, dd (2.0, 8.0) | 121.9 |
| 3 | 3.32, m 2.75, dd (2.0, 17.0) | 42.0 | 1′′ | 4.97, d (7.5) | 99.6 |
| 4 | 197.3 | 2′′ | 3.2–3.4, m | 73.1 | |
| 5 | 162.8 | 3′′ | 3.2–3.4, m | 76.1 | |
| 6 | 6.14, d (2.0) | 96.6 | 4′′ | 3.2–3.4, m | 69.5 |
| 7 | 165.4 | 5′′ | 3.2–3.4, m | 77.1 | |
| 8 | 6.17, d (2.0) | 95.5 | 6′′ | 3.67, m 3.47, m | 60.6 |
| 9 | 163.0 | 1′′′ | 4.73, d (7.0) | 101.9 | |
| 10 | 103.3 | 2′′′ | 3.2–3.4, m | 73.4 | |
| 1′ | 129.3 | 3′′′ | 3.2–3.4, m | 76.4 | |
| 2′ | 7.38, d (2.0) | 115.4 | 4′′′ | 3.2–3.4, m | 69.9 |
| 3′ | 145.2 | 5′′′ | 3.2–3.4, m | 77.3 | |
| 4′ | 147.3 | 6′′′ | 3.67, m 3.47, m | 60.8 | |
| 5′ | 6.85, d (8.0) | 115.8 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.-F.; Yang, Y.-N.; He, C.-Y.; Chen, Z.-F.; Yuan, Q.-S.; Zhao, S.-C.; Fu, Y.-F.; Zhang, P.-C.; Mao, D.-B. New Caffeoylquinic Acid Derivatives and Flavanone Glycoside from the Flowers of Chrysanthemum morifolium and Their Bioactivities. Molecules 2019, 24, 850. https://doi.org/10.3390/molecules24050850
Yang P-F, Yang Y-N, He C-Y, Chen Z-F, Yuan Q-S, Zhao S-C, Fu Y-F, Zhang P-C, Mao D-B. New Caffeoylquinic Acid Derivatives and Flavanone Glycoside from the Flowers of Chrysanthemum morifolium and Their Bioactivities. Molecules. 2019; 24(5):850. https://doi.org/10.3390/molecules24050850
Chicago/Turabian StyleYang, Peng-Fei, Ya-Nan Yang, Chun-Yu He, Zhi-Fei Chen, Qi-Shan Yuan, Sheng-Chen Zhao, Yu-Feng Fu, Pei-Cheng Zhang, and Duo-Bin Mao. 2019. "New Caffeoylquinic Acid Derivatives and Flavanone Glycoside from the Flowers of Chrysanthemum morifolium and Their Bioactivities" Molecules 24, no. 5: 850. https://doi.org/10.3390/molecules24050850
APA StyleYang, P.-F., Yang, Y.-N., He, C.-Y., Chen, Z.-F., Yuan, Q.-S., Zhao, S.-C., Fu, Y.-F., Zhang, P.-C., & Mao, D.-B. (2019). New Caffeoylquinic Acid Derivatives and Flavanone Glycoside from the Flowers of Chrysanthemum morifolium and Their Bioactivities. Molecules, 24(5), 850. https://doi.org/10.3390/molecules24050850