Bioassay-Guided Isolation and Structure Elucidation of Fungicidal and Herbicidal Compounds from Ambrosia salsola (Asteraceae)

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Structure Elucidation
2.2. HPLC Profile of A. salsola and A. dumosa
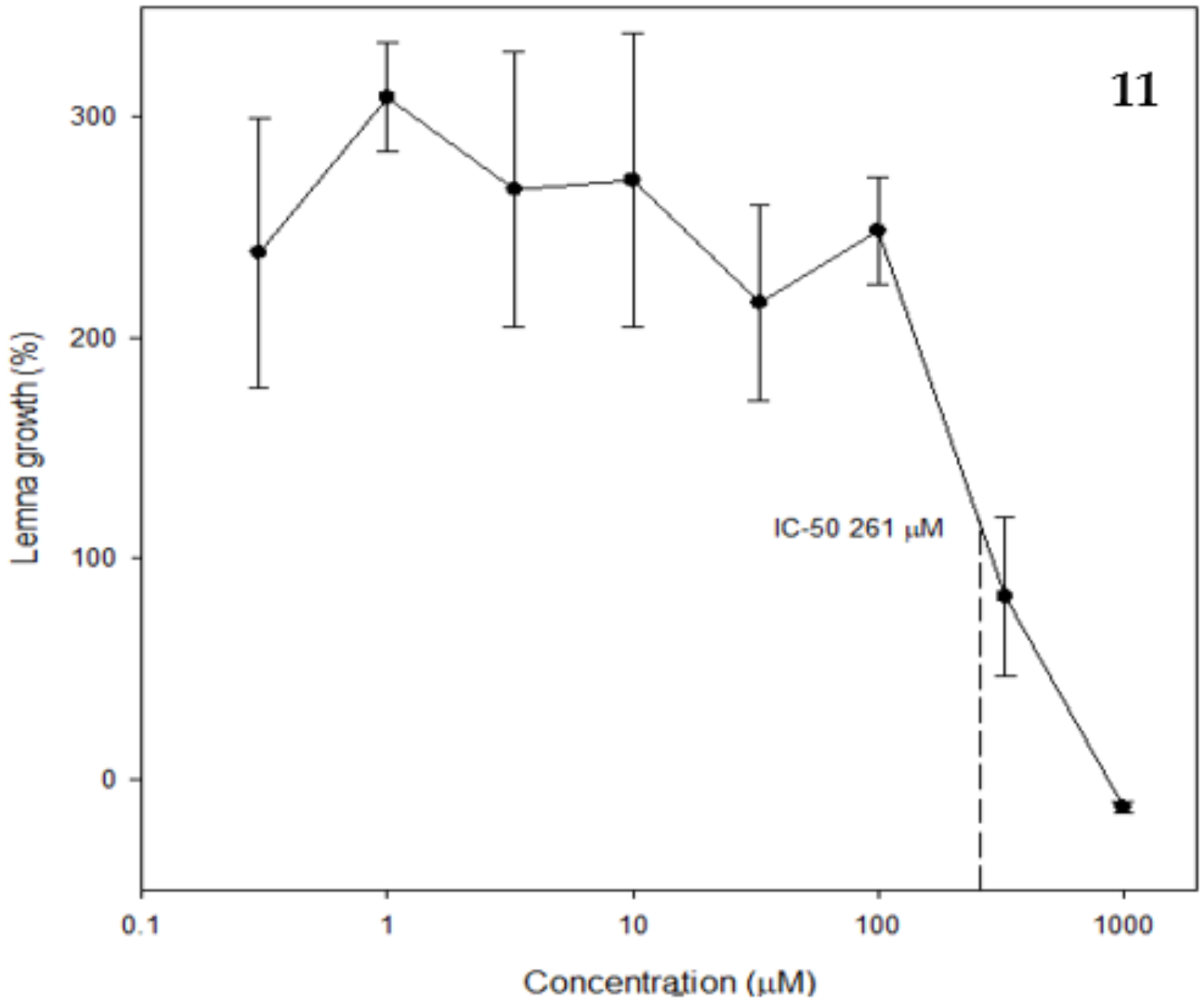
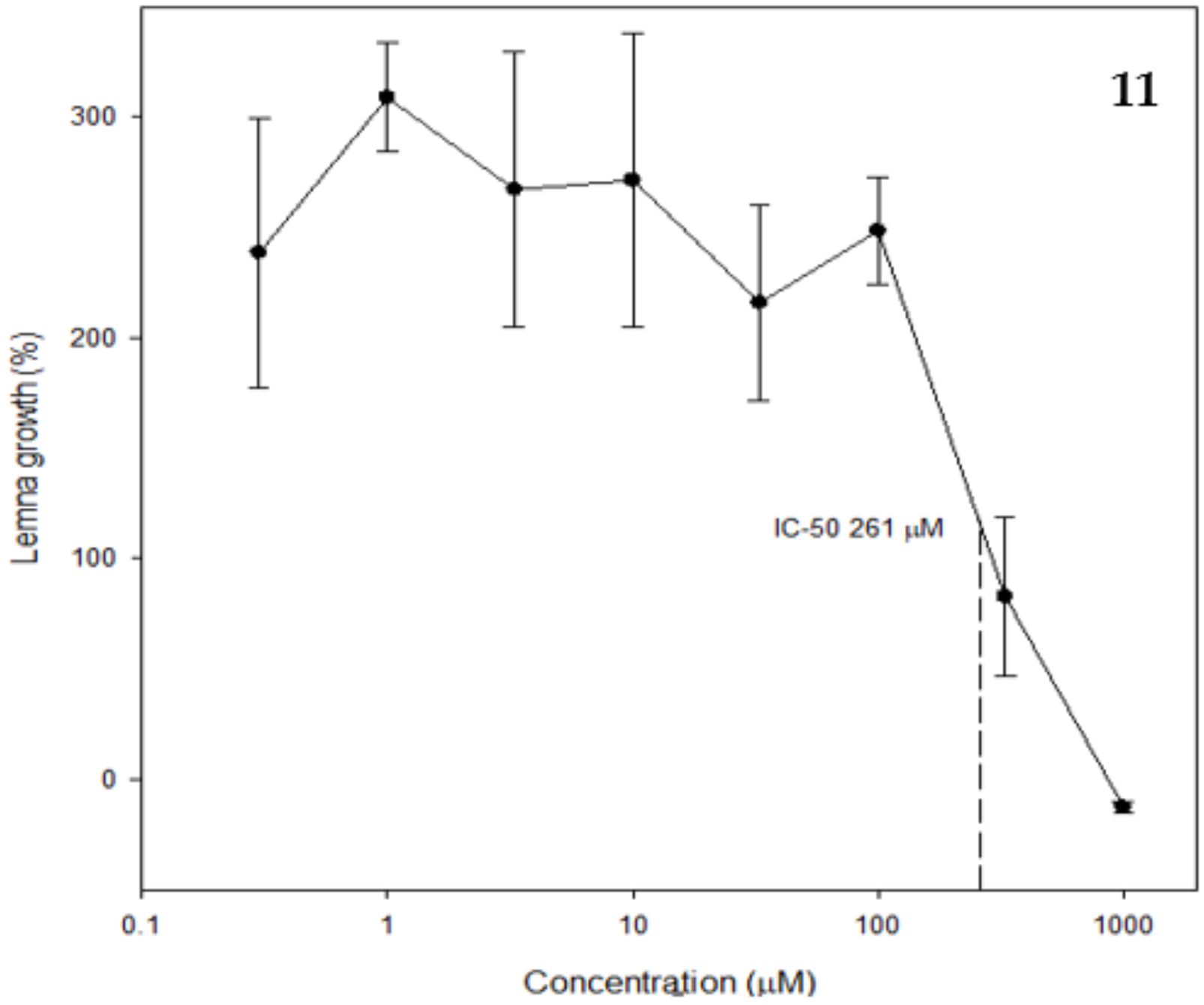
2.3. Phytotoxic and Antifungal Activity of Pure Compounds
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Plant Material and Extraction
3.3. Isolation Procedure
3.4. Physical Chemical Data
3.5. HPLC Analysis
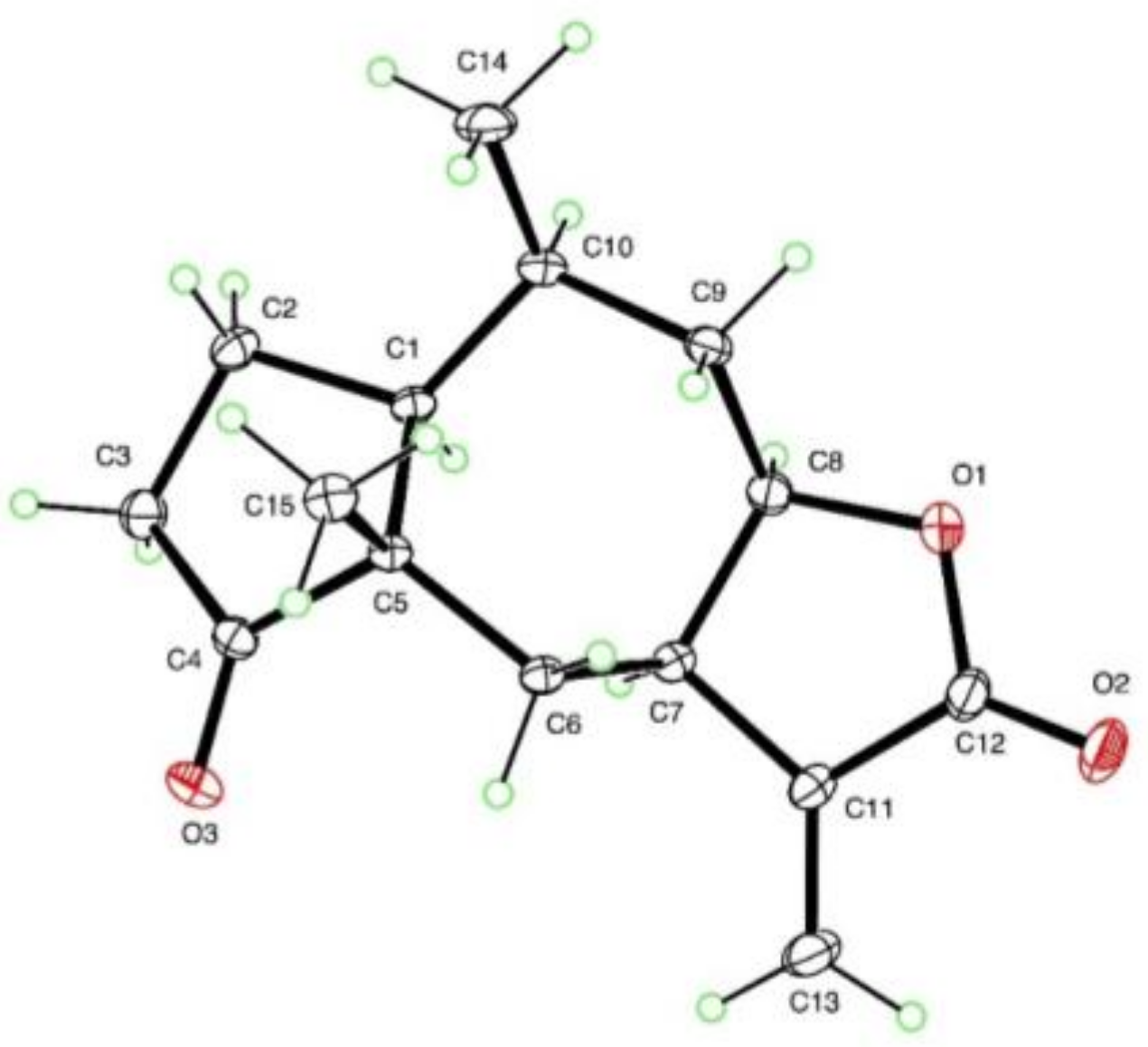
3.6. X-ray Crystallography of Compound 11
3.7. Phytotoxicity Bioassay
3.7.1. Lactuca sativa and Agrostis stolonifera
3.7.2. Lemna paucicostata
3.8. Antifungal Bioautography Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Igbedioh, S. Effects of agricultural pesticides on humans, animals, and higher plants in developing countries. Arch. Environ. Health 1991, 46, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Aktar, W.; Sengupta, D.; Chowdhury, A. Impact of pesticides use in agriculture: Their benefits and hazards. Interdiscip. Toxicol. 2009, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.E.; Duke, S.O. Natural compounds as next generation herbicides. Plant Physiol. 2014, 166, 1090–1105. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, C.L.; Dayan, F.E.; Duke, S.O. Natural products as sources for new pesticides. J. Nat. Prod. 2012, 75, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.L.; Meepagala, K.M.; Oliva, A.; Aliotta, G.; Duke, S.O. Phytotoxins from the leaves of Ruta graveolens. J. Agric. Food Chem. 2004, 52, 3345–3349. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, S.C.; Cantrell, C.L.; Duke, S.O.; Wedge, D.E.; Nandula, V.K.; Moraes, R.M.; Cerdeira, A.L. Bioassay-directed isolation and identification of phytotoxic and fungitoxic acetylenes from Conyza canadensis. J. Agric. Food Chem. 2012, 60, 5893–5898. [Google Scholar] [CrossRef] [PubMed]
- Abdelgaleil, S.A.; Hashinaga, F.J. Allelopathic potential of two sesquiterpene lactones from Magnolia grandiflora L. Biochem. Syst. Ecol. 2007, 35, 737–742. [Google Scholar] [CrossRef]
- Saad, M.M.; Abdelgaleil, S.A.; Suganuma, T. Herbicidal potential of pseudoguaninolide sesquiterpenes on wild oat, Avena fatua L. Biochem. Syst. Ecol. 2012, 44, 333–337. [Google Scholar] [CrossRef]
- Svensson, D.; Lozano, M.; Almanza, G.R.; Nilsson, B.-O.; Sterner, O.; Villagomez, R.J.P. Sesquiterpene lactones from Ambrosia arborescens Mill. inhibit pro-inflammatory cytokine expression and modulate NF-κB signaling in human skin cells. Phytomedicine 2018, 50, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.G. Anti-inflammatory activity of Ambrosia artemisiaefolia and Rhoeo spathacea. Phytomedicine 1996, 3, 163–167. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Frank, F.M.; Cazorla, S.I.; Barrera, P.; Freixa, B.; Vila, R.; Sosa, M.A.; Malchiodi, E.L.; Muschietti, L.V.; Martino, V.S. Psilostachyin C: A natural compound with trypanocidal activity. Int. J. Antimicrob. Agents 2011, 37, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Balza, F.; Towers, G.N.J.P. Structural analysis of sesquiterpene lactones from Hymenoclea salsola. Phytochemistry 1988, 27, 1421–1424. [Google Scholar] [CrossRef]
- Geissman, T.; Toribio, F.J.P. Sesquiterpene lactones: Constituents of Hymenoclea salsola T. and G. Phytochemistry 1967, 6, 1563–1567. [Google Scholar] [CrossRef]
- Proksch, P.; Wollenweber, E.; Rodriguez, E. Flavonoids from the Leaf Resin of Hymenoclea salsola T. & G. (Asteraeeae). Z. Naturforsch. 1983, 38, 668–669. [Google Scholar]
- Quang, D.N.; So, T.C.; Thanh, N.T.P.; Hoa, L.T.P.; Dien, P.H.; Luong, T.M.; Tung, N.Q.; Long, L.D.; Dai, T.D.; Tien, N.Q. Balanochalcone, a new chalcone from Balanophora laxiflora Hemsl. Nat. Prod. Res. 2018, 32, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Nel, R.J.; van Rensburg, H.; van Heerden, P.S.; Coetzee, J.; Ferreira, D.J. Stereoselective synthesis of flavonoids. Part 7. Poly-oxygenated β-hydroxydihydrochalcone derivatives. Tetrahedron 1999, 55, 9727–9736. [Google Scholar] [CrossRef]
- Muiva, L.M.; Yenesew, A.; Derese, S.; Heydenreich, M.; Peter, M.G.; Akala, H.M.; Eyase, F.; Waters, N.C.; Mutai, C.; Keriko, J.M. Antiplasmodial β-hydroxydihydrochalcone from seedpods of Tephrosia elata. Phytochem. Lett. 2009, 2, 99–102. [Google Scholar] [CrossRef]
- Kim, K.H.; Moon, E.; Choi, S.U.; Kim, S.Y.; Lee, K.R. Polyphenols from the bark of Rhus verniciflua and their biological evaluation on antitumor and anti-inflammatory activities. Phytochemistry 2013, 92, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Córdova, W.H.P.; Mesa, L.G.; Hill, A.L.P.; Lima, C.N.; Lamas, G.D.; Suárez, M.O.; Domínguez, R.S. Antimicrobial activity of crude extracts and flavonoids from leaves of Pluchea carolinensis (Jacq.) G. Don. Pharmacologyonline 2006, 3, 757–761. [Google Scholar]
- Córdova, W.H.P.; Wauters, J.-N.; Kevers, C.; Frederich, M.; Dommes, J. Antioxidant fractions and phenolic constituents from leaves of Pluchea carolinensis and Pluchea rosea. Free Radic. Antioxid. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Toribio, F.; Geissman, T. Sesquiterpene lactones of Hymenoclea monogyra. Phytochemistry 1969, 8, 313–314. [Google Scholar] [CrossRef]
- Macías, F.A.; Fernández, A.; Varela, R.M.; Molinillo, J.M.; Torres, A.; Alves, P.L. Sesquiterpene lactones as allelochemicals. J. Nat. Prod. 2006, 69, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, B.P.; Nepomuceno, M.P.; Varela, R.M.; Torres, A.N.; Molinillo, J.M.; Alves, P.L.; Macias, F.A. Phytotoxicity Study on Bidens sulphurea Sch. Bip. as a Preliminary Approach for Weed Control. J. Agric. Food Chem. 2017, 65, 5161–5172. [Google Scholar] [CrossRef] [PubMed]
- Duke, S.O.; Vaughn, K.C.; Croom, E.M.; ElSohly, H.N. Artemisinin, a constituent of annual wormwood (Artemisia annua) is a selective phytotoxin. Weed Sci. 1987, 35, 499–505. [Google Scholar]
- Michel, A.; Johnson, R.D.; Duke, S.O.; Scheffler, B.E. Dose-response relationships between herbicides with different modes of action and growth of Lemna paucicostata: An improved ecotoxicological method. Environ. Toxicol. Chem. 2004, 23, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Tielas, C.; Graña, E.; Sotelo, T.; Reigosa, M.J.; Sánchez-Moreiras, A.M. The natural compound trans-chalcone induces programmed cell death in Arabidopsis thaliana roots. Plant Cell Environ. 2012, 35, 1500–1517. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Tielas, C.; Sotelo, T.; Graña, E.; Reigosa, M.J.; Sánchez-Moreiras, A.M. Phytotoxic potential of trans-chalcone on crop plants and model species. J. Plant Growth Regul. 2014, 33, 181–194. [Google Scholar] [CrossRef]
- Díaz-Tielas, C.; Graña, E.; Maffei, M.; Reigosa, M.; Sánchez-Moreiras, A. Plasma membrane depolarization precedes photosynthesis damage and long-term leaf bleaching in (E)-chalcone-treated Arabidopsis shoots. J. Plant Physiol. 2017, 218, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.; Flack, H.D.; Wagner, T. Materials, Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.E.; Romagni, J.G.; Duke, S.O. Investigating the mode of action of natural phytotoxins. J. Chem. Ecol. 2000, 26, 2079–2094. [Google Scholar] [CrossRef]
- Stappen, I.; Alib, A.; Tabancab, N.; Khanb, I.A.; Wannerc, J.; Gochevd, V.K.; Singhe, V.; Lalf, B.; Jaitakf, V.; Kaulh, V.K.; et al. Antimicrobial and repellent activity of the essential oils of two Lamiaceae cultivated in western Himalaya. Curr. Bioact. Compd. 2015, 11, 23–30. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–12 are available from the authors. |
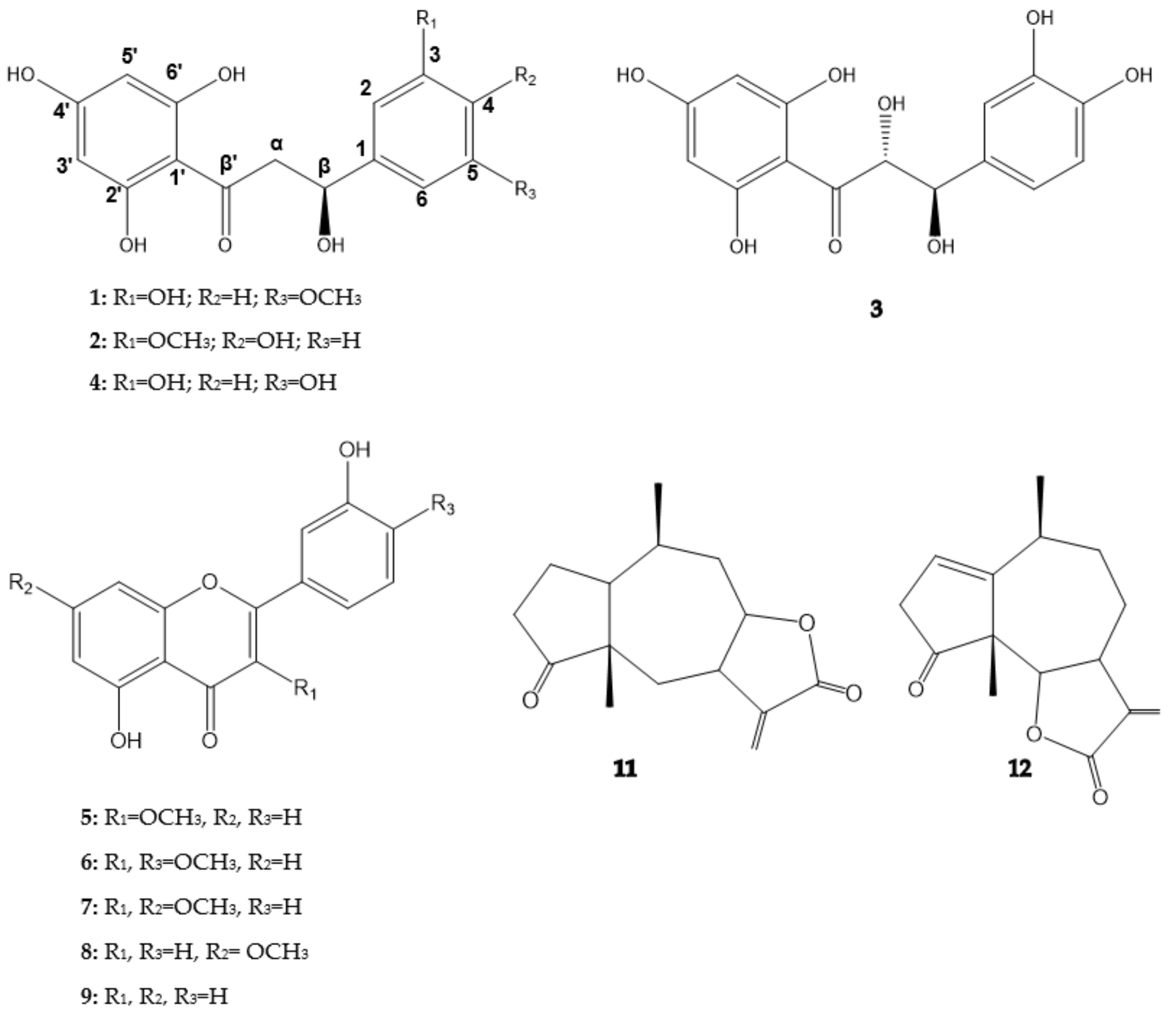


{kind=link}
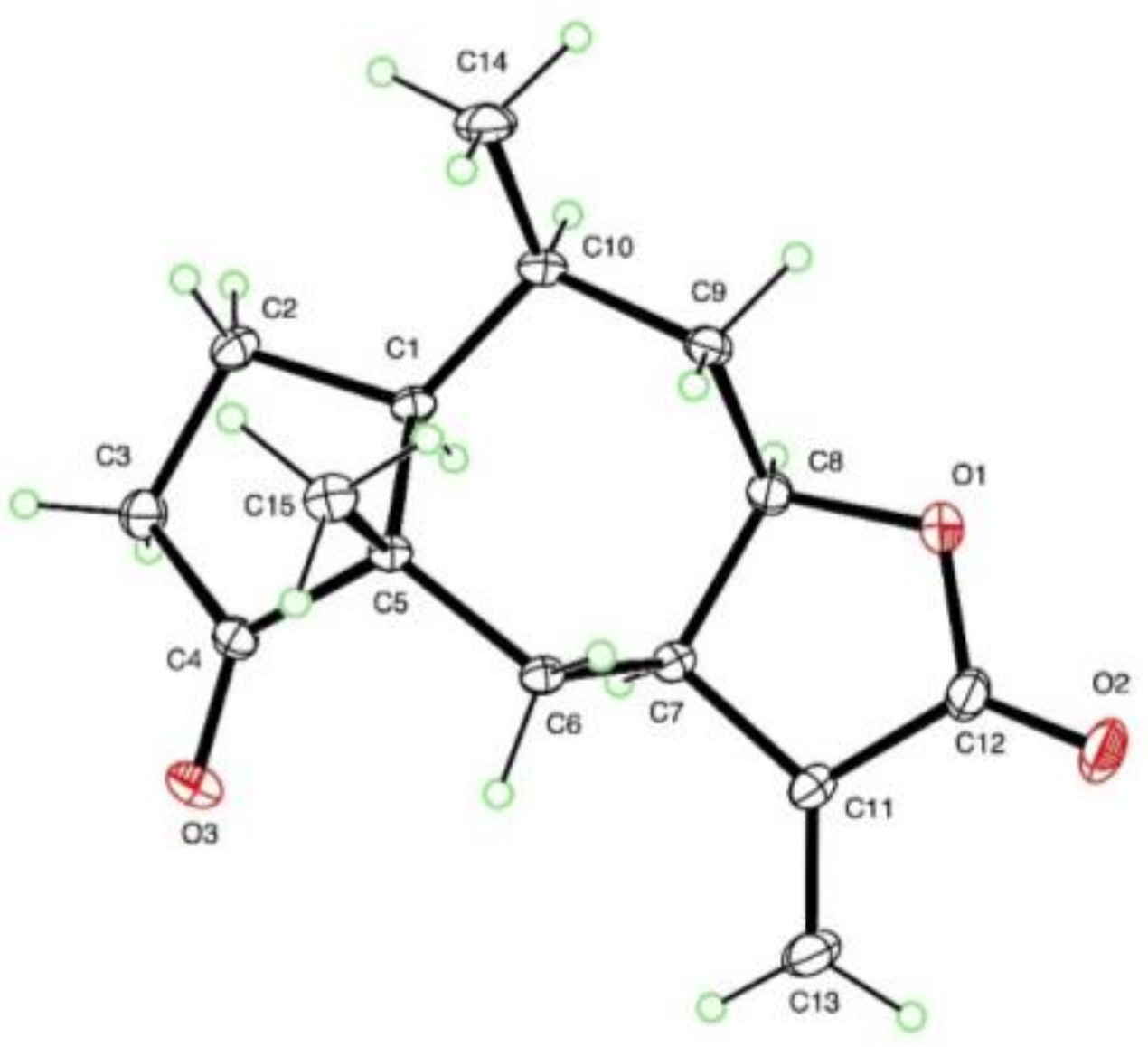{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | 3 a | |||
|---|---|---|---|---|---|---|
| δH (ppm) | δC (ppm) | δH (ppm) | δC (ppm) | δH (ppm) | δC (ppm) | |
| 1 | - | 133.3 | - | 132.0 | - | 130.0 |
| 2 | 6.95 d (J = 1.9 Hz, 1H) | 114.7 | 7.07 d (J = 2.0 Hz, 1H) | 111.5 | 6.97 d (J = 1.9 Hz, 1H) | 116.2 |
| 3 | - | 147.9 | - | 149.3 | - | 146.5 |
| 4 | 6.91 d, 1H | 112.8 | - | 148.3 | - | 147.3 |
| 5 | - | 149.5 | 6.82 d (J = 8.1 Hz, 1H) | 116.3 | 6.80 d (J = 8.1 Hz, 1H) | 116.0 |
| 6 | 6.91 d, 1H | 119.1 | 6.92 dd (J = 8.1, 2.0 Hz, 1H) | 120.7 | 6.85 dd (J = 8.1, 1.9 Hz, 1H) | 121.0 |
| β | 5.31 dd (J = 12.6, 3.0 Hz, 1H) | 80.4 | 5.34 dd (J = 12.8, 3.0 Hz, 1H) | 80.8 | 4.91 d (J = 11.4 Hz, 1H) | 85.3 |
| α | 2.71 dd (J = 17.1, 3.0 Hz,1H) 3.05 dd (J = 17.1, 12.6 Hz,1H) | 44.2 | 2.70 dd (J = 17.1, 3.0 Hz,1H) 3.12 dd (J = 17.1, 12.8 Hz,1H) | 44.3 | 4.50 d (J = 11.4 Hz,1H) | 73.8 |
| β′ | - | 197.7 | - | 197.6 | - | 198.5 |
| 1′ | - | 103.5 | - | 103.2 | - | 102.0 |
| 2′ | - | 164.9 | - | 165.0 | - | 164.6 |
| 3′ | 5.89 d (J = 2.2 Hz, 1H) | 97.3 | 5.89 d (J = 2.2 Hz, 1H) | 97.6 | 5.92 d (J = 2.0 Hz, 1H) | 97.5 |
| 4′ | - | 168.7 | - | 169.8 | - | 168.8 |
| 5′ | 5.87 d (J = 2.2 Hz, 1H) | 96.4 | 5.87 d (J = 2.2 Hz, 1H) | 96.1 | 5.89 d (J = 2.0 Hz, 1H) | 96.4 |
| 6′ | - | 165.6 | - | 165.7 | - | 165.4 |
| 3-OCH3 | - | - | 3.88 s (3H) | 56.7 | - | - |
| 5-OCH3 | 3.86 s (3H) | 56.6 | - | - | ||
| Compounds | C (µM) | L. sativa | A. stolonifera |
|---|---|---|---|
| Confertin | 10 | 0 | 0 |
| 100 | 0 | 3 | |
| 1000 | 4 | 5 | |
| Neoambrosin | 10 | 0 | 0 |
| 100 | 0 | 0 | |
| 1000 | 0 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, W.H.; Meepagala, K.M.; Fronczek, F.R.; Cook, D.D.; Wedge, D.E.; Duke, S.O. Bioassay-Guided Isolation and Structure Elucidation of Fungicidal and Herbicidal Compounds from Ambrosia salsola (Asteraceae). Molecules 2019, 24, 835. https://doi.org/10.3390/molecules24050835
Perera WH, Meepagala KM, Fronczek FR, Cook DD, Wedge DE, Duke SO. Bioassay-Guided Isolation and Structure Elucidation of Fungicidal and Herbicidal Compounds from Ambrosia salsola (Asteraceae). Molecules. 2019; 24(5):835. https://doi.org/10.3390/molecules24050835
Chicago/Turabian StylePerera, Wilmer H., Kumudini M. Meepagala, Frank R. Fronczek, Daniel D. Cook, David E. Wedge, and Stephen O. Duke. 2019. "Bioassay-Guided Isolation and Structure Elucidation of Fungicidal and Herbicidal Compounds from Ambrosia salsola (Asteraceae)" Molecules 24, no. 5: 835. https://doi.org/10.3390/molecules24050835
APA StylePerera, W. H., Meepagala, K. M., Fronczek, F. R., Cook, D. D., Wedge, D. E., & Duke, S. O. (2019). Bioassay-Guided Isolation and Structure Elucidation of Fungicidal and Herbicidal Compounds from Ambrosia salsola (Asteraceae). Molecules, 24(5), 835. https://doi.org/10.3390/molecules24050835