
An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin


Abstract
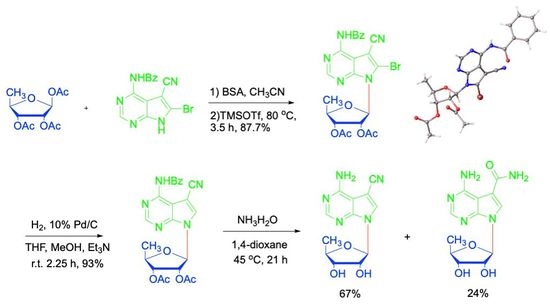
{kind=link}
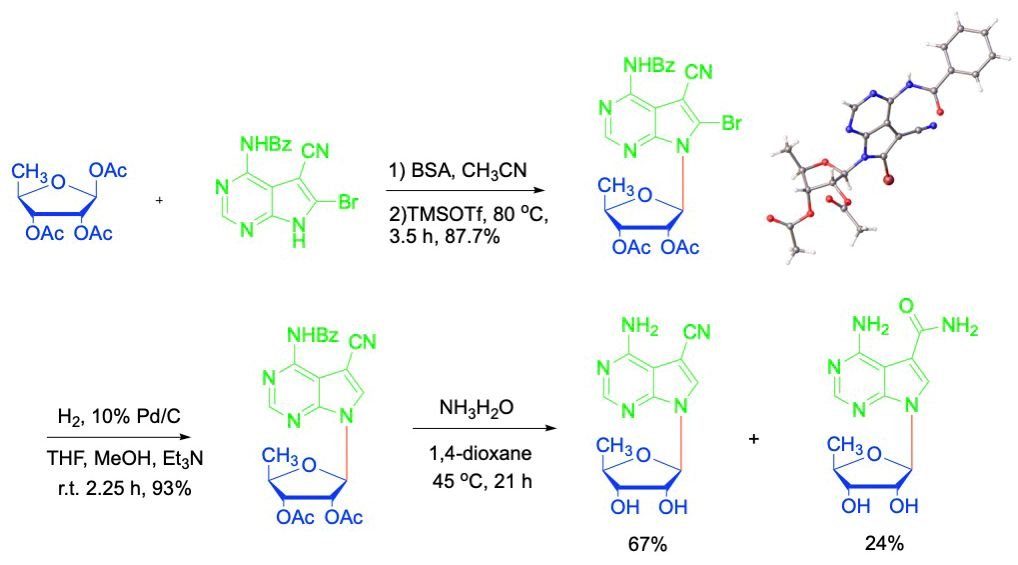
{kind=link}
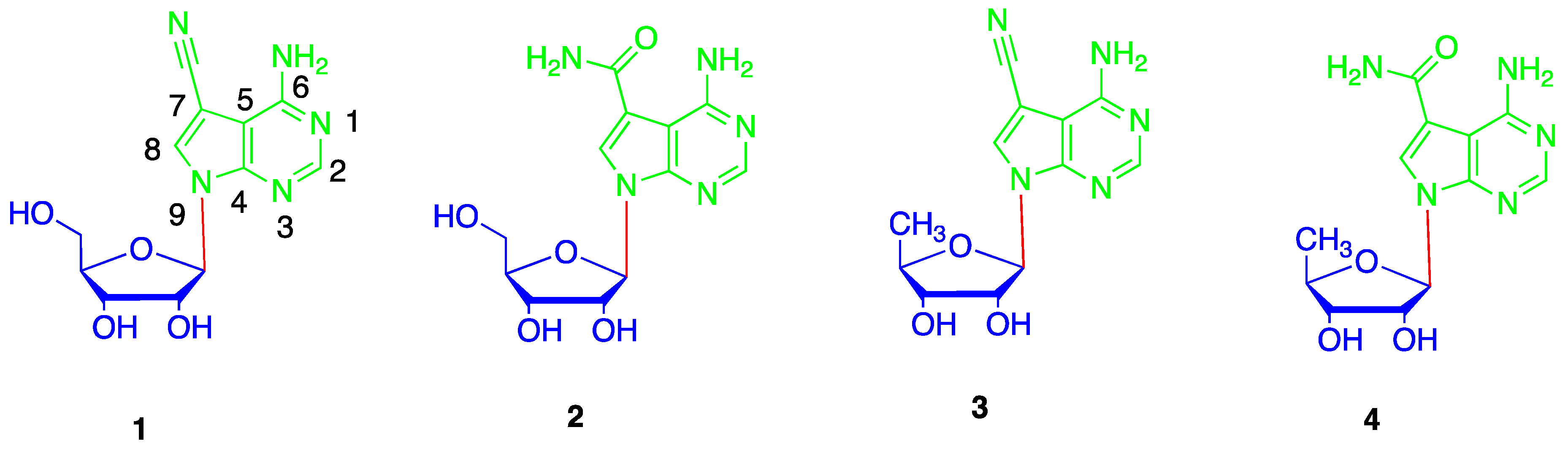
{kind=link}
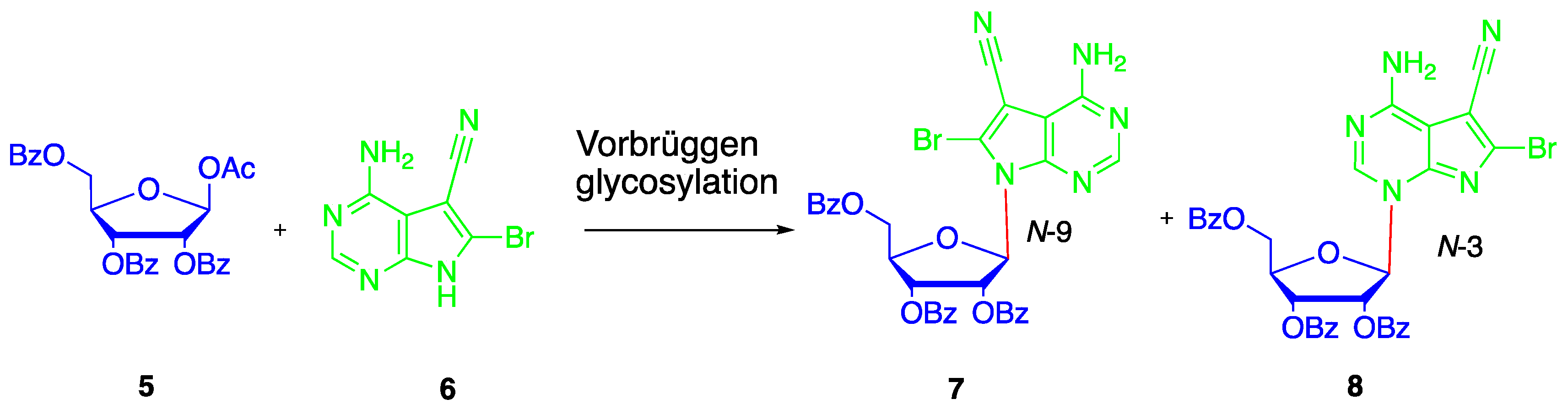
{kind=link}
{kind=link}
{kind=link}
1. Introduction
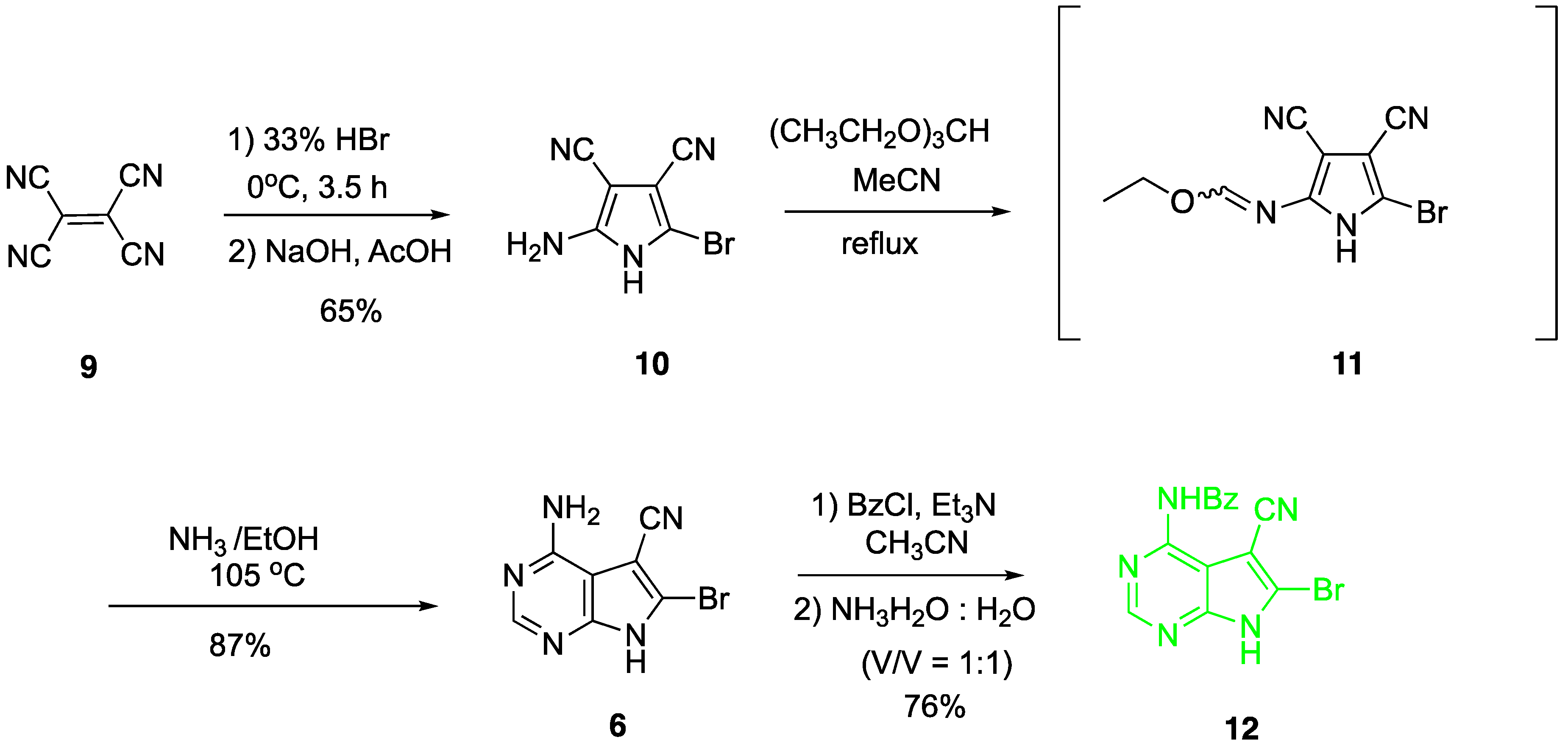
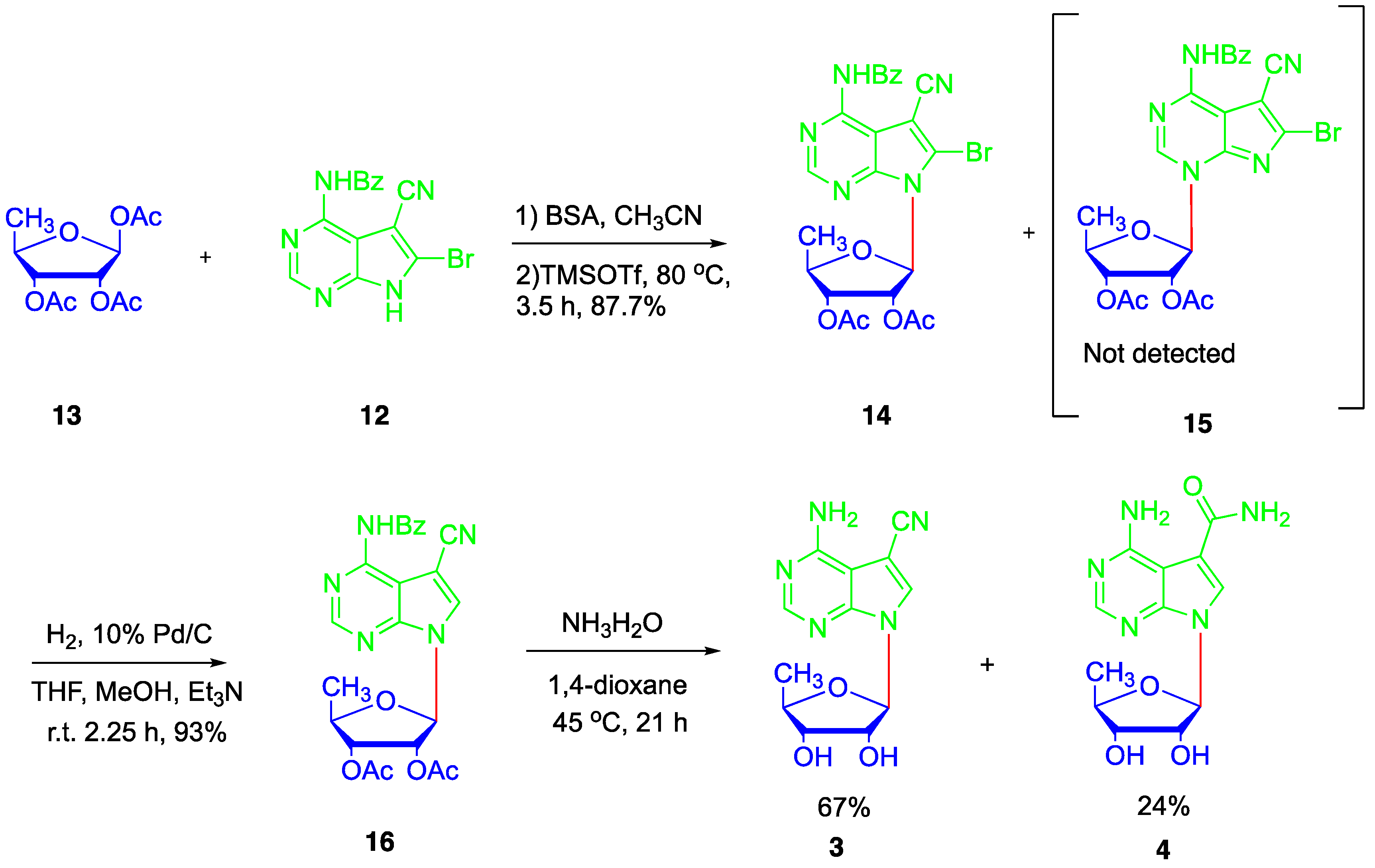
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Materials and Instruments
4.2. Synthesis of 2-amino-5-bromo-3,4-dicyano-1H-pyrrole (10)
4.3. Synthesis of 4-amino-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine (6)
4.4. Synthesis of N4-benzoyl-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine (12)
4.5. Synthesis of N4-benzoyl-5-cyano-6-bromo-7-(2′,3′-bi-O-acetyl-5′-deoxy-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (14)
4.6. Synthesis of N4-benzoyl-5-cyano-7-(2′,3′-di-O-acetyl-5′-deoxy-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (16)
4.7. Synthesis of 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4)
4.8. 5′-Deoxytoyocamycin (3)
4.9. 5′-Deoxysangivamycin (4)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Note
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wnuk, S.F. Modification of Purine and Pyrimidine Nucleosides by Direct C-H Bond Activation. Molecules 2015, 20, 4874–4901. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Littler, E. Nucleoside analogs as anti-HBV agents. Curr. Top. Med. Chem. 2006, 6, 851–865. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G.D. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Perlikova, P.; Hocek, M. Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. [Google Scholar] [CrossRef]
- Townsend, L.B.; Drach, J.C.; Wotring, L.L.; Vittori, S.; Pudlo, J.S.; Swayze, E.E.; Gupta, P.; Maruyama, T.; Saxena, N.; Coleman, L.A. Design, synthesis, and studies on the structure-activity relationships of certain pyrrolo[2,3-d]pyrimidine nucleosides and structurally related analogs as potential antineoplastic and antiviral agents. Farmaco 1991, 46, 113–139. [Google Scholar] [CrossRef]
- Dholakia, S.P.; Patel, M.M.; Patel, J.S. Role of pyrrolopyrimidine derivatives as anticancer agent: Mini review. Indo Am. J. Pharm. Res. 2015, 5, 858–867. [Google Scholar]
- De Coen, L.M.; Heugebaert, T.S.A.; Garcia, D.; Stevens, C.V. Synthetic Entries to and Biological Activity of Pyrrolopyrimidines. Chem. Rev. 2016, 116, 80–139. [Google Scholar] [CrossRef]
- Cahova, H.; Panattoni, A.; Kielkowski, P.; Fanfrlik, J.; Hocek, M. 5-Substituted Pyrimidine and 7-Substituted 7-Deazapurine dNTPs as Substrates for DNA Polymerases in Competitive Primer Extension in the Presence of Natural dNTPs. ACS Chem. Biol. 2016, 11, 3165–3171. [Google Scholar] [CrossRef] [PubMed]
- Seela, F.; Sirivolu, V.R. DNA containing side chains with terminal triple bonds: Base-pair stability and functionalization of alkynylated pyrimidines and 7-deazapurines. Chem. Biodivers. 2006, 3, 509–514. [Google Scholar] [CrossRef]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Sanna, M.; Grossi, G.; Brullo, C.; Fallacara, A.L.; Schenone, S. Pyrrolo[2,3-d]Pyrimidines as Kinase Inhibitors. Curr. Med. Chem. 2017, 24, 2059–2085. [Google Scholar] [CrossRef]
- Musumeci, F.; Sanna, M.; Greco, C.; Giacchello, I.; Fallacara, A.L.; Amato, R.; Schenone, S. Pyrrolo[2,3-d]pyrimidines active as Btk inhibitors. Expert Opin. Ther. Pat. 2017, 27, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hogenkamp, H.P.C.; Long, R.A.; Revankar, G.R.; Robins, R.K. A convenient synthesis of 5’-deoxyribonucleosides. Carbohydr. Res. 1977, 59, 449–457. [Google Scholar] [CrossRef]
- Porcari, A.R.; Ptak, R.G.; Borysko, K.Z.; Breitenbach, J.M.; Vittori, S.; Wotring, L.L.; Drach, J.C.; Townsend, L.B. Deoxy Sugar Analogues of Triciribine: Correlation of Antiviral and Antiproliferative Activity with Intracellular Phosphorylation. J. Med. Chem. 2000, 43, 2438–2448. [Google Scholar] [CrossRef] [PubMed]
- Vorbrueggen, H.; Ruh-Pohlenz, C. Synthesis of nucleosides. Org. React. (Hobokennju. S.) 2000, 55. [Google Scholar] [CrossRef]
- Seela, F.; Peng, X. Progress in 7-deazapurine-pyrrolo[2,3-d]pyrimidine-ribonucleoside synthesis. Curr. Top. Med. Chem. 2006, 6, 867–892. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bloch, A.; Bobek, M. A practical synthesis of the antibiotic toyocamycin. Nucleosides Nucleotides 1993, 12, 643–648. [Google Scholar] [CrossRef]
- Porcari, A.R.; Townsend, L.B. Total synthesis of the naturally occurring antibiotic toyocamycin using new and improved synthetic procedures. Nucleosides Nucleotides 1999, 18, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Gong, Z.; Xiao, Q.; Yang, R. An improved total synthesis of 5’-deoxytoyocamycin. Adv. Mater. Res. 2014, 997, 200–202. [Google Scholar] [CrossRef]
- Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. A total synthesis of mycalisine A. Chin. Chem. Lett. 2013, 24, 379–382. [Google Scholar] [CrossRef]
- Hu, C.; Ruan, Z.Z.; Ding, H.X.; Zhou, Y.R.; Xiao, Q. An Expedient Total Synthesis of Triciribine. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Y.; Dou, Y.H.; Ding, H.X.; Yang, R.C.; Sun, Q.; Xiao, Q. First Total Synthesis of a Naturally Occurring Iodinated 5 ‘-Deoxyxylofuranosyl Marine Nucleoside. Mar. Drugs 2012, 10, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P.; Ingale, S.A.; Ding, P.; Ming, X.; Seela, F. Studies on the glycosylation of pyrrolo[2,3-d]pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose: The formation of regioisomers during toyocamycin and 7-deazainosine syntheses. Nucleosidesnucleotides Nucleic Acids 2009, 28, 678–694. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Kwon, S.H.; Bae, I.H.; Kim, B.M. Selectivity between N-1 and N-7 nucleosides: Regioselective synthesis of BMK-Y101, a potent cdk7 and 9 inhibitor. Tetrahedron Lett. 2013, 54, 5484–5488. [Google Scholar] [CrossRef]
- Zhou, L.; Amblard, F.; Zhang, H.; McBrayer, T.R.; Detorio, M.A.; Whitaker, T.; Coats, S.J.; Schinazi, R.F. Synthesis and evaluation of Janus type nucleosides as potential HCV NS5B polymerase inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 3385–3388. [Google Scholar] [CrossRef]
- Tolman, R.L.; Robins, R.K.; Townsend, L.B. Pyrrolopyrimidine nucleosides. III. Total synthesis of toyocamycin, sangivamycin, tubercidin, and related derivatives. J. Am. Chem. Soc. 1969, 91, 2102–2108. [Google Scholar] [CrossRef]
- Tolman, R.L.; Robins, R.K.; Townsend, L.B. Pyrrolo[2,3-d]pyrimidine nucleoside antibiotics. Total synthesis and structure of toyocamycin, unamycin B, vengicide, antibiotic E-212, and sangivamycin (BA-90912). J. Am. Chem. Soc. 1968, 90, 524–526. [Google Scholar] [CrossRef]
- Xiao, C.; Sun, C.; Han, W.W.; Pan, F.; Dan, Z.; Li, Y.; Song, Z.G.; Jin, Y.H. Synthesis of 6-(het) ary Xylocydine analogues and evaluating their inhibitory activities of CDK1 and CDK2 in vitro. Bioorganic Med. Chem. 2011, 19, 7100–7110. [Google Scholar] [CrossRef] [PubMed]
- Porcari, A.R.; Townsend, L.B. An improved synthesis of the versatile heterocycle, 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine. Synth. Commun. 1998, 28, 3835–3843. [Google Scholar] [CrossRef]
- Suh, H.; Choi, K.-W.; Lee, J.; Ryou, C.; Rhee, H.; Lee, C.-H. Effects of a novel carbocyclic analog of pyrrolo[2,3-d]pyrimidine nucleoside on pleiotropic induction of cell death in prostate cancer cells with different androgen responsiveness. Bioorganic Med. Chem. Lett. 2016, 26, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, H.; Yoon, S.; Choi, K.; Lee, C.-H.; Rhee, H. The synthesis and evaluation of new carbocyclic pyrrolo[2,3-d]pyrimidine nucleoside analogs. Heterocycles 2014, 89, 1606–1619. [Google Scholar] [CrossRef]
- Wilding, B.; Winkler, M.; Petschacher, B.; Kratzer, R.; Glieder, A.; Klempier, N. Nitrile Reductase from Geobacillus kaustophilus: A Potential Catalyst for a New Nitrile Biotransformation Reaction. Adv. Synth. Catal. 2012, 354, 2191–2198. [Google Scholar] [CrossRef]
- Zemlicka, J.; Owens, J. Nucleic Acid Chemistry, Improved and New Synthetic Procedures, Methods and Techniques, Part 4; Wiley-Interscience: New York, NY, USA, 1991; pp. 16–18. [Google Scholar]
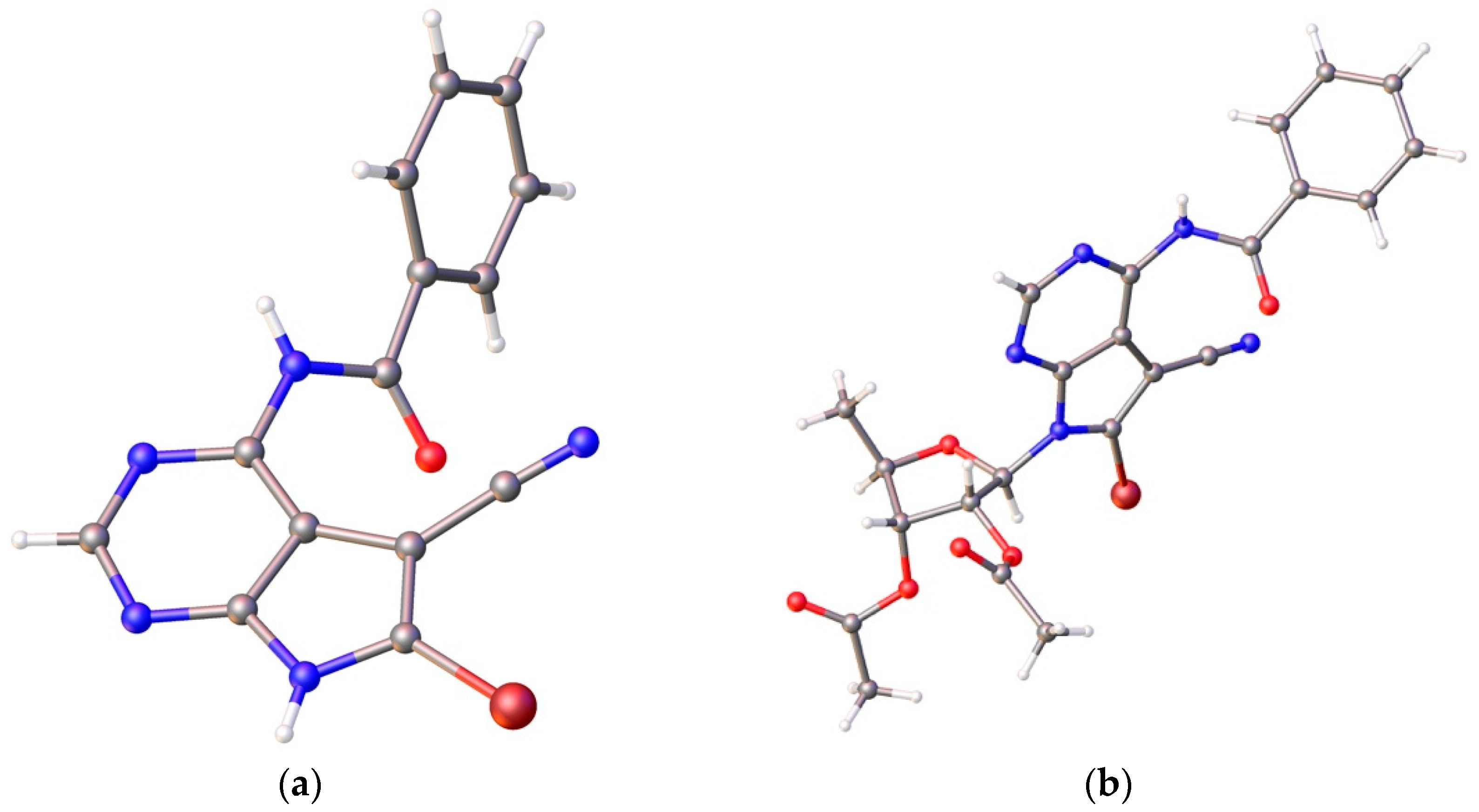
- Crystallographic data for nucleobase 12: C14H8BrN5O, M = 324.16, crystal dimensions 0.08 × 0.12 × 0.20 mm, colorless block, space group, C2/c (No. 15). CCDC 1525337 contains the supplementary crystallographic data. Crystallographic data for nucleoside 14: C23H20BrN5O6, M = 542.35, crystal dimensions 0.07 × 0.20 × 0.22 mm, monoclinic, space group, P21 (No. 4). CCDC 1895393 contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Date Centre via www.ccdc.cam.ac.uk/date_request/cif.
- Prusiner, P.; Sundaralingam, M. The crystal and molecular structure of toyocamycin monohydrate, a nucleoside antibiotic. Acta Crystallogr. Sect. B 1978, B34, 517–523. [Google Scholar] [CrossRef]
- Isaac, B.G.; Ayer, S.W.; Letendre, L.J.; Stonard, R.J. Herbicidal nucleosides from microbial sources. J. Antibiot. 1991, 44, 729–732. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3–16 are available from Prof. Dr. Qiang Xiao. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, X.; Tang, J.; Hu, C.; Bai, J.; Ding, H.; Xiao, Q. An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin. Molecules 2019, 24, 737. https://doi.org/10.3390/molecules24040737
Dong X, Tang J, Hu C, Bai J, Ding H, Xiao Q. An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin. Molecules. 2019; 24(4):737. https://doi.org/10.3390/molecules24040737
Chicago/Turabian StyleDong, Xiangyou, Jie Tang, Chen Hu, Jiang Bai, Haixin Ding, and Qiang Xiao. 2019. "An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin" Molecules 24, no. 4: 737. https://doi.org/10.3390/molecules24040737
APA StyleDong, X., Tang, J., Hu, C., Bai, J., Ding, H., & Xiao, Q. (2019). An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin. Molecules, 24(4), 737. https://doi.org/10.3390/molecules24040737