Molecular Mechanisms of Colistin-Induced Nephrotoxicity

Abstract
1. Introduction
2. Drug-Induced Nephrotoxicity: An Overview
3. Clinical and Histopathological Manifestations of Colistin-Induced Nephrotoxicity
4. The Detergent Theory
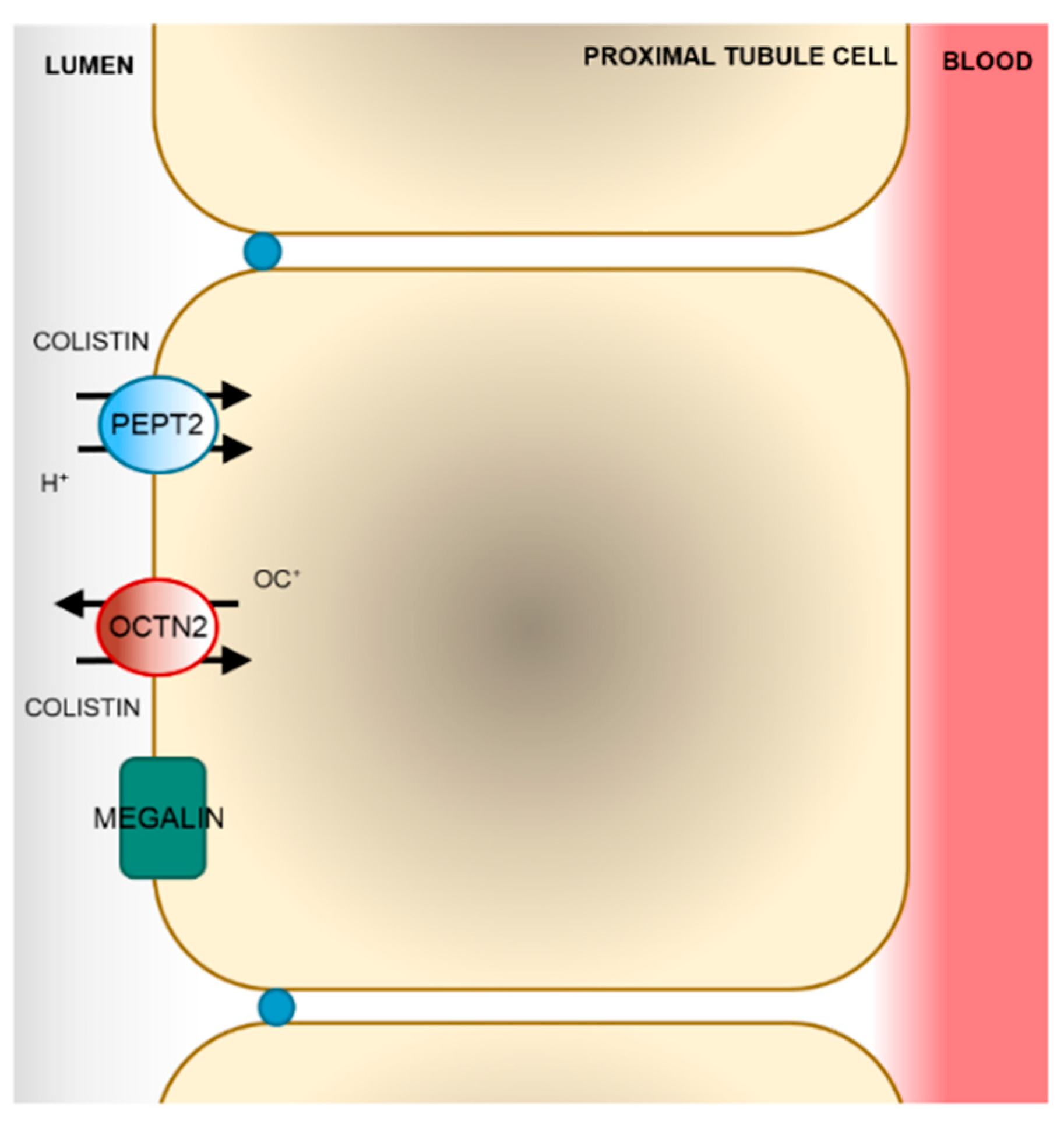
5. Membrane Transport of Colistin
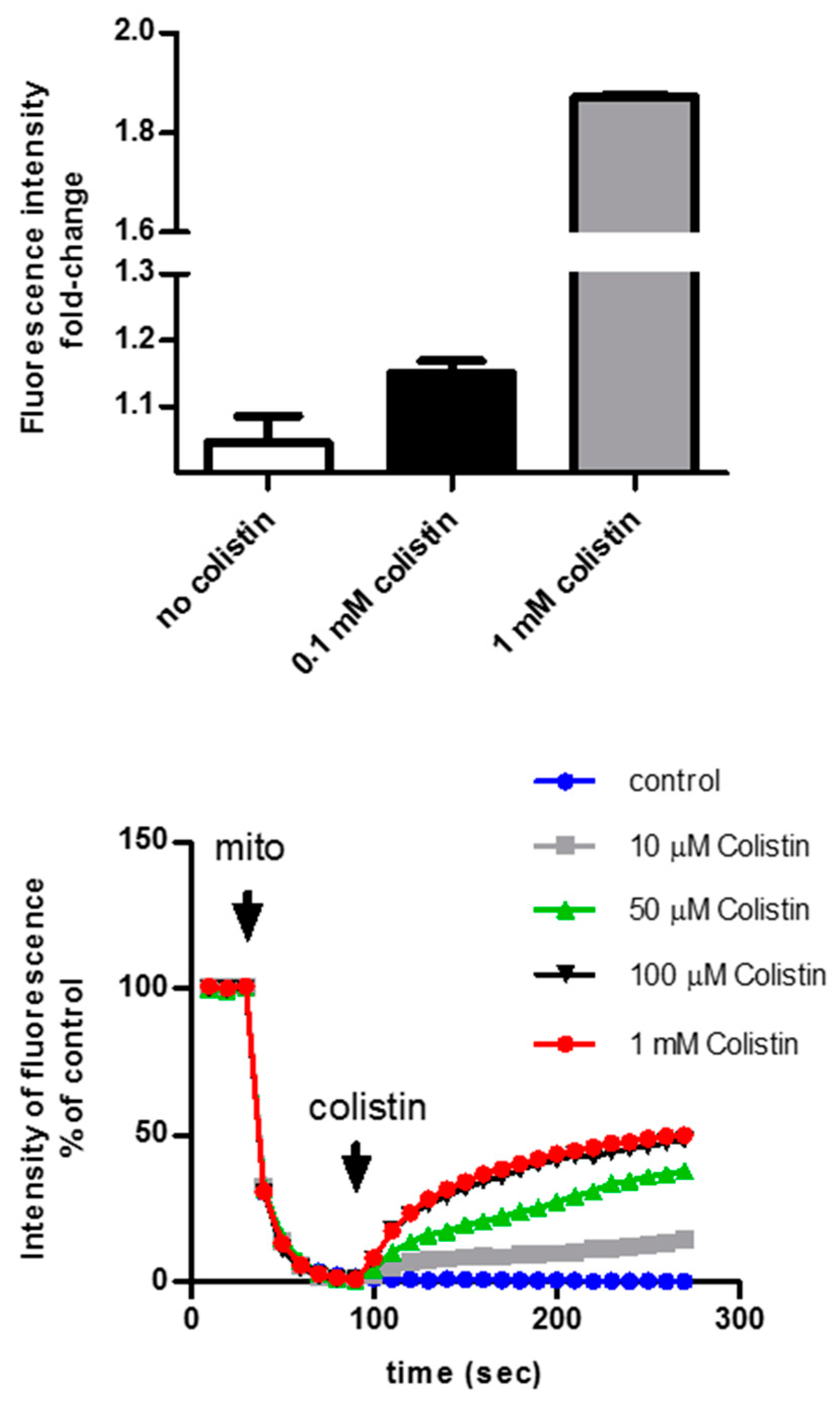
6. The Intracellular Fate of Colistin
7. Protective Strategies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Koyama, Y. A new antibiotic “colistin” produced by spore-forming soil bacteria. J. Antibiot. 1950, 3, 457–458. [Google Scholar]
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed. Res. Int. 2015, 2015, 679109. [Google Scholar] [CrossRef] [PubMed]
- Deris, Z.Z.; Akter, J.; Sivanesan, S.; Roberts, K.D.; Thompson, P.E.; Nation, R.L.; Li, J.; Velkov, T. A secondary mode of action of polymyxins against Gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J. Antibiot. 2014, 67, 147–151. [Google Scholar] [PubMed]
- Kirby, W.M.M.; Evans Roberts, C., Jr. Colistin. JAMA 1961, 177, 854. [Google Scholar]
- Kunin, C.M. Nephrotoxicity of antibiotics. JAMA 1967, 202, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Nord, N.M.; Hoeprich, P.D. Polymyxin B and Colistin. A Critical Comparison. N. Engl. J. Med. 1964, 270, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119, S3–S10, discussion S62–S70. [Google Scholar] [CrossRef]
- Palevsky, P.M.; Liu, K.D.; Brophy, P.D.; Chawla, L.S.; Parikh, C.R.; Thakar, C.V.; Tolwani, A.J.; Waikar, S.S.; Weisbord, S.D. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for acute kidney injury. Am. J. Kidney Dis. 2013, 61, 649–672. [Google Scholar] [CrossRef]
- Kellum, J.A.; Lameire, N.; Group, K.A.G.W. Diagnosis, evaluation, and management of acute kidney injury: A KDIGO summary (Part 1). Crit. Care 2013, 17, 204. [Google Scholar] [CrossRef]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Pascual, M.T.; Soroko, S.; Savage, B.R.; Himmelfarb, J.; Ikizler, T.A.; Paganini, E.P.; Chertow, G.M.; Program to Improve Care in Acute Renal Disease. Spectrum of acute renal failure in the intensive care unit: The PICARD experience. Kidney Int. 2004, 66, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Moffett, B.S.; Goldstein, S.L. Acute kidney injury and increasing nephrotoxic-medication exposure in noncritically-ill children. Clin. J. Am. Soc. Nephrol. 2011, 6, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Awdishu, L.; Mehta, R.L. The 6R’s of drug induced nephrotoxicity. BMC Nephrol. 2017, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Preuss, H.G. Basics of renal anatomy and physiology. Clin. Lab. Med 1993, 13, 1–11. [Google Scholar] [CrossRef]
- Zager, R.A. Pathogenetic mechanisms in nephrotoxic acute renal failure. Semin. Nephrol. 1997, 17, 3–14. [Google Scholar]
- Ferguson, M.A.; Vaidya, V.S.; Bonventre, J.V. Biomarkers of nephrotoxic acute kidney injury. Toxicology 2008, 245, 182–193. [Google Scholar] [CrossRef]
- Schetz, M.; Dasta, J.; Goldstein, S.; Golper, T. Drug-induced acute kidney injury. Curr. Opin. Crit. Care 2005, 11, 555–565. [Google Scholar] [CrossRef]
- Ghlissi, Z.; Hakim, A.; Mnif, H.; Ayadi, F.M.; Zeghal, K.; Rebai, T.; Sahnoun, Z. Evaluation of colistin nephrotoxicity administered at different doses in the rat model. Renal Failure 2013, 35, 1130–1135. [Google Scholar] [CrossRef]
- Falagas, M.E.; Rafailidis, P.I. Nephrotoxicity of colistin: New insight into an old antibiotic. Clin. Infectious Dis. 2009, 48, 1729–1731. [Google Scholar] [CrossRef]
- Shields, R.K.; Anand, R.; Clarke, L.G.; Paronish, J.A.; Weirich, M.; Perone, H.; Kieserman, J.; Freedy, H.; Andrzejewski, C.; Bonilla, H. Defining the incidence and risk factors of colistin-induced acute kidney injury by KDIGO criteria. PLoS ONE 2017, 12, e0173286. [Google Scholar] [CrossRef]
- Pogue, J.M.; Ortwine, J.K.; Kaye, K.S. Optimal Usage of Colistin: Are We Any Closer? Clin. Infectious Dis. 2015, 61, 1778–1780. [Google Scholar] [CrossRef]
- Dudhani, R.V.; Turnidge, J.D.; Coulthard, K.; Milne, R.W.; Rayner, C.R.; Li, J.; Nation, R.L. Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob. Agents Chemother. 2010, 54, 1117–1124. [Google Scholar] [CrossRef]
- Hartzell, J.D.; Neff, R.; Ake, J.; Howard, R.; Olson, S.; Paolino, K.; Vishnepolsky, M.; Weintrob, A.; Wortmann, G. Nephrotoxicity associated with intravenous colistin (colistimethate sodium) treatment at a tertiary care medical center. Clin. Infectious Dis. 2009, 48, 1724–1728. [Google Scholar] [CrossRef]
- Florescu, D.F.; Qiu, F.; McCartan, M.A.; Mindru, C.; Fey, P.D.; Kalil, A.C. What is the efficacy and safety of colistin for the treatment of ventilator-associated pneumonia? A systematic review and meta-regression. Clin. Infectious Dis. 2012, 54, 670–680. [Google Scholar] [CrossRef]
- Garnacho-Montero, J.; Ortiz-Leyba, C.; Jimenez-Jimenez, F.J.; Barrero-Almodovar, A.E.; Garcia-Garmendia, J.L.; Bernabeu-Wittel, I.M.; Gallego-Lara, S.L.; Madrazo-Osuna, J. Treatment of multidrug-resistant Acinetobacter baumannii ventilator-associated pneumonia (VAP) with intravenous colistin: A comparison with imipenem-susceptible VAP. Clin. Infectious Dis. 2003, 36, 1111–1118. [Google Scholar] [CrossRef]
- Kim, J.; Lee, K.H.; Yoo, S.; Pai, H. Clinical characteristics and risk factors of colistin-induced nephrotoxicity. Int. J. Antimicrob. Agents 2009, 34, 434–438. [Google Scholar] [CrossRef]
- Kofteridis, D.P.; Alexopoulou, C.; Valachis, A.; Maraki, S.; Dimopoulou, D.; Georgopoulos, D.; Samonis, G. Aerosolized plus intravenous colistin versus intravenous colistin alone for the treatment of ventilator-associated pneumonia: A matched case-control study. Clin. Infectious Dis. 2010, 51, 1238–1244. [Google Scholar] [CrossRef]
- Linden, P.K.; Kusne, S.; Coley, K.; Fontes, P.; Kramer, D.J.; Paterson, D. Use of parenteral colistin for the treatment of serious infection due to antimicrobial-resistant Pseudomonas aeruginosa. Clin. Infectious Dis. 2003, 37, e154–e160. [Google Scholar] [CrossRef]
- Linden, P.K.; Paterson, D.L. Parenteral and inhaled colistin for treatment of ventilator-associated pneumonia. Clin. Infectious Dis. 2006, 43, S89–S94. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.S.; Ronco, C.; Katz, N.; Cruz, D.N. Early diagnosis of acute kidney injury: The promise of novel biomarkers. Blood Purif. 2009, 28, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Keirstead, N.D.; Wagoner, M.P.; Bentley, P.; Blais, M.; Brown, C.; Cheatham, L.; Ciaccio, P.; Dragan, Y.; Ferguson, D.; Fikes, J.; et al. Early prediction of polymyxin-induced nephrotoxicity with next-generation urinary kidney injury biomarkers. Toxicol. Sci. 2014, 137, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Yang, L.; Weiss, T.M.; Lehrer, R.I.; Huang, H.W. Crystallization of antimicrobial pores in membranes: Magainin and protegrin. Biophys. J. 2000, 79, 2002–2009. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Kunin, C.M. Binding of antibiotics to tissue homogenates. J. Infectious Dis. 1970, 121, 55–64. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, Y.F.; Abou-Shleib, H.M.; Khalil, A.M.; El-Guink, N.M.; El-Nakeeb, M.A. Membrane permeabilization of colistin toward pan-drug resistant Gram-negative isolates. Braz. J. Microbiol. 2016, 47, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Visentin, M.; Hiller, C.; Krajnc, E.; Li, T.; Zhen, J.; Kullak-Ublick, G.A. Organic Cation Transporter 2 (OCT2-SLC22A2) overexpression may confer an increased risk of gentamicin-induced nephrotoxicity. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Smeaton, T.C.; Coulthard, K. Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob. Agents Chemother. 2003, 47, 1766–1770. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Ogura, J.; Kobayashi, M.; Yamada, T.; Iseki, K. Megalin contributes to kidney accumulation and nephrotoxicity of colistin. Antimicrob. Agents Chemother. 2013, 57, 6319–6324. [Google Scholar] [CrossRef]
- Moestrup, S.K.; Cui, S.; Vorum, H.; Bregengard, C.; Bjorn, S.E.; Norris, K.; Gliemann, J.; Christensen, E.I. Evidence that epithelial glycoprotein 330/megalin mediates uptake of polybasic drugs. J. Clin. Invest. 1995, 96, 1404–1413. [Google Scholar] [CrossRef]
- Schmitz, C.; Hilpert, J.; Jacobsen, C.; Boensch, C.; Christensen, E.I.; Luft, F.C.; Willnow, T.E. Megalin deficiency offers protection from renal aminoglycoside accumulation. Int. J. Biol. Chem. 2002, 277, 618–622. [Google Scholar] [CrossRef]
- Lu, X.; Chan, T.; Xu, C.; Zhu, L.; Zhou, Q.T.; Roberts, K.D.; Chan, H.K.; Li, J.; Zhou, F. Human oligopeptide transporter 2 (PEPT2) mediates cellular uptake of polymyxins. J. Antimicrob. Chemother. 2016, 71, 403–412. [Google Scholar] [CrossRef]
- Visentin, M.; Gai, Z.; Torozi, A.; Hiller, C.; Kullak-Ublick, G.A. Colistin is substrate of the carnitine/organic cation transporter 2 (OCTN2, SLC22A5). Drug Metab. Dispos. 2017, 45, 1240–1244. [Google Scholar] [CrossRef]
- Visentin, M.; Stieger, B.; Merz, M.; Kullak-Ublick, G.A. Octreotide Inhibits the Bilirubin Carriers Organic Anion Transporting Polypeptides 1B1 and 1B3 and the Multidrug Resistance-Associated Protein 2. J. Pharmacol. Exp. Ther. 2015, 355, 145–151. [Google Scholar] [CrossRef]
- Tamai, I. Pharmacological and pathophysiological roles of carnitine/organic cation transporters (OCTNs: SLC22A4, SLC22A5 and Slc22a21). Biopharm. Drug Dispos. 2013, 34, 29–44. [Google Scholar] [CrossRef]
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 1999, 21, 91–94. [Google Scholar] [CrossRef]
- Jeong, E.S.; Kim, G.; Moon, K.S.; Kim, Y.B.; Oh, J.H.; Kim, H.S.; Jeong, J.; Shin, J.G.; Kim, D.H. Characterization of urinary metabolites as biomarkers of colistin-induced nephrotoxicity in rats by a liquid chromatography/mass spectrometry-based metabolomics approach. Toxicol Lett. 2016, 248, 52–60. [Google Scholar] [CrossRef]
- Lu, K.; Nishimori, H.; Nakamura, Y.; Shima, K.; Kuwajima, M. A missense mutation of mouse OCTN2, a sodium-dependent carnitine cotransporter, in the juvenile visceral steatosis mouse. Biochem. Biophys. Res. Commun. 1998, 252, 590–594. [Google Scholar] [CrossRef]
- Rubio-Aliaga, I.; Boll, M.; Daniel, H. Cloning and characterization of the gene encoding the mouse peptide transporter PEPT2. Biochem. Biophys. Res. Commun. 2000, 276, 734–741. [Google Scholar] [CrossRef]
- Tamai, I.; China, K.; Sai, Y.; Kobayashi, D.; Nezu, J.; Kawahara, E.; Tsuji, A. Na(+)-coupled transport of L-carnitine via high-affinity carnitine transporter OCTN2 and its subcellular localization in kidney. Biochim. Biophys. Acta 2001, 1512, 273–284. [Google Scholar] [CrossRef]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. The Int. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef]
- Shen, H.; Smith, D.E.; Yang, T.; Huang, Y.G.; Schnermann, J.B.; Brosius, F.C. Localization of PEPT1 and PEPT2 proton-coupled oligopeptide transporter mRNA and protein in rat kidney. Am. J. Physiol. 1999, 276, F658–F665. [Google Scholar] [CrossRef]
- Christensen, E.I.; Verroust, P.J.; Nielsen, R. Receptor-mediated endocytosis in renal proximal tubule. Eur. J. Physiol. 2009, 458, 1039–1048. [Google Scholar] [CrossRef]
- Yun, B.; Azad, M.A.; Wang, J.; Nation, R.L.; Thompson, P.E.; Roberts, K.D.; Velkov, T.; Li, J. Imaging the distribution of polymyxins in the kidney. J. Antimicrob. Chemother. 2015, 70, 827–829. [Google Scholar] [CrossRef]
- Dai, C.; Li, J.; Tang, S.; Li, J.; Xiao, X. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob. Agents Chemother. 2014, 58, 4075–4085. [Google Scholar] [CrossRef]
- Mogi, T.; Murase, Y.; Mori, M.; Shiomi, K.; Omura, S.; Paranagama, M.P.; Kita, K. Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: Quinone oxidoreductase from the Gram-positive bacterium Mycobacterium smegmatis. J. Biochem. 2009, 146, 491–499. [Google Scholar] [CrossRef]
- Tochikubo, K.; Yasuda, Y.; Kozuka, S. Decreased particulate NADH oxidase activity in Bacillus subtilis spores after polymyxin B treatment. J. Gen. Microbiol. 1986, 132, 277–287. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, Y.J. Enzymatic properties of the membrane-bound NADH oxidase system in the aerobic respiratory chain of Bacillus cereus. J. Biochem. Mol. Biol. 2004, 37, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, S.; Drose, S.; Zickermann, V.; Brandt, U. The three families of respiratory NADH dehydrogenases. Results Probl. Cell Differ. 2008, 45, 185–222. [Google Scholar] [PubMed]
- Yagi, T.; Yano, T.; Di Bernardo, S.; Matsuno-Yagi, A. Procaryotic complex I (NDH-1), an overview. Biochim. Biophys. Acta 1998, 1364, 125–133. [Google Scholar] [CrossRef]
- Mogi, T.; Matsushita, K.; Murase, Y.; Kawahara, K.; Miyoshi, H.; Ui, H.; Shiomi, K.; Omura, S.; Kita, K. Identification of new inhibitors for alternative NADH dehydrogenase (NDH-II). FEMS Microbiol. Lett. 2009, 291, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Marreiros, B.C.; Sena, F.V.; Sousa, F.M.; Batista, A.P.; Pereira, M.M. Type II NADH:quinone oxidoreductase family: Phylogenetic distribution, structural diversity and evolutionary divergences. Environ. Microbiol. 2016, 18, 4697–4709. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.R.; Gong, M.; Wodke, L.; Lamb, J.H.; Jones, D.J.; Farmer, P.B.; Scrutton, N.S.; Munro, A.W. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. J. Biol. Chem. 2005, 280, 30735–30740. [Google Scholar] [CrossRef]
- Elguindy, M.M.; Nakamaru-Ogiso, E. Apoptosis-inducing Factor (AIF) and Its Family Member Protein, AMID, Are Rotenone-sensitive NADH:Ubiquinone Oxidoreductases (NDH-2). J. Biol. Chem. 2015, 290, 20815–20826. [Google Scholar] [CrossRef]
- Temocin, F.; Erdinc, S.; Tulek, N.; Demirelli, M.; Bulut, C.; Ertem, G. Incidence and Risk Factors for Colistin-Associated Nephrotoxicity. Jpn. J. Infect. Dis. 2015, 68, 318–320. [Google Scholar] [CrossRef]
- Gauthier, T.P.; Wolowich, W.R.; Reddy, A.; Cano, E.; Abbo, L.; Smith, L.B. Incidence and predictors of nephrotoxicity associated with intravenous colistin in overweight and obese patients. Antimicrob. Agents Chemother. 2012, 56, 2392–2396. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.C.; Whitesell, R.R. Ascorbate is the major electron donor for a transmembrane oxidoreductase of human erythrocytes. Biochim. Biophys. Acta 1995, 1238, 127–136. [Google Scholar] [CrossRef]
- Niki, E. Action of ascorbic acid as a scavenger of active and stable oxygen radicals. Am. J. Clin. Nutr. 1991, 54, 1119S–1124S. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Mongan, P.D. Ascorbate reduces superoxide production and improves mitochondrial respiratory chain function in human fibroblasts with electron transport chain deficiencies. Mitochondrion 2001, 1, 191–198. [Google Scholar] [CrossRef]
- Sirijatuphat, R.; Limmahakhun, S.; Sirivatanauksorn, V.; Nation, R.L.; Li, J.; Thamlikitkul, V. Preliminary clinical study of the effect of ascorbic acid on colistin-associated nephrotoxicity. Antimicrob. Agents Chemother. 2015, 59, 3224–3232. [Google Scholar] [CrossRef]
- Yousef, J.M.; Chen, G.; Hill, P.A.; Nation, R.L.; Li, J. Ascorbic acid protects against the nephrotoxicity and apoptosis caused by colistin and affects its pharmacokinetics. J. Antimicrob. Chemother. 2012, 67, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Bader, Z.; Waheed, A.; Bakhtiar, S.; Khan, I.M. Alpha-tocopherol ameliorates nephrotoxicity associated with the use of colistin in rabbits. Pak. J. Pharm. Sci. 2018, 31, 463–467. [Google Scholar] [PubMed]
- Dai, C.; Tang, S.; Wang, Y.; Velkov, T.; Xiao, X. Baicalein acts as a nephroprotectant that ameliorates colistin-induced nephrotoxicity by activating the antioxidant defence mechanism of the kidneys and down-regulating the inflammatory response. J. Antimicrob. Chemother. 2017, 72, 2562–2569. [Google Scholar] [CrossRef]
- Hori, Y.; Aoki, N.; Kuwahara, S.; Hosojima, M.; Kaseda, R.; Goto, S.; Iida, T.; De, S.; Kabasawa, H.; Kaneko, R.; et al. Megalin Blockade with Cilastatin Suppresses Drug-Induced Nephrotoxicity. JASN 2017, 28, 1783–1791. [Google Scholar] [CrossRef]
- Edrees, N.E.; Galal, A.A.A.; Abdel Monaem, A.R.; Beheiry, R.R.; Metwally, M.M.M. Curcumin alleviates colistin-induced nephrotoxicity and neurotoxicity in rats via attenuation of oxidative stress, inflammation and apoptosis. Chem. Biol. Interact. 2018, 294, 56–64. [Google Scholar] [CrossRef]
- Sivanesan, S.S.; Azad, M.A.K.; Schneider, E.K.; Ahmed, M.U.; Huang, J.; Wang, J.; Li, J.; Nation, R.L.; Velkov, T. Gelofusine Ameliorates Colistin-Induced Nephrotoxicity. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Ozkan, G.; Ulusoy, S.; Orem, A.; Alkanat, M.; Mungan, S.; Yulug, E.; Yucesan, F.B. How does colistin-induced nephropathy develop and can it be treated? Antimicrob. Agents Chemother. 2013, 57, 3463–3469. [Google Scholar] [CrossRef]
- Arslan, B.Y.; Arslan, F.; Erkalp, K.; Alagol, A.; Sevdi, M.S.; Yildiz, G.; Kucuk, S.H.; Altinay, S. Luteolin ameliorates colistin-induced nephrotoxicity in the rat models. Renal Failure 2016, 38, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Tang, S.; Deng, S.; Zhang, S.; Zhou, Y.; Velkov, T.; Li, J.; Xiao, X. Lycopene attenuates colistin-induced nephrotoxicity in mice via activation of the Nrf2/HO-1 pathway. Antimicrob. Agents Chemother. 2015, 59, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Yousef, J.M.; Chen, G.; Hill, P.A.; Nation, R.L.; Li, J. Melatonin attenuates colistin-induced nephrotoxicity in rats. Antimicrob. Agents Chemother. 2011, 55, 4044–4049. [Google Scholar] [CrossRef] [PubMed]
- Ozyilmaz, E.; Ebinc, F.A.; Derici, U.; Gulbahar, O.; Goktas, G.; Elmas, C.; Oguzulgen, I.K.; Sindel, S. Could nephrotoxicity due to colistin be ameliorated with the use of N-acetylcysteine? Intensive Care Med. 2011, 37, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Ceylan, B.; Ozansoy, M.; Kilic, U.; Yozgat, Y.; Ercan, C.; Yildiz, P.; Aslan, T. N-acetylcysteine suppresses colistimethate sodium-induced nephrotoxicity via activation of SOD2, eNOS, and MMP3 protein expressions. Renal Failure 2018, 40, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.; Thomann, C.; Ettarh, R.; Ahmad, Z. Possible protective role of silybin against polymyxin E-induced toxic effect in rat kidneys: A biochemical approach. Neurourol. Urodyn 2017, 36, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Heidari, R.; Behnamrad, S.; Khodami, Z.; Ommati, M.M.; Azarpira, N.; Vazin, A. The nephroprotective properties of taurine in colistin-treated mice is mediated through the regulation of mitochondrial function and mitigation of oxidative stress. Biomed. Pharmacother. 2018, 109, 103–111. [Google Scholar] [CrossRef]
- Ghlissi, Z.; Hakim, A.; Sila, A.; Mnif, H.; Zeghal, K.; Rebai, T.; Bougatef, A.; Sahnoun, Z. Evaluation of efficacy of natural astaxanthin and vitamin E in prevention of colistin-induced nephrotoxicity in the rat model. Environ. Toxicol. Pharmacol. 2014, 37, 960–966. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Species | Colistin PK | Kidney Function | Reference |
|---|---|---|---|---|
| Ascorbate | rat | ⇑ AUC ⇑ vd | ⇓ NGAL ⇓ apoptosis | [74] |
| human | ⇔ AUC | ⇔ NGAL ⇔ NAG | [73] | |
| α-Tocopherol | rabbit | ND | ⇓ sCRE ⇓ BUN | [75] |
| Baicalein | mouse | ND | ⇓ BUN ⇓ sCRE | [76] |
| Cilastatin | mouse | ND | ⇓ NGAL ⇓ KIM-1 | [77] |
| Curcumin | rat | ND | ⇓ MDA ⇓ NO ⇓ inflammation | [78] |
| Gelofusin | mouse | ND | ⇓ necrosis ⇓ inflammation | [79] |
| rat | ⇔ AUC ⇔ vd | ND | ||
| Proanthocyanidin | rat | ND | ⇓ BUN ⇓ sCRE ⇓ iNOS | [80] |
| Luteolin | rat | ND | ⇓ sCRE ⇓ apoptosis | [81] |
| Lycopene | mouse | ND | ⇓ sCRE ⇓ BUN ⇓ apoptosis ⇓ necrosis | [82] |
| Melatonin | rat | ⇑ AUC ⇑ vd | ⇓ NGAL ⇓ sCRE | [83] |
| N-acetylcysteine | rat | ND | ⇓ SOD ⇓ NO | [84,85] |
| Sylibin | rat | ND | ⇓ NAG ⇓ Urea ⇓ sCRE ⇓ Urate ⇓Na+, K+ | [86] |
| Taurin | mouse | ND | ⇓ sCRE ⇓ BUN | [87] |
| Vitamin E | rat | ND | ⇓ uGGT ⇓ MDA | [88] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules 2019, 24, 653. https://doi.org/10.3390/molecules24030653
Gai Z, Samodelov SL, Kullak-Ublick GA, Visentin M. Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules. 2019; 24(3):653. https://doi.org/10.3390/molecules24030653
Chicago/Turabian StyleGai, Zhibo, Sophia L. Samodelov, Gerd A. Kullak-Ublick, and Michele Visentin. 2019. "Molecular Mechanisms of Colistin-Induced Nephrotoxicity" Molecules 24, no. 3: 653. https://doi.org/10.3390/molecules24030653
APA StyleGai, Z., Samodelov, S. L., Kullak-Ublick, G. A., & Visentin, M. (2019). Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules, 24(3), 653. https://doi.org/10.3390/molecules24030653