
Microwave-Assisted Synthesis of Mono- and Disubstituted 4-Hydroxyacetophenone Derivatives via Mannich Reaction: Synthesis, XRD and HS-Analysis

 ,
,
Abstract
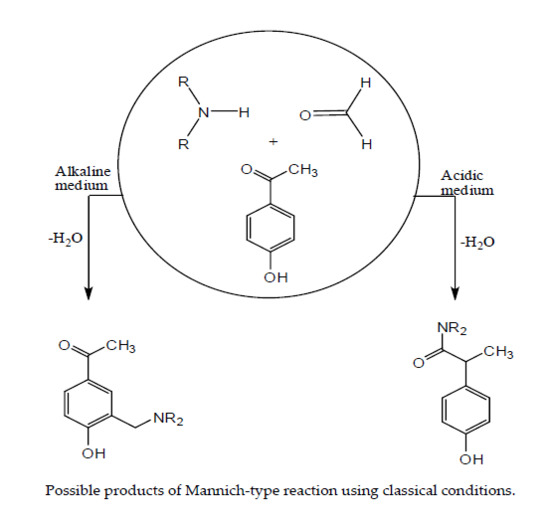
1. Introduction
2. Results and Discussion
2.1. General Synthesis
2.2. Crystal Structure Description
2.3. Hirshfeld Analysis
3. Materials and Methods
3.1. General Synthesis of Mannich Bases (2–5)
3.1.1. 1-{4-Hydroxy-3-[(morpholin-4-yl)methyl]phenyl}ethan-1-one (2a)
3.1.2. 1-{4-Hydroxy-3,5-bis[(morpholin-4-yl) methyl]phenyl}ethan-1-one (2b)
3.1.3. 1-{4-Hydroxy-3-[(pyrrolidin-1-yl) methyl]phenyl}ethan-1-one (3a)
3.1.4. 1-{4-Hydroxy-3,5-bis[(pyrrolidin-1-yl) methyl] phenyl} ethan-1-one (3b)
3.1.5. 1-{4-Hydroxy-3-[(piperidin-1-yl) methyl] phenyl} ethan-1-one (4a)
3.1.6. 1-{4-Hydroxy-3,5-bis[(piperidin-1-yl) methyl] phenyl} ethan-1-one (4b)
3.1.7. 1-{3-[(Diethylamino) methyl]-4-hydroxyphenyl} ethan-1-one (5a)
3.2. X-ray Structure Determination
3.3. Computations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tramontini, M. Advances in the Chemistry of Mannich Bases. Synthesis 1973, 12, 703–775. [Google Scholar] [CrossRef]
- Filho, J.F.A.; Lemos, B.C.; de Souza, A.S.; Pinheiro, S.; Greco, S.J. Multicomponent Mannich reactions: General aspects, methodologies and applications. Tetrahedron 2017, 73, 6977–7004. [Google Scholar] [CrossRef]
- Teissier, E.; Zandomeneghi, G.; Loquet, A.; Lavillette, D.; Lavergne, J.-P.; Montserret, R.; Cosset, F.-L.; Böckmann, A.; Meier, B.H.; Penin, F.; et al. Mechanism of Inhibition of Enveloped Virus Membrane Fusion by the Antiviral Drug Arbidol. PLoS ONE 2011, 6, e15874. [Google Scholar] [CrossRef] [PubMed]
- Boriskin, Y.; Leneva, I.; Pecheur, E.-I.; Polyak, S. Arbidol: A Broad-Spectrum Antiviral Compound that Blocks Viral Fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel Tacrine−8-Hydroxyquinoline Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease, with Neuroprotective, Cholinergic, Antioxidant, and Copper-Complexing Properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Lan, W.; Liu, Z.; Huang, J.; Tang, H.; Wang, H. Synthesis and biological evaluation of 1, 3-dihydroxyxanthone mannich base derivatives as anticholinesterase agents. Chem. Cent. J. 2013, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Dorronsoro, I.; Rubio, L.; Muñoz, P.; García-Palomero, E.; Del Monte, M.; Bidon-Chanal, A.; Orozco, M.; Luque, F.J.; Castro, A.; et al. Donepezil–tacrine hybrid related derivatives as new dual binding site inhibitors of AChE. Bioorg. Med. Chem. 2005, 13, 6588–6597. [Google Scholar] [CrossRef]
- Malhotra, M.; Sharma, R.; Sanduja, M.; Kumar, R.; Jain, E.; Deep, A. Synthesis, characterization and evaluation of Mannich bases as potent antifungal and hydrogen peroxide scavenging agents. Acta Pol. Pharm. Drug Res. 2012, 69, 355–361. [Google Scholar]
- Buravlev, E.V.; Shevchenko, O.G.; Kutchin, A.V. Synthesis and membrane-protective activity of novel derivatives of α-mangostin at the C-4 position. Bioorg. Med. Chem. Lett. 2015, 25, 826–829. [Google Scholar] [CrossRef]
- Aeluri, R.; Alla, M.; Polepalli, S.; Jain, N. Synthesis and antiproliferative activity of imidazo[1,2-a]pyrimidine Mannich bases. Eur. J. Med. Chem. 2015, 100, 18–23. [Google Scholar] [CrossRef]
- Issa, S.; Walchshofer, N.; Kassab, I.; Termoss, H.; Chamat, S.; Geahchan, A.; Bouaziz, Z. Synthesis and antiproliferative activity of oxazinocarbazole and N,N-bis(carbazolylmethyl)amine derivatives. Eur. J. Med. Chem. 2010, 45, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Joseph, T.; Halligudi, S.B. Mannich reaction in Brönsted acidic ionic liquid: A facile synthesis of β-amino carbonyl compounds. J. Mol. Catal. A Chem. 2006, 244, 179–182. [Google Scholar] [CrossRef]
- Müller, R.; Goesmann, H.; Waldmann, H. N,N-Phthaloylamino Acids as Chiral Auxiliaries in Asymmetric Mannich-Type Reactions. Angew. Chem. Int. Ed. 1999, 38, 184–187. [Google Scholar] [CrossRef]
- Xie, L.-H.; Cheng, J.; Luo, Z.-W.; Lu, G. Mannich Reaction of Indole with Cyclic Imines in Water. Tetrahedron Lett. 2018, 59, 457–461. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, S.; Qiu, R.; Xia, J.; Dai, W.; Yu, Z.; Au, C.-T.; Wong, W.-Y. Synthesis and structure of an air-stable hypervalent organobismuth (III) perfluorooctanesulfonate and its use as high-efficiency catalyst for Mannich-type reactions in water. J. Organomet. Chem. 2009, 694, 3559–3564. [Google Scholar] [CrossRef]
- Abedini-Torghabeh, J.; Eshghi, H.; Bakavoli, M.; Rahimizadeh, M. PPh3-catalyzed Mannich reaction: A facile one-pot synthesis of β-amino carbonyl compounds under solvent-free conditions at room temperature. Res. Chem. Intermed. 2015, 41, 3649–3658. [Google Scholar] [CrossRef]
- Wei, C.; Li, C.J. A highly efficient three-component coupling of aldehyde, alkyne, and amines via C-H activation catalyzed by gold in water. J. Am. Chem. Soc. 2003, 125, 9584–9585. [Google Scholar] [CrossRef]
- Li, Z.; Ma, X.; Liu, J.; Feng, X.; Tian, G.; Zhu, A. Silica-supported aluminum chloride: A recyclable and reusable catalyst for one-pot three-component Mannich-type reactions. J. Mol. Catal. A Chem. 2007, 272, 132–135. [Google Scholar] [CrossRef]
- Nagarajan, S.; Kandasamy, E. Reusable 1,2,4-Triazolium Based Brønsted Acidic Room Temperature Ionic Liquids as Catalyst for Mannich Base Reaction. Catal. Lett. 2014, 144, 1507–1514. [Google Scholar] [CrossRef]
- Zhao, G.; Jiang, T.; Gao, H.; Han, B.; Huang, J.; Sun, D. Mannich reaction using acidic ionic liquids as catalysts and solvents. Green Chem. 2004, 6, 75–77. [Google Scholar] [CrossRef]
- Lehmann, F.; Pilotti, Å.; Luthman, K. Efficient large scale microwave assisted Mannich reactions using substituted acetophenones. Mol. Divers. 2003, 7, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kabalka, G.W.; Zhou, L.-L.; Wang, L.; Pagni, R.M. A microwave-enhanced, solventless Mannich condensation of terminal alkynes and secondary amines with para-formaldehyde on cuprous iodide doped alumina. Tetrahedron 2006, 62, 857–867. [Google Scholar] [CrossRef]
- Rao, G.V.; Reddy, P.N.; Reddy, Y.T.; Kumar, V.N.; Rajitha, B. Synthesis of benzo [b] furan Mannich bases under solventless, PTSA/PTC catalytic conditions assisted by microwave irradiation. Indian J. Chem. 2005, 44, 1109–1111. [Google Scholar] [CrossRef]
- McLean, N.J.; Tye, H.; Whittaker, M. Microwave assisted Petasis boronic-Mannich reactions. Tetrahedron Lett. 2004, 45, 993–995. [Google Scholar] [CrossRef]
- Mete, E.; Gul, H.I.; Bilginer, S.; Algul, O.; Topaloglu, M.E.; Gulluce, M.; Kazaz, C. Synthesis and Antifungal Evaluation of 1-Aryl-2-dimethyl-aminomethyl-2-propen-1-one Hydrochlorides. Molecules 2011, 16, 4660–4671. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.; Habibullah, S.; Hasan, S.M.; Alam, M.M.; Akhter, N.; Shaquiquzzaman, M. Synthesis of some new 3,4-dihydro-2H-1,3-benzoxazines under microwave irradiation in solvent-free conditions and their biological activity. Med. Chem. Res. 2011, 20, 1147–1153. [Google Scholar] [CrossRef]
- Sreevalli, W.; Ramachandran, G.; Madhuri, W.; Sathiyanarayanan, K.I. ChemInform Abstract: Green Trends in Mannich Reaction. ChemInform 2015, 46, 97–115. [Google Scholar] [CrossRef]
- Mavandadi, F.; Pilotti, Å. The impact of microwave-assisted organic synthesis in drug discovery. Drug Discov. Today 2006, 11, 165–174. [Google Scholar] [CrossRef]
- Dong, X.; Liu, T.; Chen, J.; Ying, H.; Hu, Y. Microwave-Assisted Mannich Reaction of 2-Hydroxy-chalcones. Synth. Commun. 2009, 39, 733–742. [Google Scholar] [CrossRef]
- Reddy, M.V.B.; Su, C.R.; Chiou, W.F.; Liu, Y.N.; Chen, R.Y.H.; Bastow, K.F.; Lee, K.H.; Wu, T.S.; Bhaskar, M.V.; Su, C.R.; et al. Design, synthesis, and biological evaluation of Mannich bases of heterocyclic chalcone analogs as cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 7358–7370. [Google Scholar] [CrossRef]
- APEX2 (Version 2.1), SAINTPlus. Data Reduction and Correction Program (Version 7.31A, Bruker Advansed X-ray Solutions; BrukerAXS Inc.: Madison, WI, USA, 2006.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Sheldrick SHELXS-97—Program for the Solution of Crystal Structures; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Johnson, C.K. ORTEP II, Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976. [Google Scholar]
- Farrugia, L.J. UCr ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. Diamond-Crystal and Molecular Structure Visualization; Crystal Impact: Bonn, Germany, 2012. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Taylor, R.; Kennard, O. Comparison of X-ray and Neutron Diffraction Results for the N--H…O--C Hydrogen Bond. Acta Crystallogr. Sect. B 1983, 39, 133–138. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Lewis, L. Cooperative aspects of hydrogen bonding in carbohydrates. Carbohydr Res. 1978, 60, 179–182. [Google Scholar] [CrossRef]
- Rivera, A.; Duarte, Y.; González-Salas, D.; Ríos-Motta, J.; Zaragoza, G. X-ray and hydrogen-bonding Properties of 1-((1H-benzotriazol-1-yl)methyl) naphthalen-2-ol. Molecules 2009, 14, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Mozzchukhin, A.O.; Macharashvili, A.A.; Shklover, V.E.; Struchkov, Y.T.; Shipov, A.G.; Sergeev, V.N.; Artamkin, S.A.; Pestunovich, S.V.; Baukov, Y.I. Crystal and molecular structures of (O-Ge)-chelate 1-(dimethylchlorogermylmethyl)pyrrolidone-2 and 1-(dimethylchlorogermylmethyl)piperidone-2, and of (N-Ge)-chelate O-(dimethylchlorogermylmethyl)-δ-valerolactim and 2-(chlorodimethylgermylmethylthio)-pyrro. J. Organomet. Chem. 1991, 408, 305–322. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J. Crystal Explorer 3.0; University of Westren Australia: Perth, Australia, 2007. [Google Scholar]
Sample Availability: Atomic coordinates, bond lengths, bond angles and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre (CCDC). These data can be obtained free of charge via www.ccdc.cam.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336 033; or deposit@ ccdc.cam.ac.uk). Any requests to the CCDC for data should quote the full literature citation and CCDC reference numbers for 2a and 3a 1832359, 1832328 respectively. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | a | b | Microwave Yield % Reactant Ratio 1:1.5:1.5 | Microwave Yield % Reactant Ratio 1:2:2 | Time (min) | ||
|---|---|---|---|---|---|---|---|
| a | b | a | b | ||||
| 2 |  |  | 80 | 20 | - | quantitative yield | 30 |
| 3 |  |  | 49 | 15 | - | 86 | 15 |
| 4 |  |  | 58 | 14 | - | 94 | 15 |
| 5 |  | - | 77 | - | quantitative yield | - | 15 |
| Compound | 2a | 3a |
|---|---|---|
| Chemical formula | C13H17NO3 | C13H17NO2 |
| Mr | 235.27 | 219.27 |
| Crystal system, space group | Monoclinic, P21/n | Orthorhombic, Pbca |
| Temperature (K) | 100 (2) | 100 (2) |
| a, b, c (Å) | 7.297 (6), 11.375 (8), 14.571 (12) | 10.8735 (8), 10.9988 (9), 19.0645 (15) |
| β (°) | 93.79 (3) | |
| V (Å3) | 1206.9 (16) | 2280.0 (3) |
| Z | 4 | 8 |
| Radiation type | Mo Kα | Mo Kα |
| μ (mm−1) | 0.09 | 0.09 |
| Crystal size (mm) | 0.50 × 0.40 × 0.35 | 0.44 × 0.18 × 0.17 |
| Data collection | ||
| Diffractometer | Bruker D8 Venture Photon | Bruker D8 Venture Photon |
| Absorption correction | Multi-scan SADABS2016/2 (Bruker AXS) | Multi-scan SADABS2016/2 (Bruker AXS) |
| Tmin, Tmax | 0.879, 0.928 | 0.879, 0.928 |
| No. Of measured, independent and observed [I > 2σ(I)] reflections | 28902, 3719, 3447 | 86662, 3500, 3186 |
| Rint | 0.025 | 0.034 |
| (sin ϴ/λ)max (Å−1) | 0.717 | 0.650 |
| Refinement | ||
| R[F2 > 4σ(F2)], wR(F2), S | 0.034, 0.095, 1.03 | 0.037, 0105, 1.054 |
| No. Of reflection | 3719 | 3500 |
| No. Of parameters | 157 | 147 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e·−3) | 0.41, −0.20 | 0.39 and −0.308 |
| O1–C1 | 1.3539 (13) | C11–C10 | 1.5160 (14) |
| O1–H1 | 0.8400 | N1–C11 | 1.4675 (15) |
| O2–C10 | 1.4209 (12) | N1–C7 | 1.4759 (13) |
| O2–C9 | 1.4241 (15) | C1–C6 | 1.3959 (14) |
| O3–C12 | 1.2208 (13) | C1–C2 | 1.4048 (13) |
| N1–C8 | 1.4633 (12) | C12–C13 | 1.5031 (15) |
| C10–O2–C9 | 110.06 (7) | O3–C12–C4 | 120.84 (8) |
| C8–N1–C11 | 109.45 (7) | O3–C12–C13 | 120.05 (9) |
| C8–N1–C7 | 110.34 (7) | C4–C12–C13 | 119.09 (8) |
| C11–N1–C7 | 111.90 (7) | C5–C6–C1 | 119.33 (8) |
| O1–C1–C6 | 119.13 (7) | C6–C5–C4 | 121.58 (8) |
| O1–C1–C2 | 120.61 (7) | C3–C2–C1 | 119.27 (7) |
| N1–C8–C9 | 109.95 (7) | N1–C11–C10 | 109.34 (7) |
| N1–C7–C2 | 111.38 (6) | O2–C9–C8 | 111.10 (7) |
| O1–C1 | 1.3381 (9) | C6–H6 | 0.9500 |
| O1–H1 | 0.8400 | C5–H5 | 0.9500 |
| N1–C7 | 1.4777 (10) | C8–C9 | 1.5386 (12) |
| N1–C11 | 1.4781 (10) | O2–C12 | 1.2226 (12) |
| N1–C8 | 1.4817 (10) | C3–C2 | 1.3924 (10) |
| C7–N1–C11 | 112.43 (6) | N1–C11–C10 | 102.67 (6) |
| C7–N1–C8 | 113.93 (6) | O2–C12–C4 | 120.57 (8) |
| C11–N1–C8 | 103.46 (6) | O2–C12–C13 | 120.09 (8) |
| O1–C1–C2 | 118.31 (7) | N1–C7–C2 | 112.27 (6) |
| C3–C2–C1–O1 | 177.70 (7) | C7–C2–C1–O1 | −1.46 (11) |
| C11–N1–C7–C2 | −169.16 (6) | C7–N1–C8–C9 | 162.59 (7) |
| C8–N1–C7–C2 | 73.55 (8) | C8–N1–C11–C10 | −46.12 (7) |
| C3–C2–C7–N1 | 80.26 (9) | C3–C4–C12–O2 | 173.05 (9) |
| C1–C2–C7–N1 | −100.58 (8) | C5–C4–C12–O2 | −6.28 (13) |
| Contact | Percentage in 2a | Percentage in 3a |
|---|---|---|
| H…H | 55.8% | 60.7% |
| H…O | 12.6% | 7.9% |
| O…H | 14.8% | 9.4% |
| C…C | 0.9% | 0.0% |
| N…H/H…N | 0.0% | 1.6% |
| H…C | 6.7% | 10.3% |
| C…H | 8.8% | 8.3% |
| O…C/C…O | 0.0% | 0.1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aljohani, G.; Said, M.A.; Lentz, D.; Basar, N.; Albar, A.; Alraqa, S.Y.; Ali, A.A.-S. Microwave-Assisted Synthesis of Mono- and Disubstituted 4-Hydroxyacetophenone Derivatives via Mannich Reaction: Synthesis, XRD and HS-Analysis. Molecules 2019, 24, 590. https://doi.org/10.3390/molecules24030590
Aljohani G, Said MA, Lentz D, Basar N, Albar A, Alraqa SY, Ali AA-S. Microwave-Assisted Synthesis of Mono- and Disubstituted 4-Hydroxyacetophenone Derivatives via Mannich Reaction: Synthesis, XRD and HS-Analysis. Molecules. 2019; 24(3):590. https://doi.org/10.3390/molecules24030590
Chicago/Turabian StyleAljohani, Ghadah, Musa A. Said, Dieter Lentz, Norazah Basar, Arwa Albar, Shaya Y. Alraqa, and Adeeb Al-Sheikh Ali. 2019. "Microwave-Assisted Synthesis of Mono- and Disubstituted 4-Hydroxyacetophenone Derivatives via Mannich Reaction: Synthesis, XRD and HS-Analysis" Molecules 24, no. 3: 590. https://doi.org/10.3390/molecules24030590
APA StyleAljohani, G., Said, M. A., Lentz, D., Basar, N., Albar, A., Alraqa, S. Y., & Ali, A. A.-S. (2019). Microwave-Assisted Synthesis of Mono- and Disubstituted 4-Hydroxyacetophenone Derivatives via Mannich Reaction: Synthesis, XRD and HS-Analysis. Molecules, 24(3), 590. https://doi.org/10.3390/molecules24030590