
New Rigid Polycyclic Bis(phosphane) for Asymmetric Catalysis

 , ,
, ,  ,
, 

Abstract
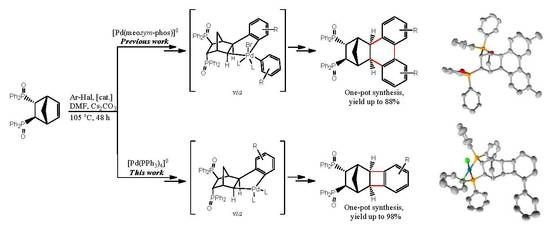
1. Introduction
2. Results and Discussion
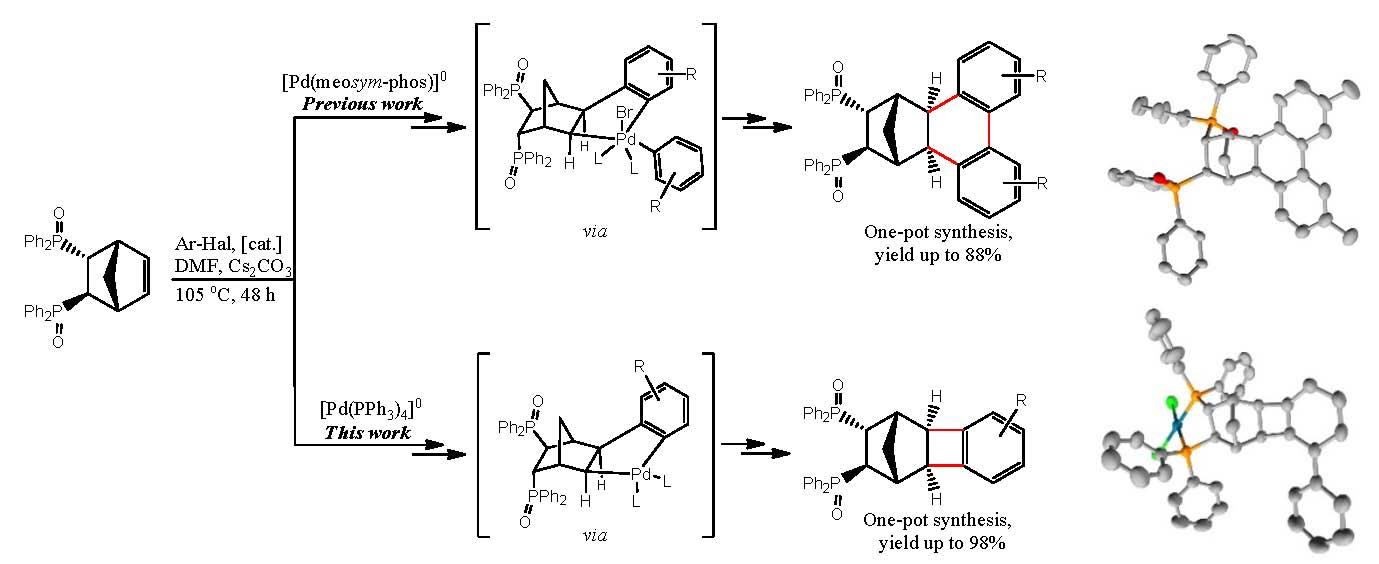
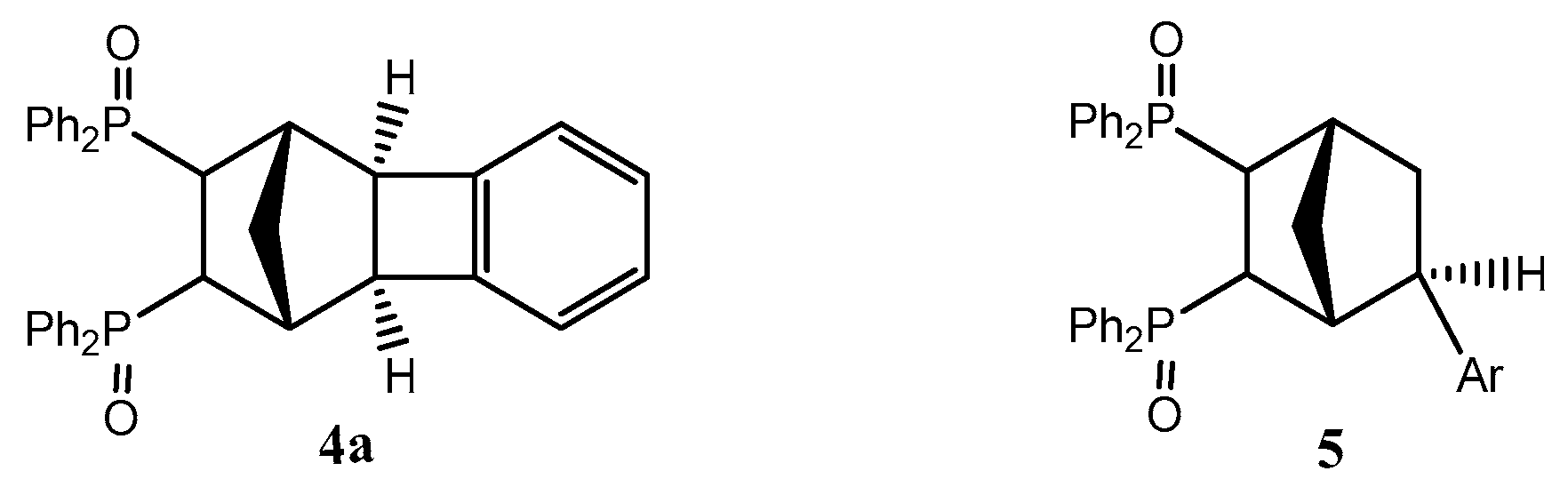
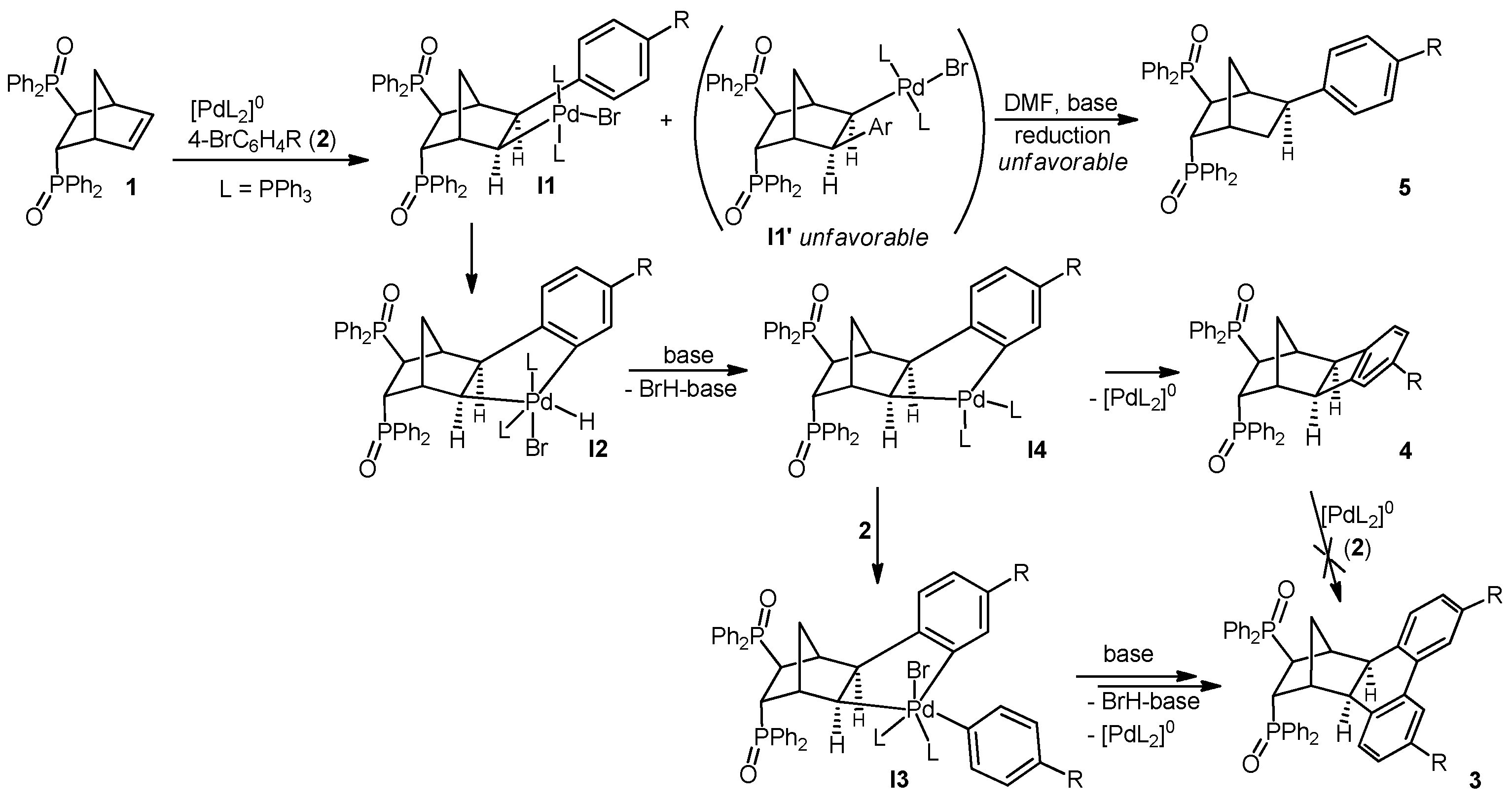
2.1. General Mechanism and Selectivity of the Reaction
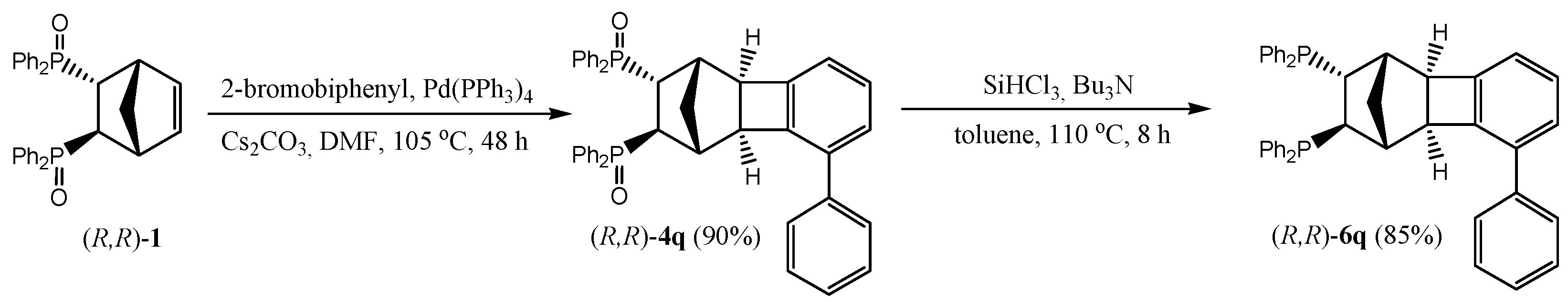
2.2. Preparation of Chiral Non-Racemic Ligand 6q
2.3. Synthesis of the Complex [Pd(6q)Cl2]
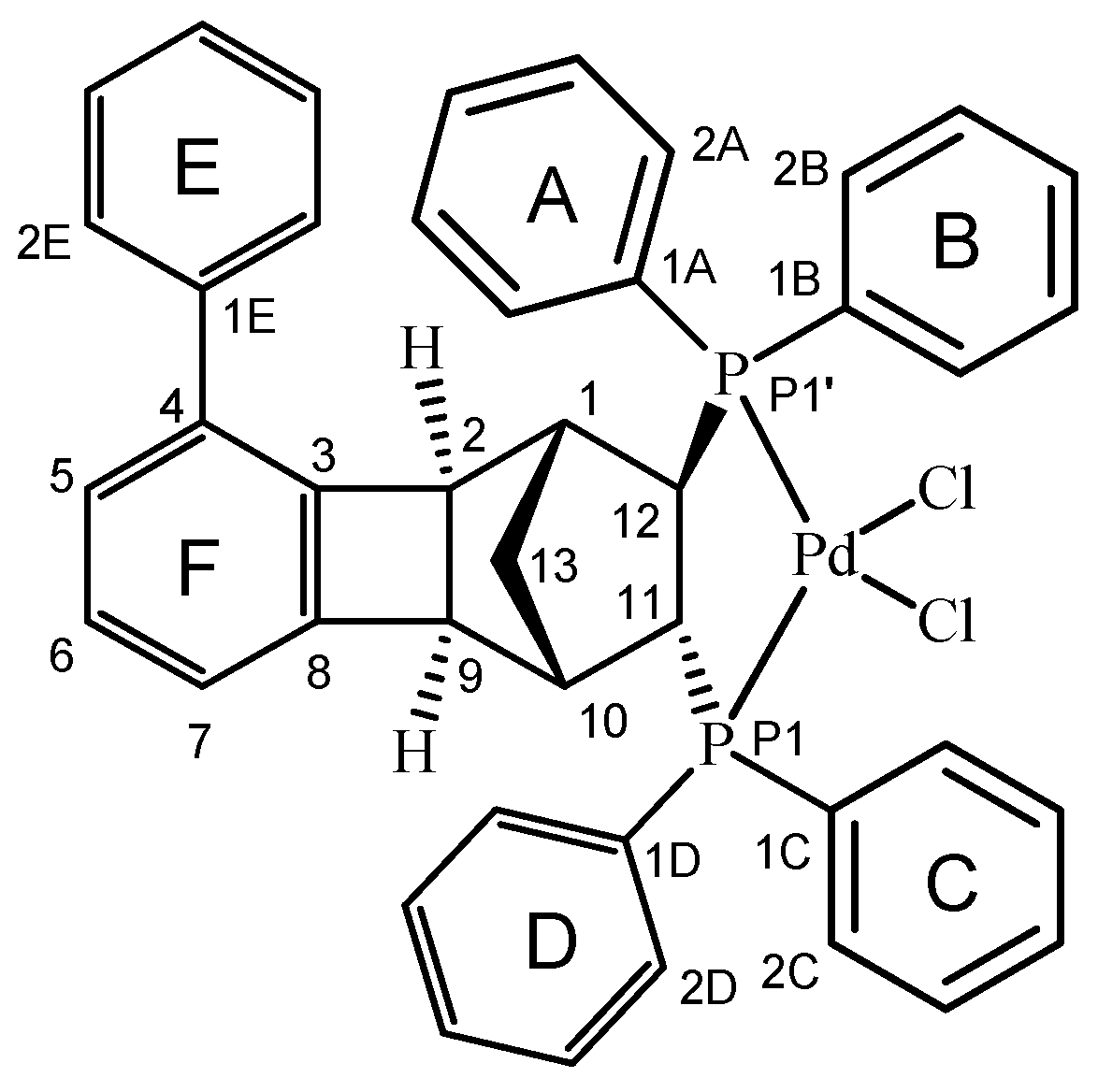
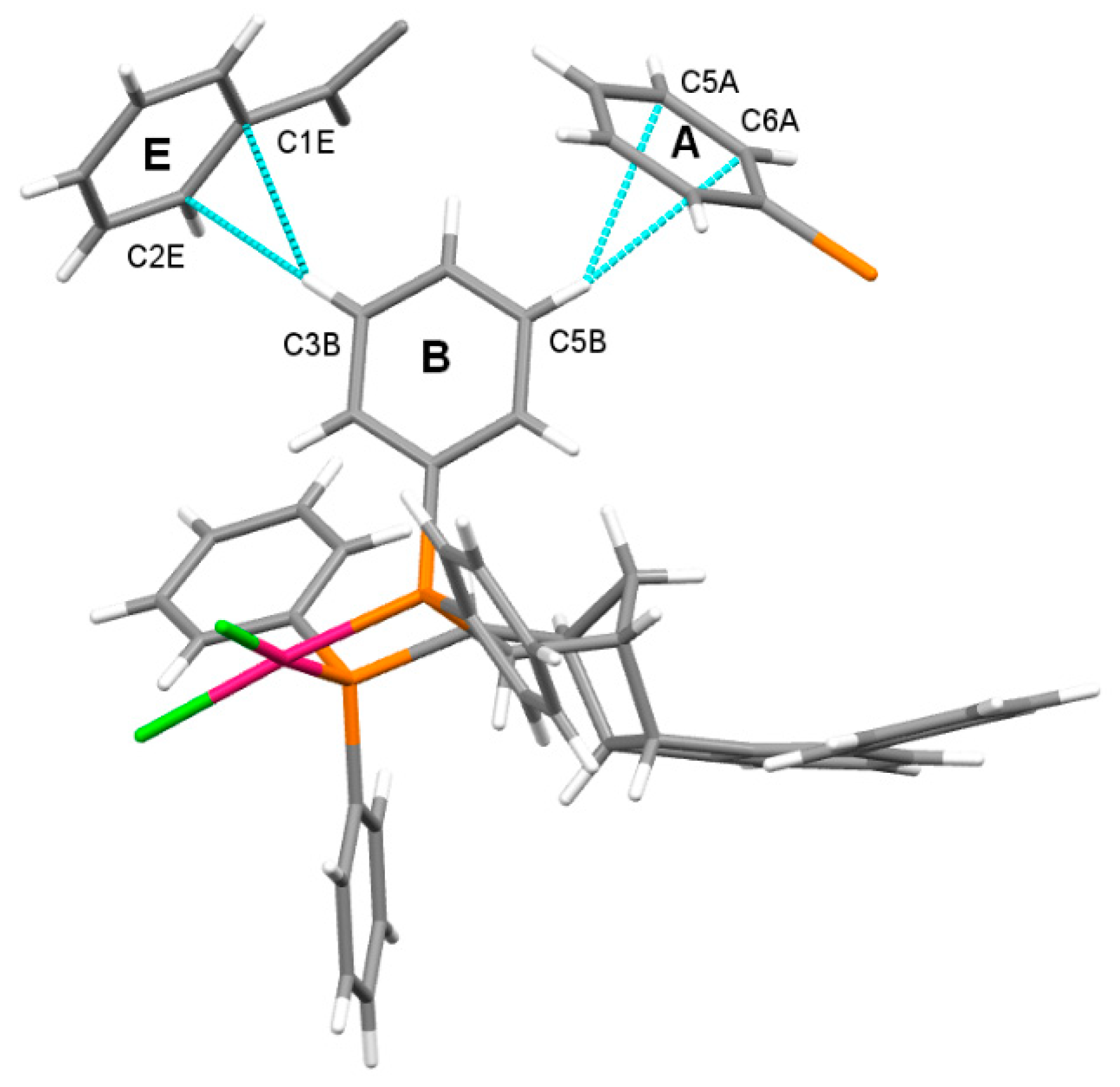
2.4. Molecular and Crystal Structure of the Complex [Pd(6q)Cl2]
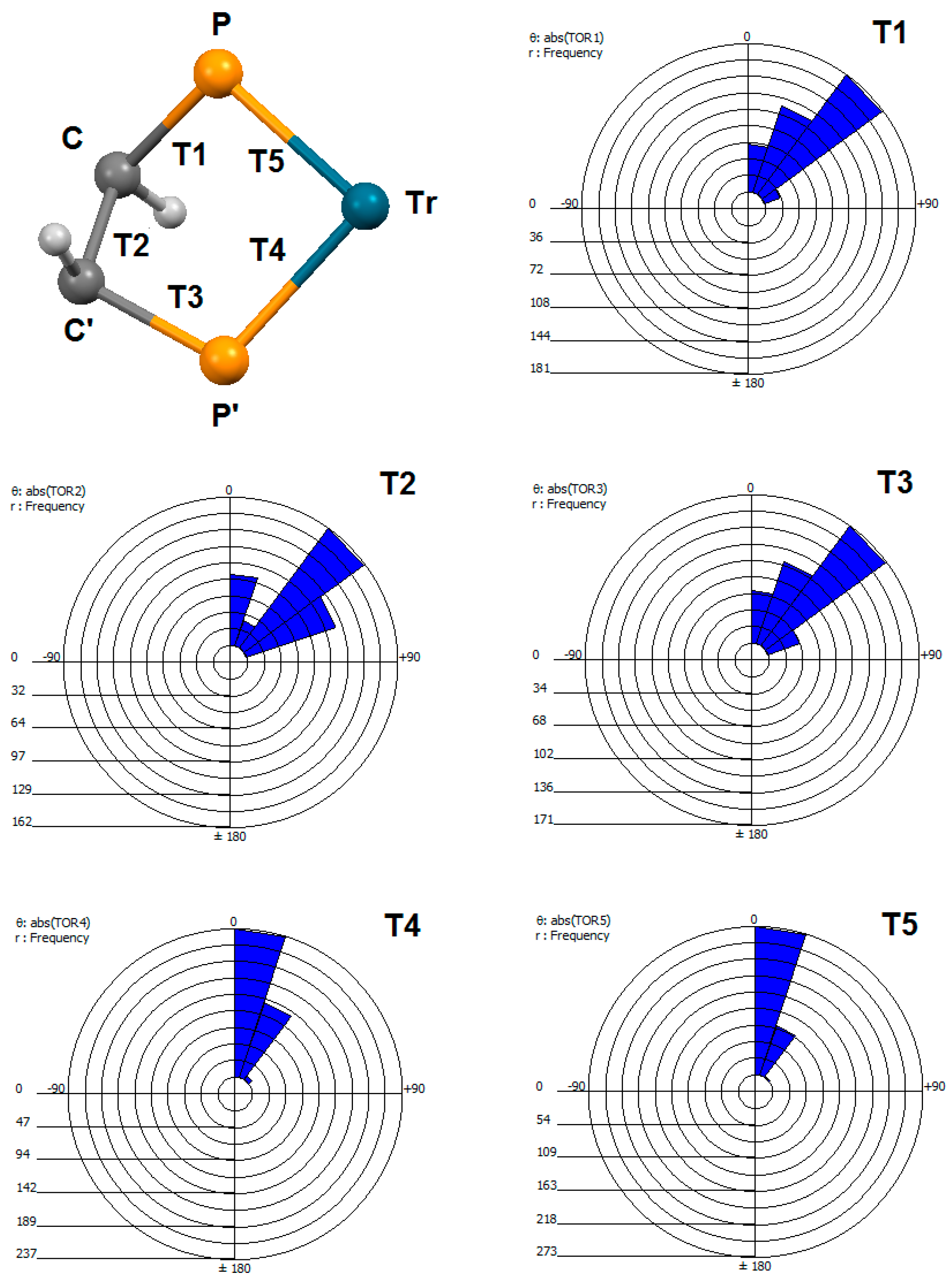
2.5. Geometry of Five-Membered Metallacycles of the Transition Metals and Bis(phosphine) Ligands Found in the Cambridge Structural Database (CSD)
2.6. Quantum Chemical Considerations
2.7. The Evaluation of the Efficiency of the Ligand 6q Palladium Complexes in the Asymmetric Catalysis
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Spectral Data
3.2.1. Typical Procedure for the Coupling Reaction Leading to the Formation of [(1R,2R,9S,10S,11R,12R)-4-phenyltetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl]bis(diphenylphosphane) dioxide (R,R-4q)
3.2.2. 1,2,3,4,4a,12b-Hexahydro-1,4-methanotriphenylene-2,3-diylbis(diphenylphosphane) dioxide (rac-3a)
3.2.3. (5-Methyltetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl)bis(diphenylphosphane) dioxide (rac-4b)
3.2.4. (4-Methoxytetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl)bis(diphenylphosphane) dioxide (rac-4c)
3.2.5. (4-Methyltetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl)bis(diphenylphosphane) dioxide (rac-4f)
3.2.6. (4-Chlorotetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl)bis(diphenylphosphane) dioxide (rac-4g)
3.2.7. (4,6-Dimethyl-5-nitrotetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl)bis(diphenylphosphane) dioxide (rac-4h)
3.2.8. (5-Methoxytetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl)bis(diphenylphosphane) dioxide (rac-4i)
3.2.9. [Methyl 11,12-bis(diphenylphosphoryl)tetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-5-carboxylate (rac-4j)
Dimethyl 2,3-bis(diphenylphosphoryl)-1,2,3,4,4a,12b-hexahydro-1,4-methanotriphenylene-7,10-dicarboxylate (rac-3j)
3.2.10. [7,10-Bis(trifluoromethyl)-1,2,3,4,4a,12b-hexahydro-1,4-methanotriphenylene-2,3-diyl]bis(diphenylphosphane) dioxide (rac-3k)
3.2.11. [7,10-Bis(trimethylsilyl)-1,2,3,4,4a,12b-hexahydro-1,4-methanotriphenylene-2,3-diyl]bis(diphenylphosphane) dioxide (rac-3l)
3.2.12. 6b,7,8,9,10,10a-Hexahydro-7,10-methanobenzo[a]biphenylene-8,9-diylbis(diphenylphosphane) dioxide (rac-4m)
[5-(naphthalen-1-yl)bicyclo[2.2.1]heptane-2,3-diyl]bis(diphenylphosphane) dioxide (rac-5m)
3.2.13. (7-Methoxy-1,2,3,4,4a,10b-hexahydro-1,4-methanobenzo[b]biphenylene-2,3-diyl)bis(diphenylphosphane) dioxide (rac-4n)
3.2.14. 8c,9,10,11,12,12a-Hexahydro-9,12-methanobenzo[3,4]cyclobuta[1,2-l]phenanthrene-10,11-diylbis(diphenylphosphane) dioxide (rac-4p)
[5-(phenanthren-9-yl)bicyclo[2.2.1]heptane-2,3-diyl]bis(diphenylphosphane) dioxide (rac-5p)
3.2.15. Synthesis of [(1R,2R,9S,10S,11R,12R)-4-Phenyltetracyclo[8.2.1.02,9.03,8]trideca-3,5,7-triene-11,12-diyl]bis(diphenylphosphane) ((R,R)-6q)
3.2.16. Synthesis of [Pd(6q)Cl2]
3.2.17. General Procedure for Allylic Alkylation Reaction
3.3. Computational Studies
3.4. Crystallographic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oestreich, M. The Mizoroki-Heck Reaction; Wiley: Hoboken, NJ, USA, 2009; 587p, ISBN 9780470033944. [Google Scholar]
- Beletskaya, I.P.; Cheprakov, A.V. The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- de Meijere, A.; Bräse, S.; Oestreich, M. Metal-Catalyzed Cross-Coupling Reactions and More; 3 Volume Set; Wiley: Hoboken, NJ, USA, 2014; 1576p, ISBN 9783527331543. [Google Scholar]
- Catellani, M.; Chiusoli, G.P. Palladium-catalyzed synthesis of 1,2,3,4,4a,12b-hexahydro-1,4-methanotriphenylenes. J. Organomet. Chem. 1985, 286, c13–c16. [Google Scholar] [CrossRef]
- Ye, J.; Lautens, M. Palladium-catalysed norbornene-mediated C−H functionalization of arenes. Nat. Chem. 2015, 7, 863–870. [Google Scholar] [CrossRef]
- Li, R.; Dong, G. Direct Annulation between Aryl Iodides and Epoxides through Palladium/Norbornene Cooperative Catalysis. Angew. Chem. Int. Ed. Engl. 2017, 57, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Casnati, A.; Fontana, M.; Coruzzi, G.; Aresta, B.M.; Corriero, N.; Maggi, R.; Maestri, G.; Motti, E.; Della Ca’, N. Enhancing Reactivity and Selectivity of Aryl Bromides: A Complementary Approach to Dibenzo[b,f]azepine Derivatives. ChemCatChem Catal. 2018, 10, 4346–4352. [Google Scholar] [CrossRef]
- Liu, C.; Liang, Y.; Zheng, N.; Zhang, B.-S.; Feng, Y.; Bi, S.; Liang, Y.-M. Synthesis of indolines via a palladium/norbornene-catalyzed reaction of aziridines with aryl iodides. Chem. Commun. 2018, 54, 3407–3410. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Jasiński, R.; Formela, A. The Halogen-Less Catalytic Transition Metal-Mediated Cross-Coupling Reactions: A Sustainable Alternative for Utilisation of Organohalides. In Chemistry Beyond Chlorine; Tundo, P., He, L.-N., Lokteva, E., Mota, C., Eds.; Springer: Cham, Switzerland, 2016; pp. 17–94. [Google Scholar]
- Brunner, H.; Pieronczyk, W. Asymmetric Hydrogenation of (Z)-(Acetylamino)-cinnamic Acid by a Rh/norphos Catalyst. Angew. Chem. Int. Ed. Engl. 1979, 18, 620–621. [Google Scholar] [CrossRef]
- Marchand, A.P.; Romanski, J.; Kumar, T.P. (R,R)-(-)-NORPHOS and (S,S)-(+)-NORPHOS. In e-EROS: Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Szwaczko, K.; Demchuk, O.M.; Mirosław, B.; Strzelecka, D.; Pietrusiewicz, K.M. Straightforward approach to norbornene core based chiral ligands by tandem cross dehydrogenative coupling reactions. Tetrahedron Lett. 2016, 57, 3491–3495. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kapłon, K.; Mazur, L.; Strzelecka, D.; Pietrusiewicz, K.M. Readily available catalysts for demanding Suzuki–Miyaura couplings under mild conditions. Tetrahedron 2016, 72, 6668–6677. [Google Scholar] [CrossRef]
- Qiu, Z.; Sun, R.; Teng, D. Synthesis of highly rigid phosphine–oxazoline ligands for palladium-catalyzed asymmetric allylic alkylation. Org. Biomol. Chem. 2018, 16, 7717–7724. [Google Scholar] [CrossRef]
- Snieckus, V.; Demchuk, O.; Yoruk, B.; Blackburn, T. A Mixed Naphthyl-Phenyl Phosphine Ligand Motif for Suzuki, Heck, and Hydrodehalogenation Reactions. Synlett 2006, 2908–2913. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kielar, K.; Pietrusiewicz, K.M. Rational design of novel ligands for environmentally benign cross-coupling reactions. Pure Appl. Chem. 2011, 83, 633–644. [Google Scholar] [CrossRef]
- Chakravarty, A.R.; Cotton, F.A.; Tocher, D.A. Displacive transfer of a phenyl group from triphenylphosphine to a metal atom: Synthesis and molecular structure of the dibenzamidediphenyldiruthenium compound Ru2Ph2(PhCONH)2[Ph2POC(Ph)N]2. J. Am. Chem. Soc. 1984, 106, 6409–6413. [Google Scholar] [CrossRef]
- Jin, L.; Qian, J.; Sun, N.; Hu, B.; Shen, Z.; Hu, X. Pd-Catalyzed reductive heck reaction of olefins with aryl bromides for Csp2–Csp3 bond formation. Chem. Commun. 2018, 54, 5752–5755. [Google Scholar] [CrossRef]
- Wang, C.; Xiao, G.; Guo, T.; Ding, Y.; Wu, X.; Loh, T.-P. Palladium-Catalyzed Regiocontrollable Reductive Heck Reaction of Unactivated Aliphatic Alkenes. J. Am. Chem. Soc. 2018, 140, 9332–9336. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D. Calculation of puckered rings with analytical gradients. J. Phys. Chem. 1990, 94, 5502–5509. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Eller, S.; Trettenbrein, B.; Fessler, M.; Haringer, S.; Ruggenthaler, M.; Gutmann, R.; van der Veer, W.E.; Kopacka, H.; Müller, T.; Obendorf, D.; et al. Surprising photochemical reactivity and visible light-driven energy transfer in heterodimetallic complexes. Dalton Trans. 2011, 40, 3815–3829. [Google Scholar] [CrossRef]
- Wilson, W.L.; Rahn, J.A.; Alcock, N.W.; Fischer, J.; Frederick, J.H.; Nelson, J.H. Thermal dimerization of 1-substituted-3,4-dimethylphospholes within the coordination sphere of platinum(II). Inorg. Chem. 1994, 33, 109–117. [Google Scholar] [CrossRef]
- Oberhauser, W.; Ienco, A.; Vizza, F.; Trettenbrein, B.; Oberhuber, D.; Strabler, C.; Ortner, T.; Brüggeller, P. Regioselective Hydromethoxycarbonylation of Terminal Alkynes Catalyzed by Palladium(II)–Tetraphos Complexes. Organometallics 2012, 31, 4832–4837. [Google Scholar] [CrossRef]
- Oberhauser, W.; Stampfl, T.; Bachmann, C.; Haid, R.; Langes, C.; Kopacka, H.; Ongania, K.-H.; Brüggeller, P. Palladium(II), platinum(II), and platinum(IV) complexes containing trans-1,2-bis(diphenylphosphino)ethene or cis,trans,cis-1,2,3,4-tetrakis(diphenylphosphino)cyclobutane: Complete X-ray structural characterization of binuclear compounds. Polyhedron 2000, 19, 913–923. [Google Scholar] [CrossRef]
- Strabler, C.; Ortner, T.; Prock, J.; Granja, A.; Gutmann, R.; Kopacka, H.; Müller, T.; Brüggeller, P. Versatile Supramolecular Coordination Behaviour of a Bis(bidentate) Tetraphosphane. Eur. J. Inorg. Chem. 2013, 2013, 5121–5132. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, UK, 2010. [Google Scholar]
- Jasiński, R.; Demchuk, O.M.; Babyuk, D. A Quantum-Chemical DFT Approach to Elucidation of the Chirality Transfer Mechanism of the Enantioselective Suzuki–Miyaura Cross-Coupling Reaction. J. Chem. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Mirosław, B.; Babyuk, D.; Łapczuk-Krygier, A.; Kącka-Zych, A.; Demchuk, O.M.; Jasiński, R. Regiospecific formation of the nitromethyl-substituted 3-phenyl-4,5-dihydroisoxazole via [3 + 2] cycloaddition. Monatshefte für Chemie (Chem. Mon.) 2018, 149, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Demchuk, O.M.; Jasiński, R.; Pietrusiewicz, K.M. New Insights into the Mechanism of Reduction of Tertiary Phosphine Oxides by Means of Phenylsilane. Heteroat. Chem. 2015, 26, 441–448. [Google Scholar] [CrossRef]
- Jasiński, R. Molecular mechanism of thermal decomposition of fluoronitroazoxy compounds: DFT computational study. J. Fluor. Chem. 2014, 160, 29–33. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Justyniak, I.; Miroslaw, B.; Jasiński, R. 2-Methoxynaphthylnaphthoquinone and its solvate: Synthesis and structure-properties relationship. J. Phys. Org. Chem. 2014, 27, 66–73. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Arlt, D.; Jasiński, R.; Pietrusiewicz, K.M. Relationship between structure and efficiency of atropisomeric phosphine ligands in homogeneous catalytic asymmetric hydrogenation. J. Phys. Org. Chem. 2012, 25, 1006–1011. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Jasiński, R. Organophosphorus ligands: Recent developments in design, synthesis, and application in environmentally benign catalysis. Phosphorus Sulfur Silicon Relat. Elem. 2015, 191, 245–253. [Google Scholar] [CrossRef]
- Issa, T.B.; Sayari, F.; Ghalla, H.; Benhamada, L. Synthesis, crystal structure, DFT calculations and molecular docking of l-pyroglutamic acid. J. Mol. Struct. 2019, 1178, 436–449. [Google Scholar] [CrossRef]
- Jin, M.-J.; Takale, V.B.; Sarkar, M.S.; Kim, Y.-M. Highly enantioselective Pd-catalyzed allylic alkylation using new chiral ferrocenylphosphinoimidazolidine ligands. Chem. Commun. 2006, 663. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Shima, T.; Yamagishi, T.; Hida, M. Palladium-catalyzed asymmetric allylic alkylation using dimethyl malonate and its derivatives as nucleophile. Tetrahedron Asymmetry 1991, 2, 663–666. [Google Scholar] [CrossRef]
- Łapczuk-Krygier, A.; Ponikiewski, Ł.; Jasiński, R. The crystal structure of (1RS,4RS,5RS,6SR)-5-cyano-5-nitro-6-phenyl-bicyclo[2.2.1]hept-2-ene. Crystallogr. Rep. 2014, 59, 961–963. [Google Scholar] [CrossRef]
- Jasiński, R.; Kwiatkowska, M.; Sharnin, V.; Barański, A. Experimental and theoretical studies of Diels–Alder reaction between methyl (Z)-2-nitro-3-(4-nitrophenyl)-2-propenoate and cyclopentadiene. Monatshefte für Chemie (Chem. Mon.) 2013, 144, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction. CrysAlis PRO ver. 1.171.38.46; Rigaku Oxford Diffraction: Yarnton, UK, 2015. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Sample Availability: Samples of compounds 1–6 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Aryl Halide | Equiv. of Aryl Halide | Product 3 | Product 3 (Yield%) | Product 4 | Product 4 (Yield%) |
|---|---|---|---|---|---|---|
| 1 |  | 3.0 |  | 3a (69) a |  | 4a (no) b |
| 2 |  | 1.2 3.0 |  | 3b (no) |  | 4b (59) 4b (83) |
| 3 |  | 1.2 |  | 3c (no) |  | 4c (76) |
| 4 |  | 1.2 3.0 |  | 3c (no) |  | 4c (35) 4c (45) |
| 5 |  | 3.0 |  | 3e (no) |  | 4e (no) |
| 6 |  | 1.2 1.2 3.0 |  | 3f (no) |  | 4f (25) a 4f (78) 4f (85) |
| 7 |  | 1.2 3.0 |  | 3g (no) |  | 4g (48) 4g (54) |
| 8 |  | 1.2 |  | 3h (no) |  | 4h (86) |
| 9 |  | 3.0 |  | 3i (no) |  | 4i (18) |
| 10 |  | 1.2 3.0 |  | 3j (<2) 3j (51) |  | 4j (84) 4j (24) |
| 11 |  | 1.2 3.0 |  | 3k (32) 3k (67) |  | 4k (no) |
| 12 |  | 3.0 |  | 3l (63) |  | 4l (no) |
| 13 |  | 1.2 |  | 3m (no) |  | 4m (68) |
| 14 |  | 1.2 |  | 3n (no) |  | 4n (53) |
| 15 |  | 1.2 |  | 3o (no) |  | 4o (no) |
| 16 |  | 1.2 |  | 3p (no) |  | 4p (54) |
| 17 |  | 1.2 |  | 3q (no) |  | 4q (98) |
| Bond | Length | Angle | Value |
|---|---|---|---|
| Pd1-Cl1 | 2.3452(12) | P1-Pd1-P1′ | 88.62(3) |
| Pd1-Cl2 | 2.3337(10) | P1-Pd1-Cl1 | 175.85(4) |
| Pd1-P1′ | 2.2463(8) | P1-Pd1-Cl2 | 89.30(4) |
| Pd1-P1 | 2.2722(9) | C1A-P1-Pd1′ | 120.58(13) |
| P1′-C1A | 1.812(4) | C1A-P1′-C12 | 107.45(17) |
| P1′-C1B | 1.802(4) | C1B-P1′-Pd1 | 113.58(12) |
| P1‘-C12 | 1.824(3) | C1B-P1′-C1A | 105.59(18) |
| P1-C1C | 1.820(4) | C1B-P1′-C12 | 108.17(16) |
| P1-C1D | 1.811(4) | C12-P1′-Pd1 | 100.75(11) |
| P1-C11 | 1.836(3) | C1C-P1-Pd1 | 118.19(13) |
| C1-C2 | 1.537(5) | C1C-P1-C11 | 105.81(16) |
| C1-C12 | 1.563(4) | C1D-P1-Pd1 | 114.56(14) |
| C1-C13 | 1.539(4) | C1D-P1-C1C | 103.96(18) |
| C1E-C4 | 1.477(6) | C1D-P1-C11 | 110.03(16) |
| C2-C3 | 1.525(4) | C11-P1-Pd1 | 103.92(11) |
| C2-C9 | 1.602(5) | ||
| C3-C4 | 1.388(5) | The valence angles of the four-membered ring | |
| C3-C8 | 1.386(5) | C2-C3-C8 | 93.9(3) |
| C4-C5 | 1.406(5) | C3-C8-C9 | 94.2(3) |
| C5-C6 | 1.389(6) | C8-C9-C2 | 85.8(3) |
| C6-C7 | 1.394(6) | C9-C2-C3 | 86.1(3) |
| C7-C8 | 1.374(5) | ||
| C8-C9 | 1.525(5) | ||
| C9-C10 | 1.540(5) | ||
| C10-C11 | 1.545(4) | ||
| C10-C13 | 1.550(5) | ||
| C11-C12 | 1.555(5) | ||
| Torsion | Value |
|---|---|
| Pd1-P1’-C12-C11 | 56.1(2) |
| P1’-C12-C11-P1 | −61.2(2) |
| C12-C11-P1-Pd1 | 36.3(2) |
| C11-P1-Pd1-P1’- | −1.8(1) |
| P1-Pd1-P1’-C12 | −26.5(1) |
| H11-C11-C12-H12 | 161 |
| C3-C4-C1E-C2E | 14.6(6) |
| D–H…A | D–H | D...A | H...A | <D–H...A |
|---|---|---|---|---|
| C2C-H2C…Cl2 | 0.93 | 3.482(4) | 2.73 | 139 |
| C6-H6…Cl2i | 0.93 | 3.557(4) | 2.87 | 132 |
| C6E-H6E…Cl1i | 0.93 | 3.623(4) | 2.76 | 154 |
| C7-H7…Cl1ii | 0.93 | 3.756(4) | 2.99 | 141 |
| C3B-H3B…Cg1ii | 0.93 | 3.689(5) | 2.77 | 169 |
| C5B-H5B…Cg2iii | 0.93 | 3.521(4) | 2.71 | 146 |
| Code | Torsion | Minimum | Maximum | Mean |
|---|---|---|---|---|
| abs(T1 or T3) | Tr–P–C–C | 0.733 | 60.136 | 36.141 |
| abs(T2) | P–C–C–P | 0.353 | 69.677 | 44.108 |
| abs(T4 or T5) | P–Tr–P–C | 0.921 | 44.906 | 17.174 |
| abs(T6) | H–C–C–H | 0.164 | 179.96 | 113.52 |

| [Pd(6q)Cl2]xray | [Pd(6q)Cl2]calc | [Pd(6a)Cl2]calc | 4qcalc | 4acalc | |
|---|---|---|---|---|---|
| Angle, (°) | |||||
| A/B | 73.3 | 81.5 | 81.3 | 68.9 | 70.0 |
| C/D | 74.3 | 67.4 | 67.6 | 66.7 | 65.5 |
| B/C | 39.6 | 39.3 | 43.7 | 4.2 | 8.2 |
| A/D | 39.3 | 45.1 | 39.9 | 45.4 | 44.5 |
| Torsion angle, (°) | |||||
| C3-C4-C1E-C2E | 14.6 | 30.4 | na | 29.3 | na |
| P1-C11-C12-P2 | −61.2 | −67.2 | −66.9 | −101.5 | −103.8 |
| Bond lengths, (Å) | |||||
| C1-C2 | 1.537(5) | 1.551 | 1.552 | 1.550 | 1.550 |
| C2-C3 | 1.525(4) | 1.529 | 1.530 | 1.532 | 1.532 |
| C3-C4 | 1.388(5) | 1.397 | 1.390 | 1.397 | 1.391 |
| C4-C5 | 1.406(5) | 1.417 | 1.415 | 1.418 | 1.415 |
| C5-C6 | 1.389(6) | 1.406 | 1.408 | 1.406 | 1.407 |
| C6-C7 | 1.394(6) | 1.414 | 1.413 | 1.414 | 1.414 |
| C7-C8 | 1.374(5) | 1.390 | 1.392 | 1.391 | 1.391 |
| C8-C3 | 1.386(5) | 1.403 | 1.406 | 1.405 | 1.407 |
| C8-C9 | 1.525(5) | 1.533 | 1.532 | 1.530 | 1.531 |
| C9-C2 | 1.602(5) | 1.606 | 1.608 | 1.600 | 1.602 |
| C9-C10 | 1.540(5) | 1.549 | 1.550 | 1.547 | 1.546 |
| C10-C11 | 1.545(4) | 1.550 | 1.549 | 1.552 | 1.551 |
| C11-C12 | 1.555(5) | 1.561 | 1.561 | 1.564 | 1.566 |
| C12-C1 | 1.563(4) | 1.560 | 1.563 | 1.555 | 1.555 |
| C1-C13 | 1.539(5) | 1.552 | 1.552 | 1.549 | 1.550 |
| C13-C10 | 1.550(5) | 1.562 | 1.562 | 1.555 | 1.554 |
| C12-P1 | 1.823(5) | 1.881 | 1.881 | 1.872 | 1.878 |
| C11-P2 | 1.836(5) | 1.883 | 1.884 | 1.859 | 1.850 |
| Pd-Cl1 | 2.345 (1) | 2.423 | 2.425 | na | na |
| Pd-Cl2 | 2.334(1) | 2.420 | 2.419 | na | na |
| Pd-P1 | 2.246 (1) | 2.381 | 2.379 | na | na |
| Pd-P2 | 2.272(1) | 2.380 | 2.379 | na | na |

| Entry | Ligand, mol% | [Pd(allyl)Cl]2, mol% | Base | Yield, % | ee, % |
|---|---|---|---|---|---|
| 1 | 2.0 (R,R-6q) | 2.0 | BSA */KOAc | 97 | 90 ** |
| 2 | 4.0 (R,R-6q) | 1.0 | BSA/KOAc | 98 | 67 |
| 3 | 2.0 (R,R-6q) | 2.0 | Cs2CO3/K2CO3 | 97 | 81 |
| 4 | 2.0 (R,R-6q) | 2.0 | Cs2CO3 | 96 | 83 |
| 5 | 2.0 (R,R-1) | 2.0 | BSA/KOAc | 95 | 87 |
| 6 | 2.0 (R,R-1) | 2.0 | Cs2CO3 | 92 | 85 |
| 7 | 1.2 (S,S-1) | 0.5 | NaH | 80 | 81 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrusiewicz, K.M.; Szwaczko, K.; Mirosław, B.; Dybała, I.; Jasiński, R.; Demchuk, O.M. New Rigid Polycyclic Bis(phosphane) for Asymmetric Catalysis. Molecules 2019, 24, 571. https://doi.org/10.3390/molecules24030571
Pietrusiewicz KM, Szwaczko K, Mirosław B, Dybała I, Jasiński R, Demchuk OM. New Rigid Polycyclic Bis(phosphane) for Asymmetric Catalysis. Molecules. 2019; 24(3):571. https://doi.org/10.3390/molecules24030571
Chicago/Turabian StylePietrusiewicz, K. Michał, Katarzyna Szwaczko, Barbara Mirosław, Izabela Dybała, Radomir Jasiński, and Oleg M. Demchuk. 2019. "New Rigid Polycyclic Bis(phosphane) for Asymmetric Catalysis" Molecules 24, no. 3: 571. https://doi.org/10.3390/molecules24030571
APA StylePietrusiewicz, K. M., Szwaczko, K., Mirosław, B., Dybała, I., Jasiński, R., & Demchuk, O. M. (2019). New Rigid Polycyclic Bis(phosphane) for Asymmetric Catalysis. Molecules, 24(3), 571. https://doi.org/10.3390/molecules24030571