
Microwave-Assisted Stereoselective Heterocyclization to Novel Ring d-fused Arylpyrazolines in the Estrone Series



Abstract
:
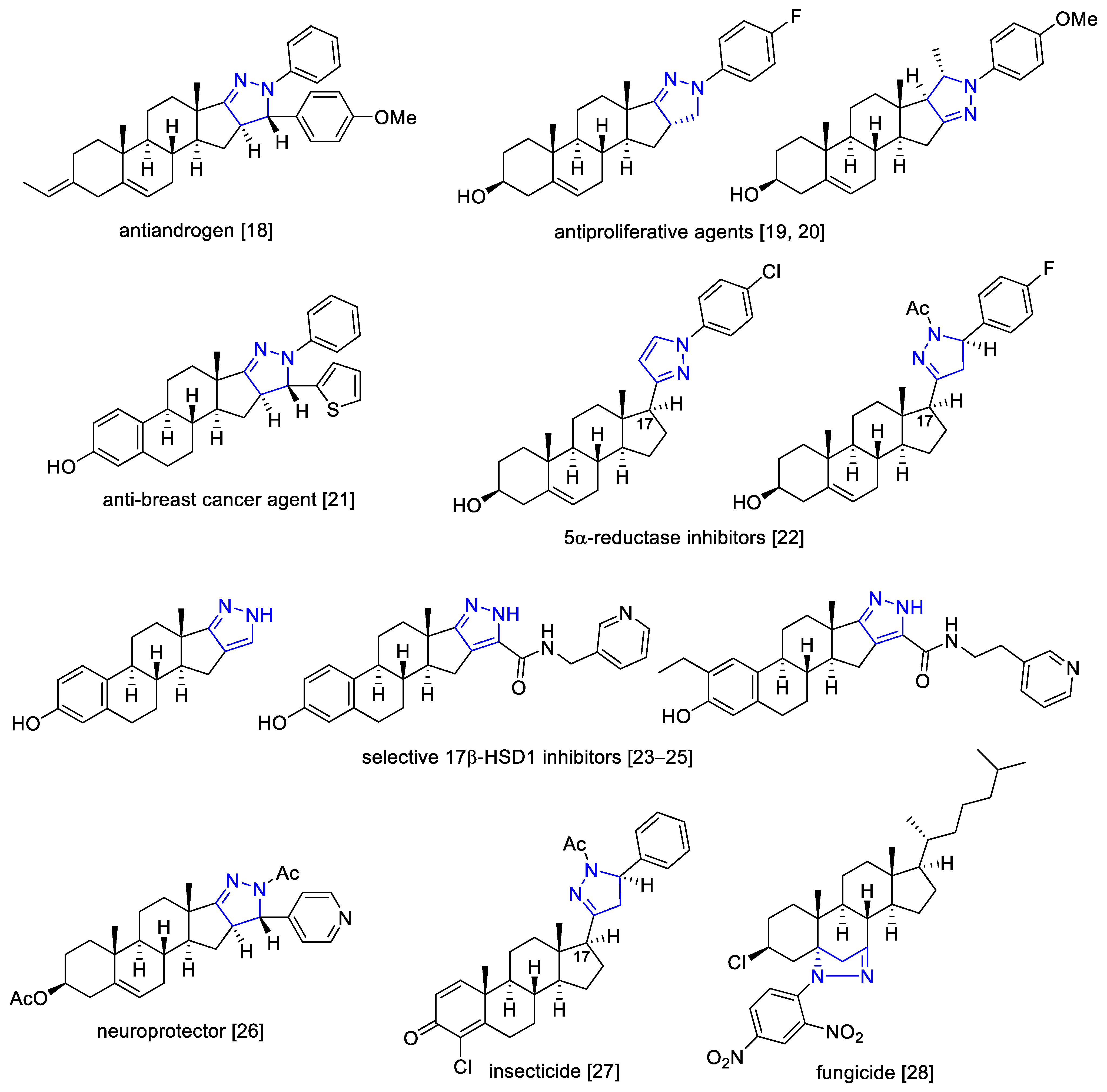
1. Introduction
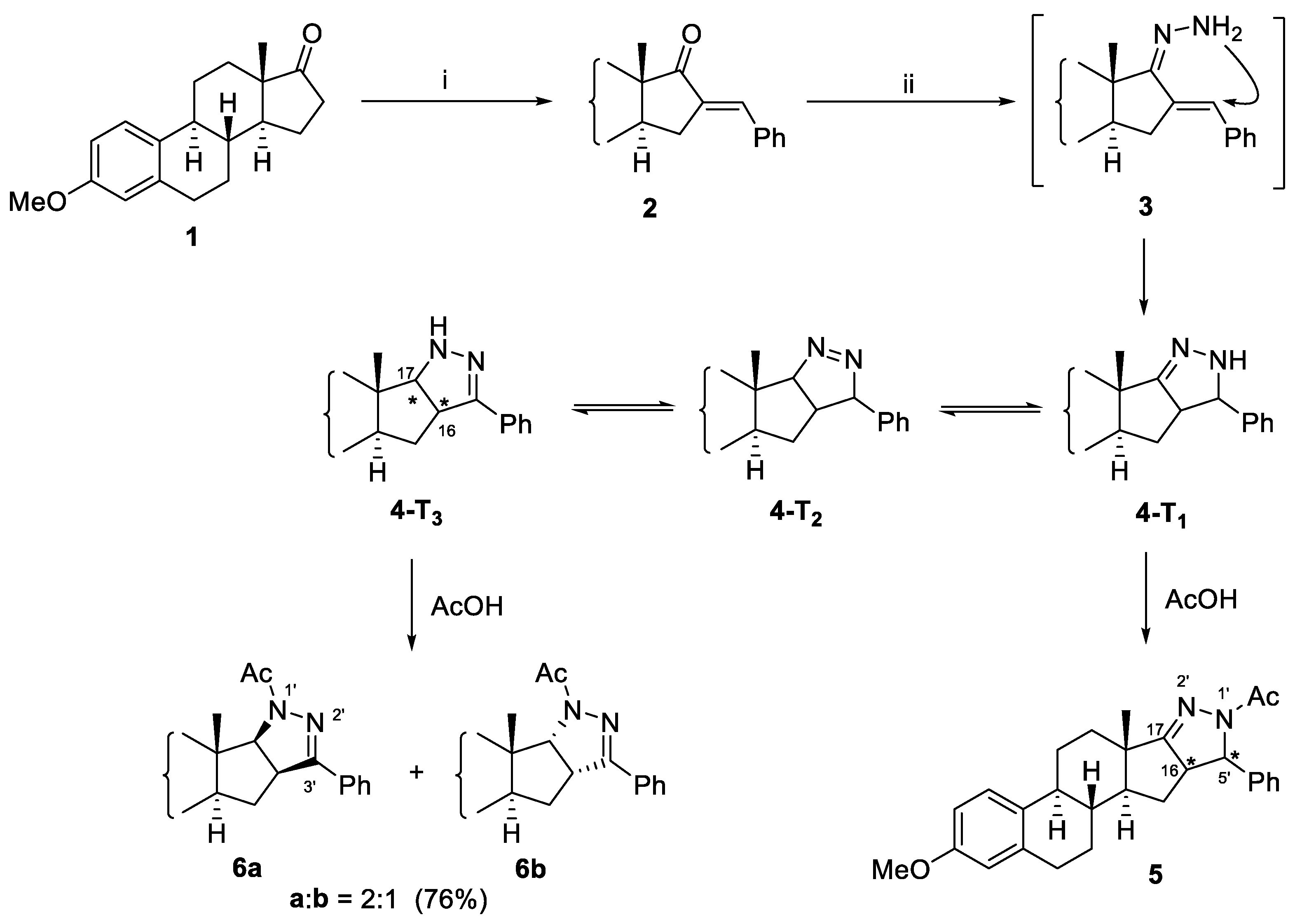
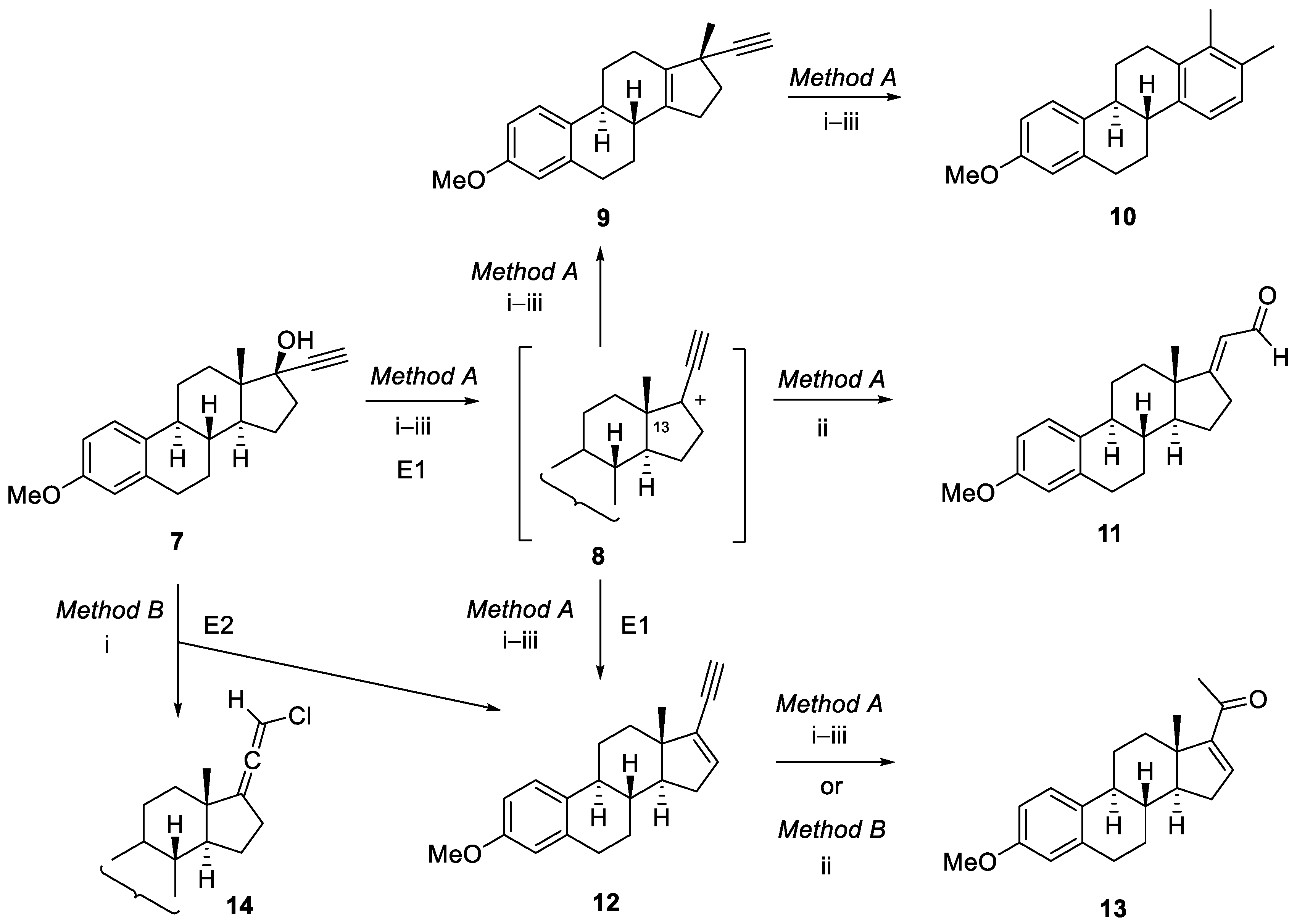
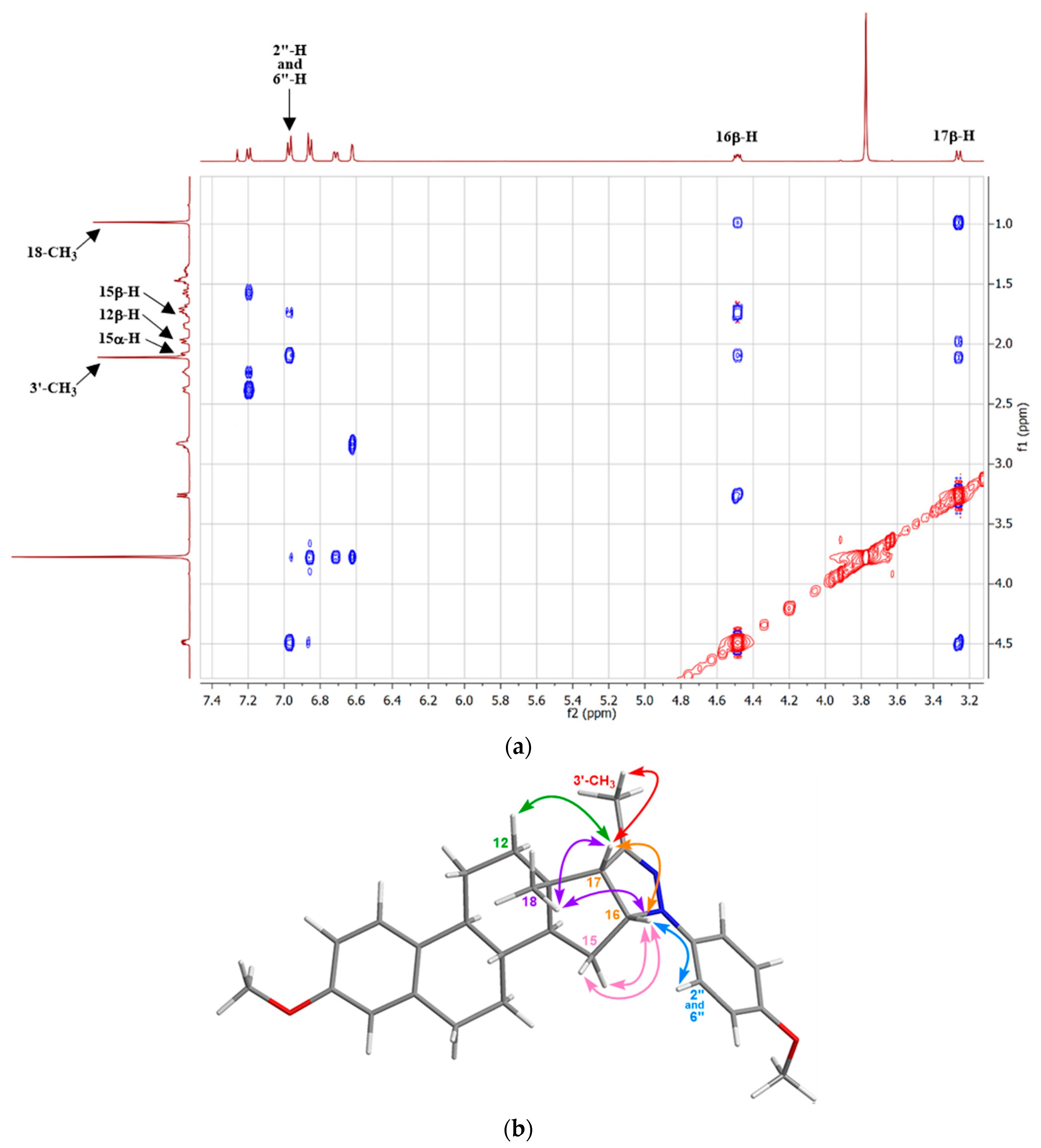
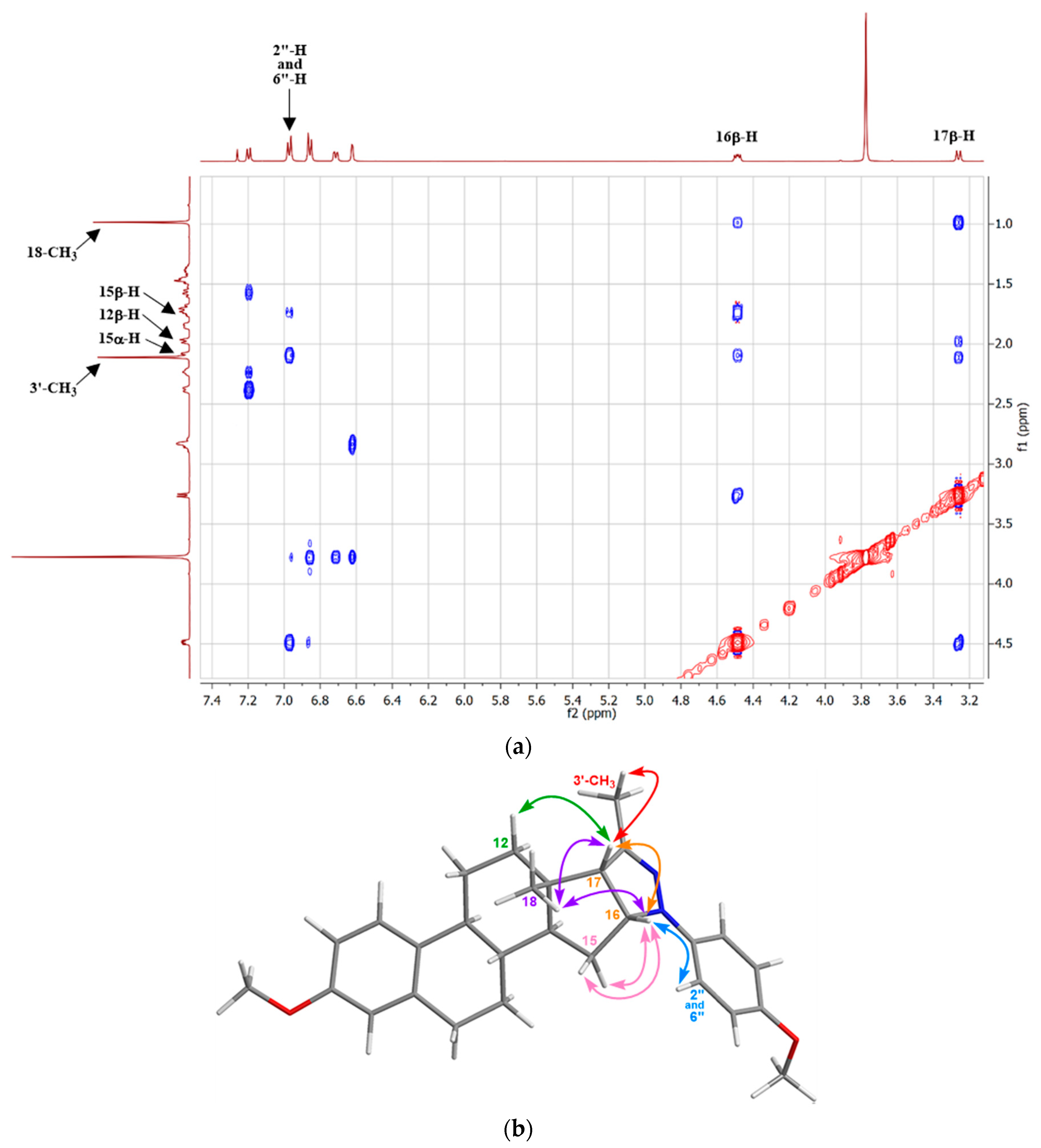
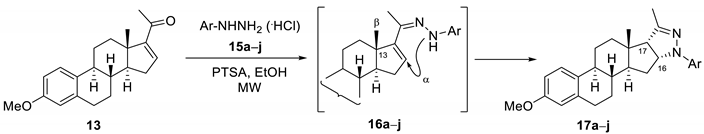
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedures
3.2.1. MW-assisted Synthesis of 3-methoxy-16-benzylidene-estra-1,3,5(10)-triene-17-one (2)
3.2.2. Cyclization of 3-methoxy-16-benzylidene-estra-1,3,5(10)-triene-17-one (2) with hydrazine hydrate
3.2.3. E2-type dehydration of mestranol to 17-ethinyl-3-methoxyestra-1,3,5(10),16-tetraene intermediate 12 by Method B
3.2.4. MW-Assisted Syntheses of 3-methoxy-19-norpregna-1,3,5(10),16-tetraene-20-one (13)
3.2.5. General Procedure for the Synthesis of Ring d-condensed Pyrazolines 17a–j under MW Irradiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Varghese, B.; Al-Busafi, S.N.; Suliman, F.O.; Al-Kindya, S.M.Z. Unveiling a versatile heterocycle: pyrazoline—A review. RSC Adv. 2017, 7, 46999–47016. [Google Scholar] [CrossRef]
- Marella, A.; Ali, M.R.; Alam, M.T.; Saha, R.; Tanwar, O.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Pyrazolines: A biological review. Mini Rev. Med. Chem. 2013, 13, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Jain, P. Synthetic and biological studies of pyrazolines and related heterocyclic compounds. Arab. J. Chem. 2014, 7, 553–596. [Google Scholar] [CrossRef]
- Ali, I.; Wani, W.A.; Khan, A.; Haque, A.; Ahmad, A.; Saleem, K.; Manzoor, N. Synthesis and synergistic antifungal activities of a pyrazoline based ligand and its copper(II) and nickel(II) complexes with conventional antifungals. Microb. Pathogenesis 2012, 53, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgın, S.; Atlı, Ö.; Demirel, R.; Kaplancıklı, Z.A. A novel series of thiazolyl–pyrazoline derivatives: Synthesis and evaluation of antifungal activity, cytotoxicity and genotoxicity. Eur. J. Med. Chem. 2015, 92, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Husain, A.; Khan, S.A.; Mujeeb, M.; Bhandarie, A. Synthesis, antimicrobial and antitubercular activities of some novel pyrazoline derivatives. J. Saudi Chem. Soc. 2016, 20, 577–584. [Google Scholar] [CrossRef]
- Hassan, S.Y. Synthesis, antibacterial and antifungal activity of some new pyrazoline and pyrazole derivatives. Molecules 2013, 18, 2683–2711. [Google Scholar] [CrossRef]
- Kaplancıklı, Z.A.; Özdemir, A.; Turan-Zitouni, G.; Altıntop, M.D.; Can, O.D. New pyrazoline derivatives and their antidepressant activity. Eur. J. Med. Chem. 2010, 45, 4383–4387. [Google Scholar] [CrossRef]
- Özdemir, Z.; Kandilci, H.B.; Gumusel, B.; Calis, U.; Bilgin, A.A. Synthesis and studies on antidepressant and anticonvulsant activities of some 3-(2-thienyl)pyrazoline derivatives. Archiv. Der Pharm. 2008, 341, 701–707. [Google Scholar] [CrossRef]
- Chandra, T.; Garg, N.; Lata, S.; Saxena, K.K.; Kumar, A. Synthesis of substituted acridinyl pyrazoline derivatives and their evaluation for anti-inflammatory activity. Eur. J. Med. Chem. 2010, 45, 1772–1776. [Google Scholar] [CrossRef]
- Amir, M.; Kumar, H.; Khan, S.A. Synthesis and pharmacological evaluation of pyrazoline derivatives as new anti-inflammatory and analgesic agents. Bioorg. Med. Chem. Lett. 2008, 18, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, M.; Altıntop, M.D.; Çiftçi, H.I.; Koga, R.; Otsuka, M.; Fujita, M.; Özdemir, A. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules 2015, 20, 19066–19084. [Google Scholar] [CrossRef] [PubMed]
- Shaharyar, M.; Abdullah, M.M.; Bakht, M.A.; Majeed, J. Pyrazoline bearing benzimidazoles: Search for anticancer agent. Eur. J. Med. Chem. 2010, 45, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Beyhan, N.; Kocyigit-Kaymakcioglu, B.; Gümrü, S.; Aricioglu, F. Synthesis and anticonvulsant activity of some 2-pyrazolines derived from chalcones. Arab. J. Chem. 2017, 10, S2073–S2081. [Google Scholar] [CrossRef]
- Bhandari, S.; Tripathi, A.C.; Saraf, S.K. Novel 2-pyrazoline derivatives as potential anticonvulsant agents. Med. Chem. Res. 2013, 22, 5290–5296. [Google Scholar] [CrossRef]
- Azarifar, D.; Ghasemnejad, H. Microwave-assisted synthesis of some 3,5-arylated 2-pyrazolines. Molecules 2003, 8, 642–648. [Google Scholar] [CrossRef]
- Azarifar, D.; Khosravi, K.; Veisi, R.-A. An efficient oxidation of 2-pyrazolines and isoxazolines by bisbromine-1,4-diazabicyclo[2.2.2]octane complex (DABCO-Br2). ARKIVOC 2010, 2010, 178–184. [Google Scholar]
- Amr, A.E.E.; Latif-Abdel, A.N.; Abdulla, M.M. Synthesis and antiandrogenic activity of some new 3-substituted androstano[17,16-c]-5′-arylpyrazoline and their derivatives. Bioorg. Med. Chem. 2006, 14, 373–384. [Google Scholar] [CrossRef]
- Mótyán, G.; Zupkó, I.; Minorics, R.; Schneider, Gy.; Wölfling, J.; Frank, É. Lewis acid-induced intramolecular access to novel steroidal ring D-condensed arylpyrazolines exerting in vitro cell-growth-inhibitory effects. Mol. Divers. 2015, 19, 511–527. [Google Scholar] [CrossRef]
- Frank, É.; Mucsi, Z.; Zupkó, I.; Réthy, B.; Falkay, G.; Schneider, Gy.; Wölfling, J. Efficient approach to androstene-fused arylpyrazolines as potent antiproliferative agents. Experimental and theoretical studies of substituent effects on BF3-catalyzed intramolecular [3+2] cycloadditions of olefinic phenylhydrazones. J. Am. Chem. Soc. 2009, 131, 3894–3904. [Google Scholar] [CrossRef]
- Amr, A.E.E.; El-Naggar, M.; Al-Omar, M.A.; Elsayed, E.A.; Abdall, M.M. In vitro and in vivo anti-breast cancer activities of some synthesized pyrazolinyl-estran-17-one candidates. Molecules 2018, 23, 1572. [Google Scholar] [CrossRef] [PubMed]
- Banday, A.H.; Shameem, S.A.; Jeelani, S. Steroidal pyrazolines and pyrazoles as potential 5α-reductase inhibitors: Synthesis and biological evaluation. Steroids 2014, 92, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.S.; Allan, G.M.; Bubert, C.; Vicker, N.; Smith, A.; Tutill, H.J.; Purohit, A.; Wood, L.; Packham, G.; Mahon, M.F.; et al. E-Ring modified steroids as novel potent inhibitors of 17β-hydroxysteroid dehydrogenase type 1. J. Med. Chem. 2005, 48, 5749–5770. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.M.; Lawrence, H.R.; Cornet, J.; Fischer, D.S.; Bubert, C.; Vicker, N.; Smith, A.; Tutill, H.J.; Purohit, A.; Day, J.M.; et al. Modification of estrone at the 6, 16, and 17 positions: Novel potent inhibitors of 17β-hydroxysteroid dehydrogenase type 1. J. Med. Chem. 2006, 49, 1325–1345. [Google Scholar] [CrossRef] [PubMed]
- Vicker, N.; Lawrence, H.R.; Allan, G.M.; Bubert, C.; Smith, A.; Tutill, H.J.; Purohit, A.; Day, J.M.; Mahon, M.F.; Reed, M.J.; et al. Focused libraries of 16-substituted estrone derivatives and modified E-ring steroids: Inhibitors of 17 β-hydroxysteroid dehydrogenase type 1. Chem. Med. Chem. 2006, 1, 464–481. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Thota, S.; Bansal, R. Studies on 16,17-pyrazoline substituted heterosteroids as anti-Alzheimer and anti-Parkinsonian agents using LPS induced neuroinflammation models of mice and rats. ACS Chem. Neurosci. 2018, 9, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.-J.; Wei, S.-P.; Gao, J.-M.; Tang, J.-J. Potential insecticidal activity of steroidal C-17 pyrazolinyl derivatives. J. Braz. Chem. Soc. 2015, 26, 389–392. [Google Scholar]
- Shamsuzzaman; Khanam, H.; Dar, A.M.; Siddiqui, N.; Rehman, S. Synthesis, characterization, antimicrobial and anticancer studies of new steroidal pyrazolines. J. Saudi Chem. Soc. 2016, 20, 7–12. [Google Scholar] [CrossRef]
- Mótyán, G.; Kovács, F.; Wölfling, J.; Gyovai, A.; Zupkó, I.; Frank, É. Microwave-assisted stereoselective approach to novel steroidal ring D-fused 2-pyrazolines and an evaluation of their cell-growth inhibitory effects in vitro. Steroids 2016, 112, 36–46. [Google Scholar] [CrossRef]
- Shamsuzzaman; Khanam, H.; Mashrai, A.; Sherwani, A.; Owais, M.; Siddiqui, N. Synthesis and anti-tumor evaluation of B-ring substituted steroidal pyrazoline derivatives. Steroids 2013, 78, 1263–1272. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis—A review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Pattanashetty, S.H.; Hosamani, K.M.; Barretto, D.A. Microwave assisted synthesis, computational study and biological evaluation of novel quinolin-2(1H)-one based pyrazoline hybrids. Chem. Data Collect. 2018, 15, 184–196. [Google Scholar] [CrossRef]
- Kulathooran, S.; Vadivel, T.; Dhamodaran, M.; Selvakumar, B. Microwave Solvent-free Synthesis of Some Bioactive 3-(2,5-Dimethylfuran-3-yl)-pyrazoline Derivatives and their Antimicrobial Activity. Orient. J. Chem. 2015, 32, 1067–1073. [Google Scholar] [CrossRef]
- Patel, N.B.; Shaikh, F.M.; Patel, H.R.; Rajani, D. Synthesis of 2-pyrazolines from pyridine based chalcone by conventional and microwave techniques: Their comparison and antimicrobial studies. J. Saudi Chem. Soc. 2016, 20, S451–S456. [Google Scholar] [CrossRef]
- Frank, É.; Schneider, G. Synthesis of sex hormone-derived modified steroids possessing antiproliferative activity. J. Steroid Biochem. Mol. Biol. 2013, 137, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Ispán, D.; Szánti-Pintér, E.; Papp, M.; Wouters, J.; Tumanov, N.; Zsirka, B.; Gömöry, Á.; Kollár, L.; Skoda-Földes, R. The use of switchable polarity solvents for the synthesis of 16-arylidene steroids via Claisen-Schmidt condensation. Eur. J. Org. Chem. 2018, 2018, 3236–3244. [Google Scholar] [CrossRef]
- Romero-López, A.; Montiel-Smith, S.; Meza-Reyes, S.; Merino-Montiel, P. Synthesis of steroidal derivatives containing substituted, fused and spiro pyrazolines. Steroids 2014, 87, 86–92. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. The tautomerism of pyrazolines (dihydropyrazoles). J. Chil. Chem. Soc. 2015, 60, 2966–2970. [Google Scholar] [CrossRef]
- Paquette, L.A.; Stevens, K.E. Stereocontrolled total synthesis of the triquinane marine sesquiterpene Δ9(12)-capnellene. Can. J. Chem. 1984, 62, 2415–2420. [Google Scholar] [CrossRef]
- Li, J.J. Rupe rearrangement. In Name Reactions. A Collection of Detailed Mechanisms and Synthetic Applications; Springer: Berlin/Heidelberg, Germany, 2009; pp. 480–481. [Google Scholar]
- Pindur, U.; Schall, T. Proton acid-induced rearrangements of α-alkynylestradiol methyl ethers. Liebigs Ann. Chem. 1993, 1099–1103. [Google Scholar] [CrossRef]
- Vincze, I.; Lőkös, M.; Bakos, M.; Dancsi, A.; Mák, M. Investigations on the dehydration of 17α-ethynyl-17β-hydroxysteroid. Steroids 1993, 58, 220–224. [Google Scholar] [CrossRef]
- Kovács, D.; Kádár, Z.; Mótyán, G.; Schneider, Gy.; Wölfling, J.; Zupkó, I.; Frank, É. Synthesis, characterization and biological evaluation of some novel 17-isoxazoles in the estrone series. Steroids 2012, 77, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Nadaraia, N.S.; Kakhabrishvili, M.L.; Onashvili, E.O.; Barbakadze, N.N.; Getia, M.Z.; Pichette, A.; Sikharulidze, M.I.; Makhmudov, U.S. Synthesis of several 5α-androstano[17,16-d]pyrazolines from tigogenin. Chem. Nat. Compd. 2014, 50, 1024–1028. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Ar-NH-NH2 | Ar | Product | Yield 1 (%) |
| 1 | 15a | Ph | 17a | 89 |
| 2 | 15b | 4-CH3-C6H4 | 17b | 95 |
| 3 | 15c | 2-CH3-C6H4 | 17c | 75 |
| 4 | 15d | 2,4-diCH3-C6H3 | 17d | 80 |
| 5 | 15e | 4-F-C6H4 | 17e | 88 |
| 6 | 15f | 4-Cl-C6H4 | 17f | 81 |
| 7 | 15g | 4-Br-C6H4 | 17g | 83 |
| 8 | 15h | 4-CN-C6H4 | 17h | 85 |
| 9 | 15i | 4-NO2-C6H4 | 17i | 80 |
| 10 | 15j | 4-MeO-C6H4 | 17j | 82 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mótyán, G.; Molnár, B.; Wölfling, J.; Frank, É. Microwave-Assisted Stereoselective Heterocyclization to Novel Ring d-fused Arylpyrazolines in the Estrone Series. Molecules 2019, 24, 569. https://doi.org/10.3390/molecules24030569
Mótyán G, Molnár B, Wölfling J, Frank É. Microwave-Assisted Stereoselective Heterocyclization to Novel Ring d-fused Arylpyrazolines in the Estrone Series. Molecules. 2019; 24(3):569. https://doi.org/10.3390/molecules24030569
Chicago/Turabian StyleMótyán, Gergő, Barnabás Molnár, János Wölfling, and Éva Frank. 2019. "Microwave-Assisted Stereoselective Heterocyclization to Novel Ring d-fused Arylpyrazolines in the Estrone Series" Molecules 24, no. 3: 569. https://doi.org/10.3390/molecules24030569
APA StyleMótyán, G., Molnár, B., Wölfling, J., & Frank, É. (2019). Microwave-Assisted Stereoselective Heterocyclization to Novel Ring d-fused Arylpyrazolines in the Estrone Series. Molecules, 24(3), 569. https://doi.org/10.3390/molecules24030569