
Coupling the Antimalarial Cell Penetrating Peptide TP10 to Classical Antimalarial Drugs Primaquine and Chloroquine Produces Strongly Hemolytic Conjugates

 ,
,  , ,
, ,  ,
, 
 and
and 
Abstract
:
1. Introduction
2. Results
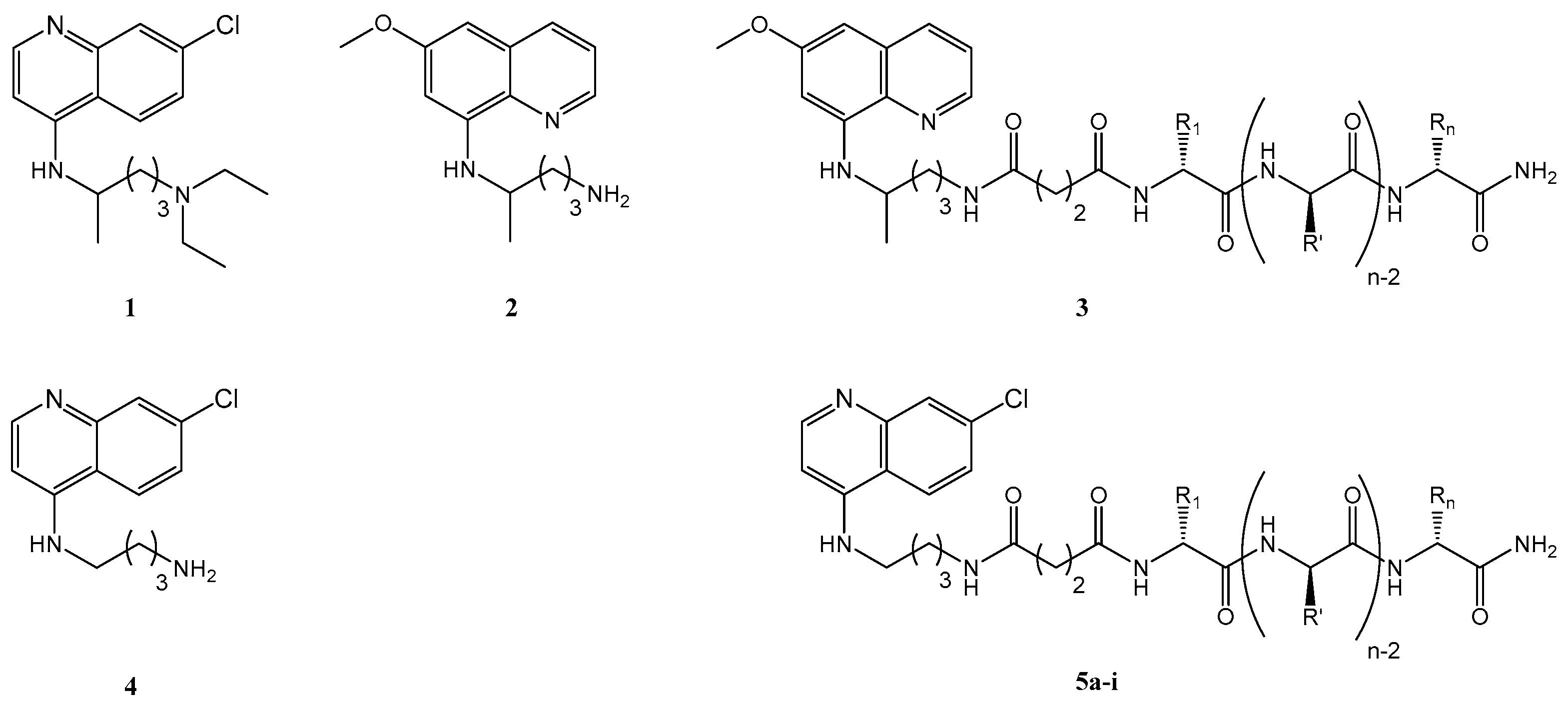
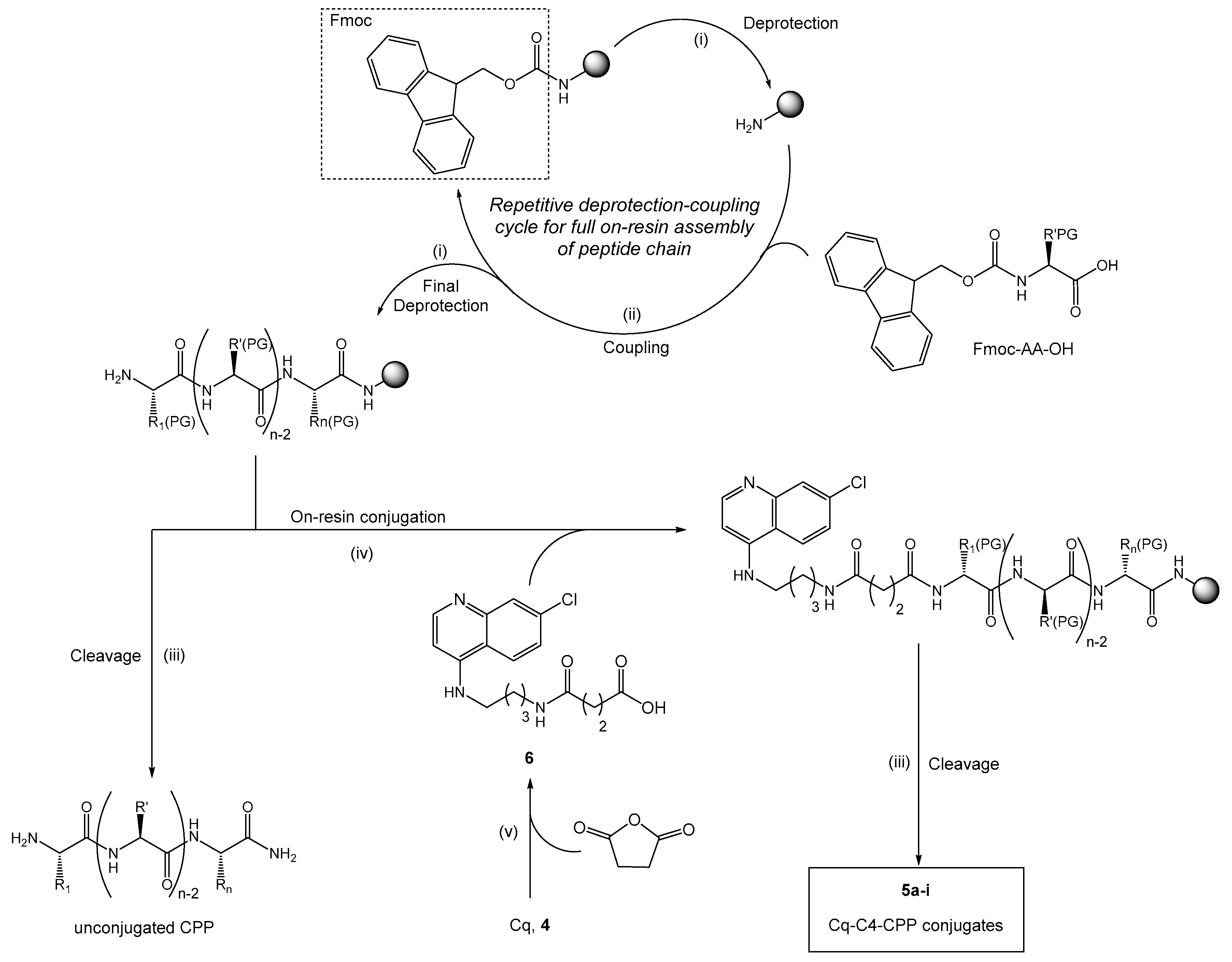
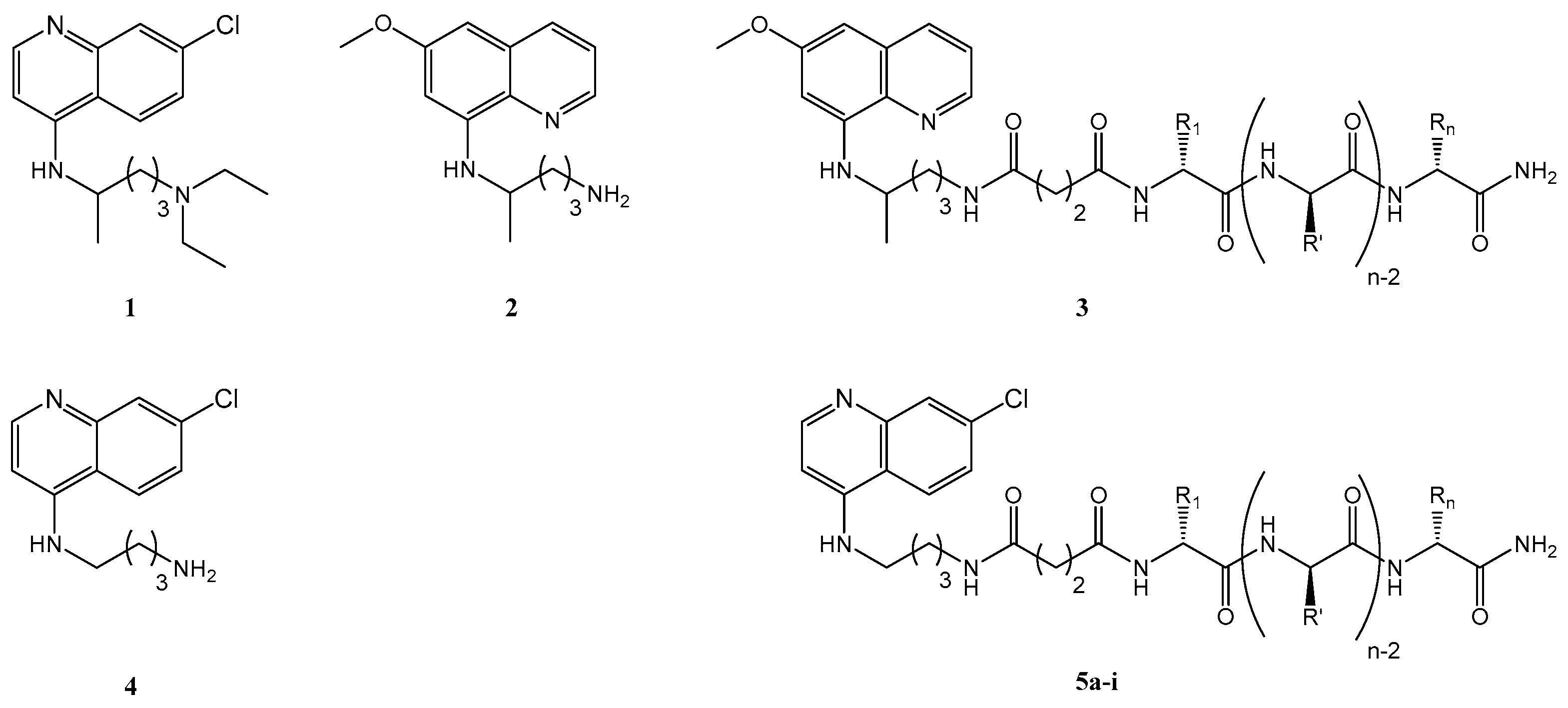
2.1. Synthesis and Antiplasmodial Activity of First-Generation Drug-Peptide Conjugates 5a–5i
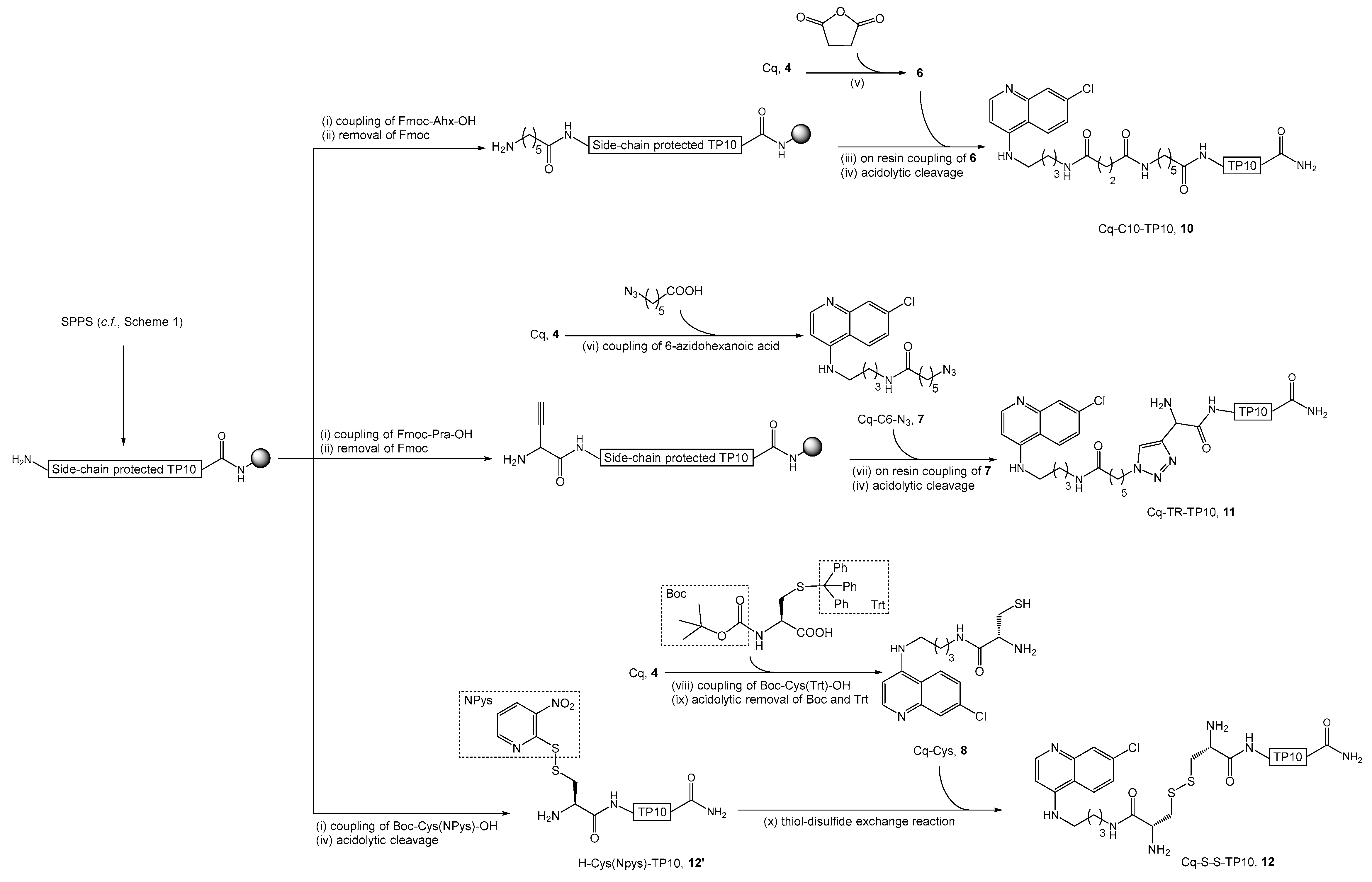
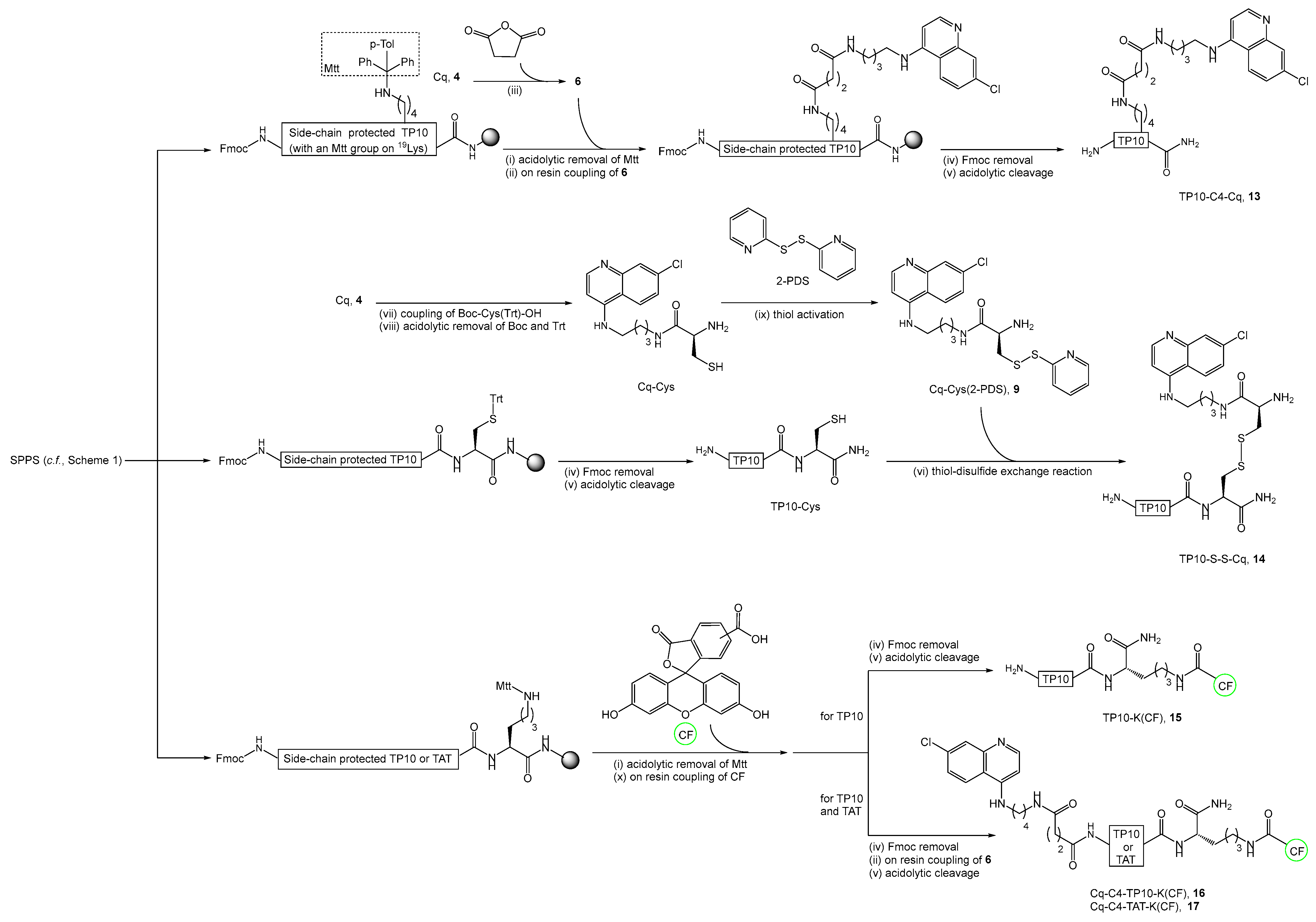
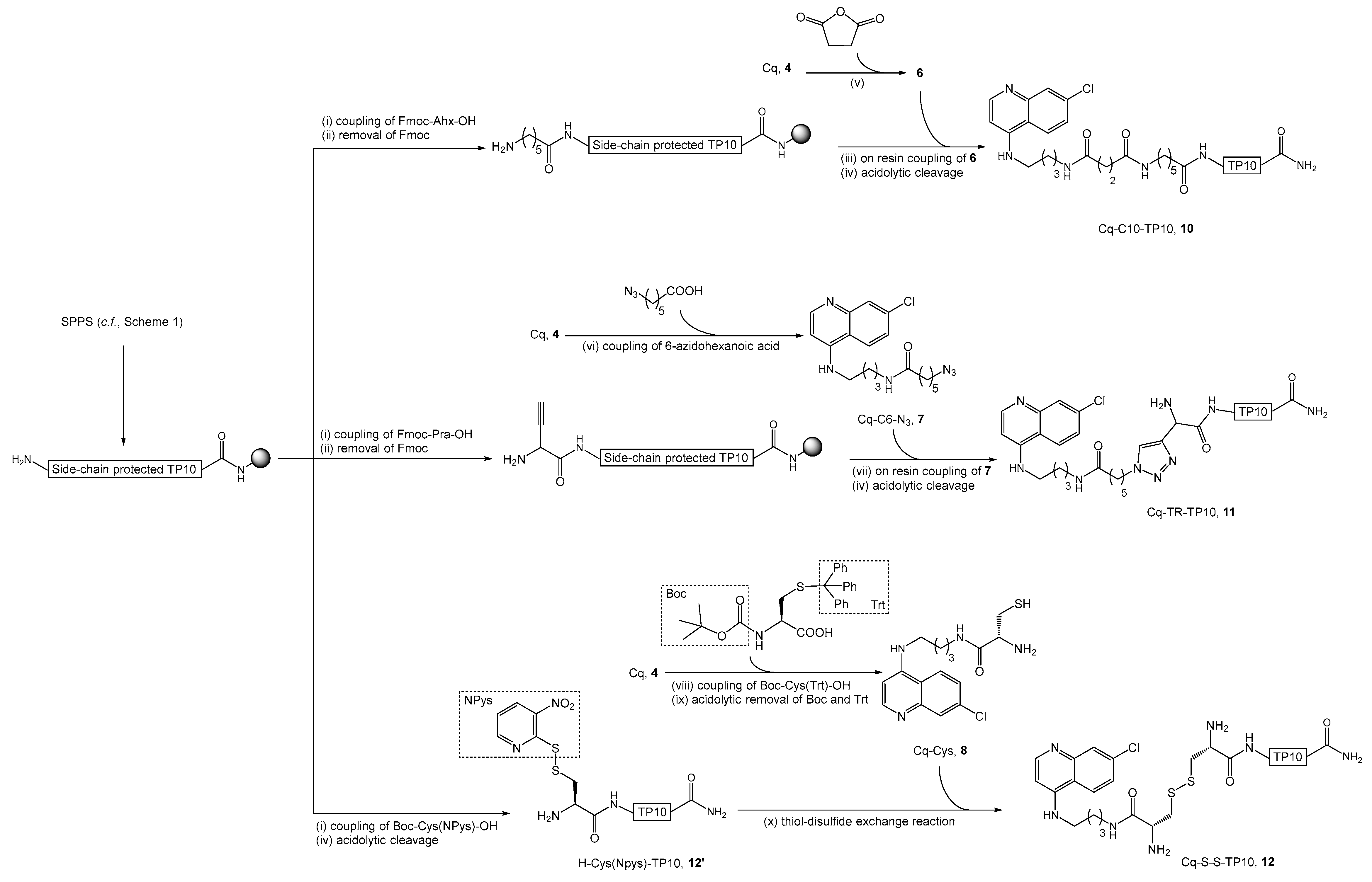
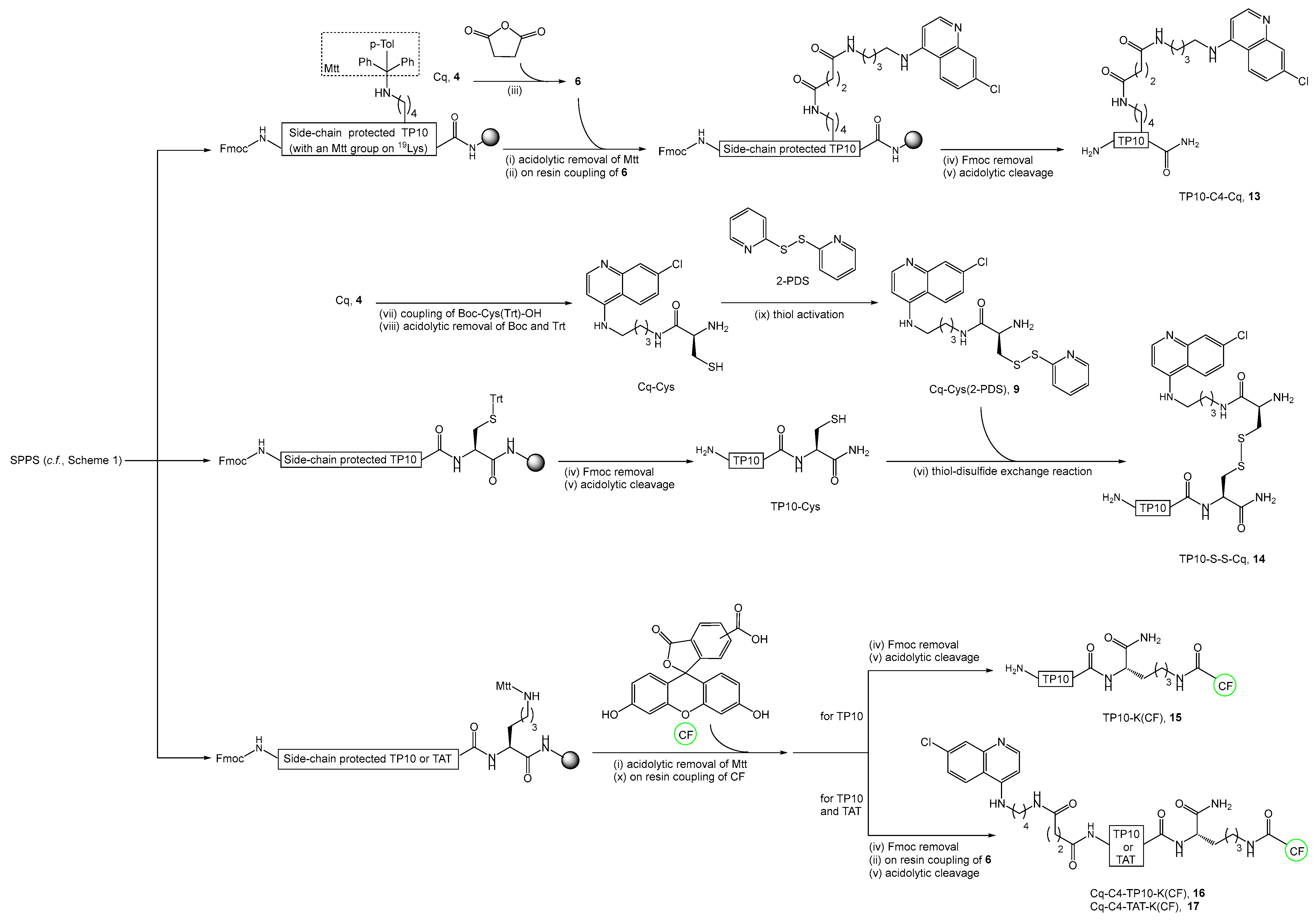
2.2. Synthesis, Antiplasmodial, and Hemolytic Activity of Second-Generation Conjugates 10–14
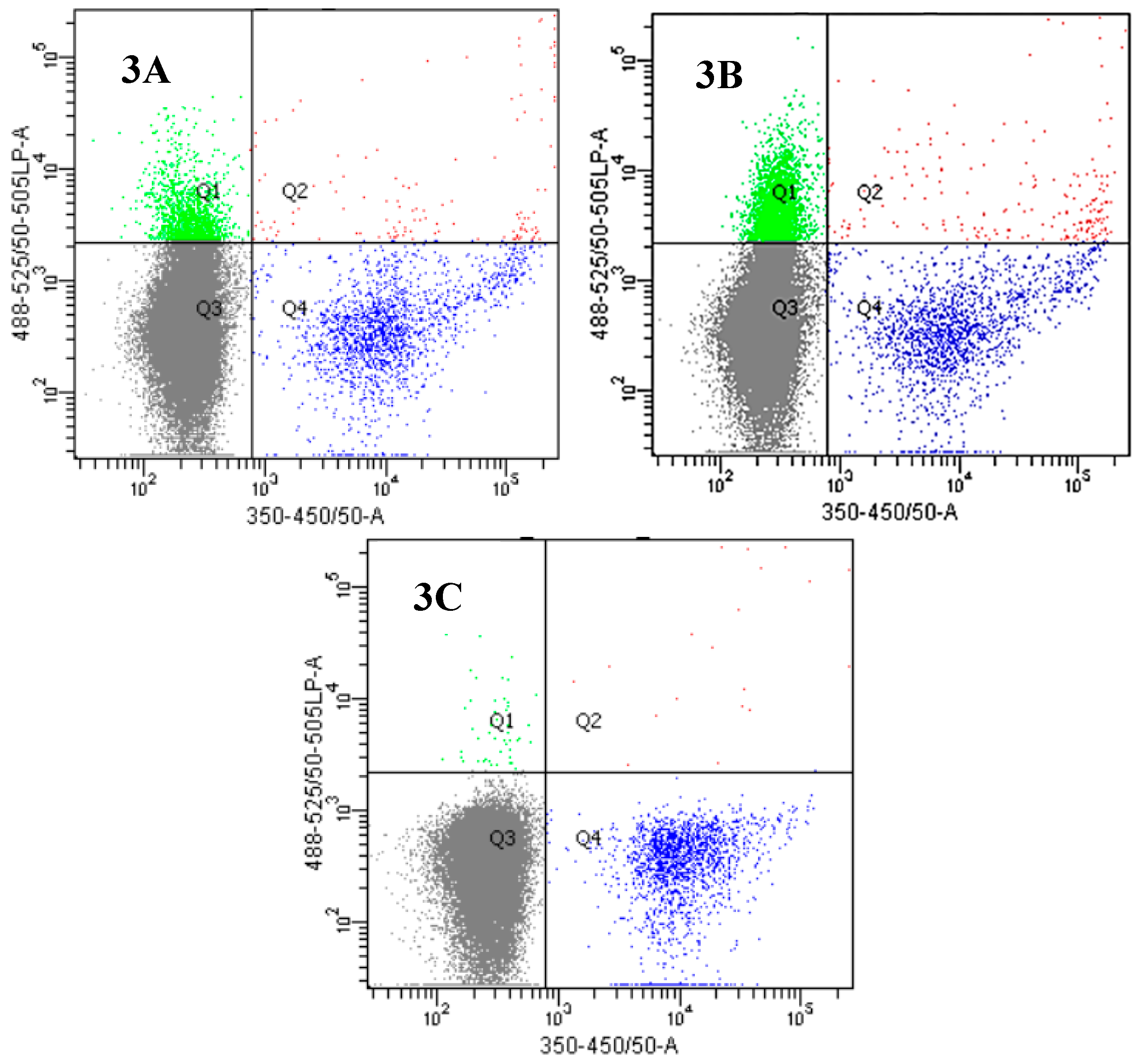
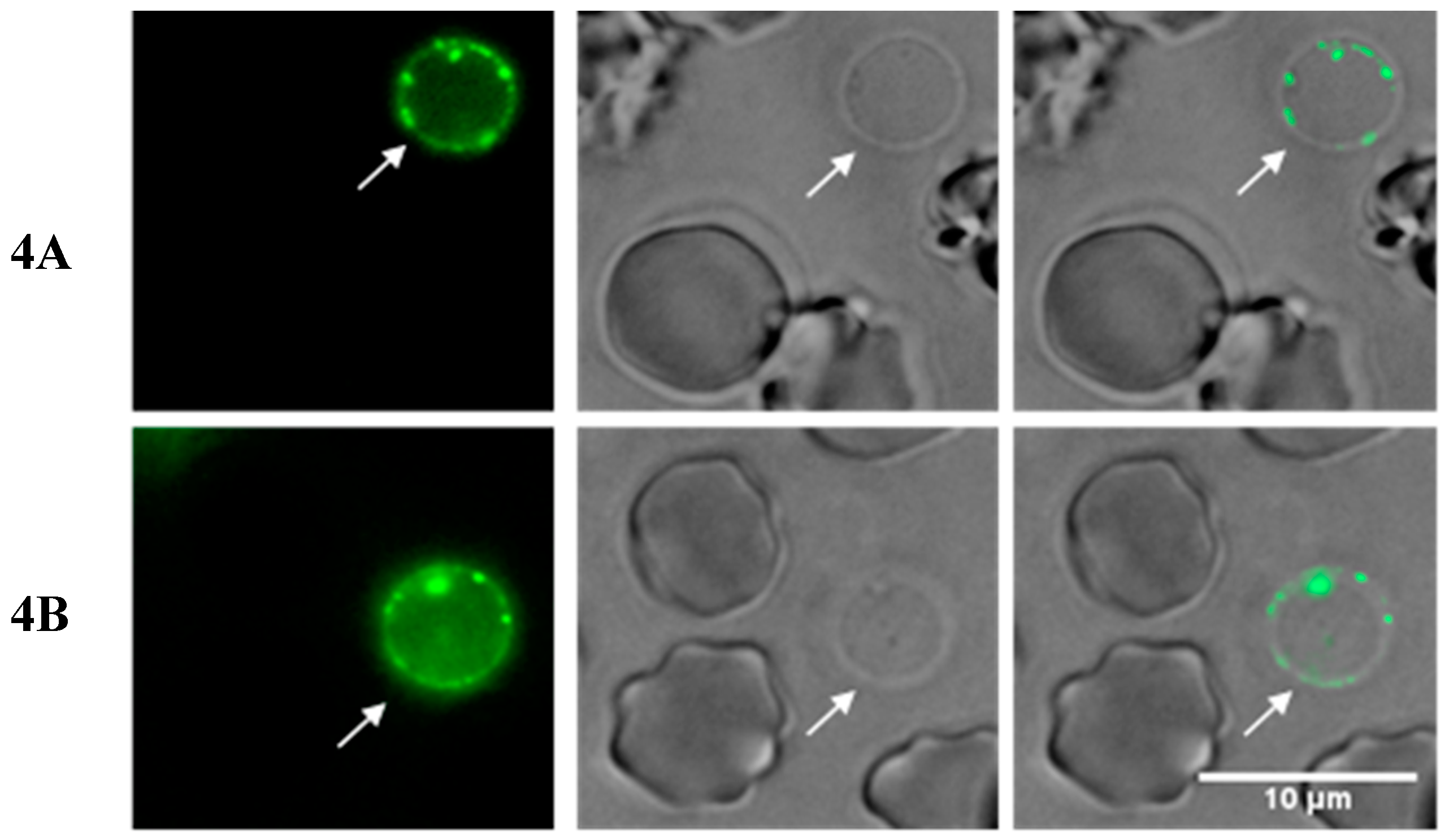
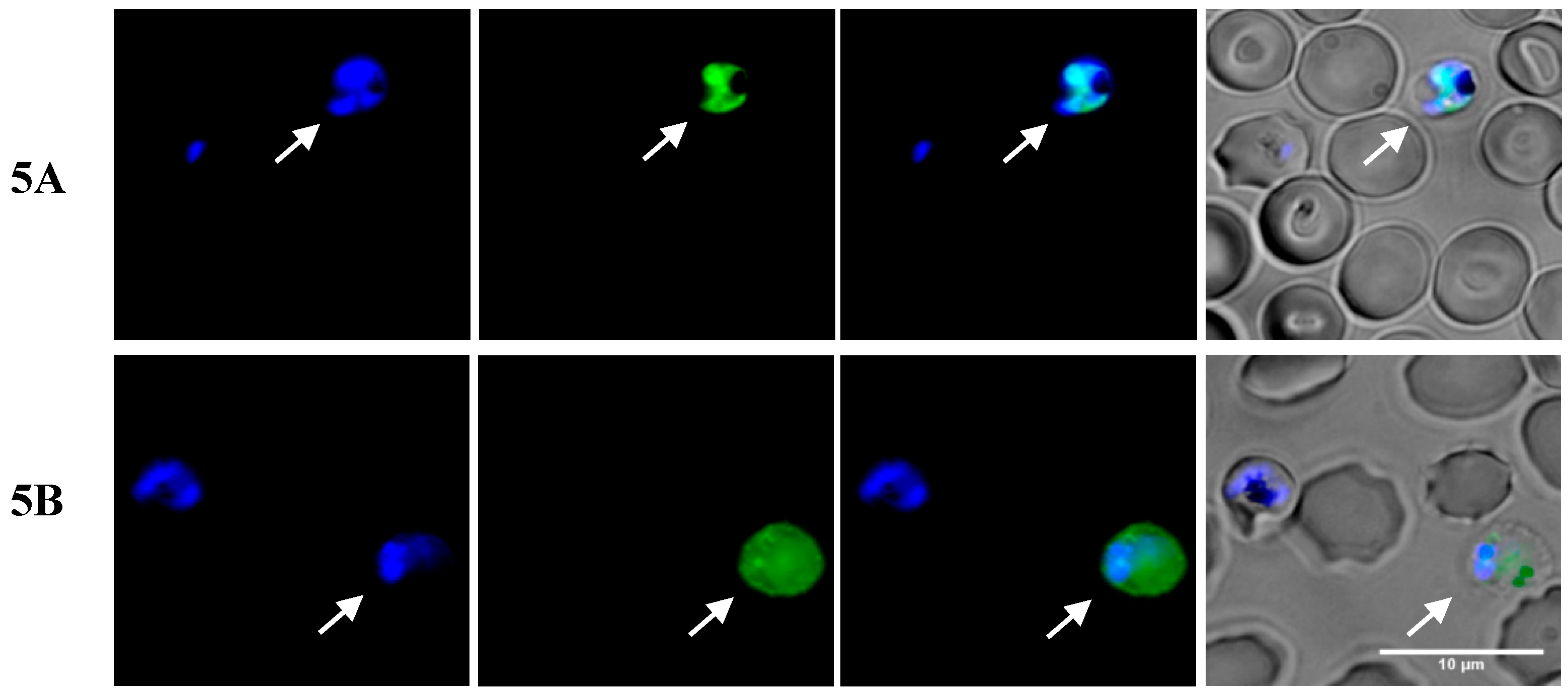
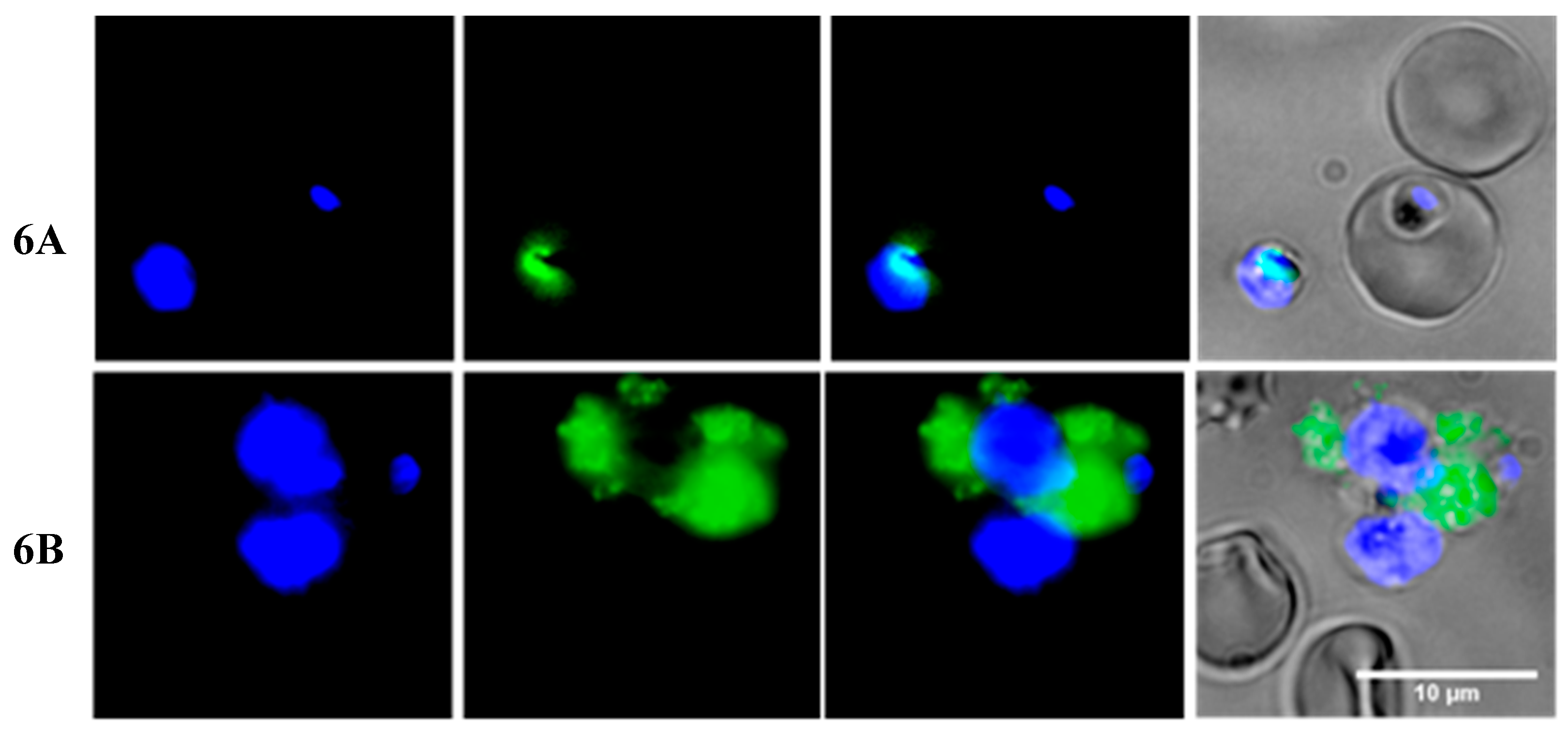
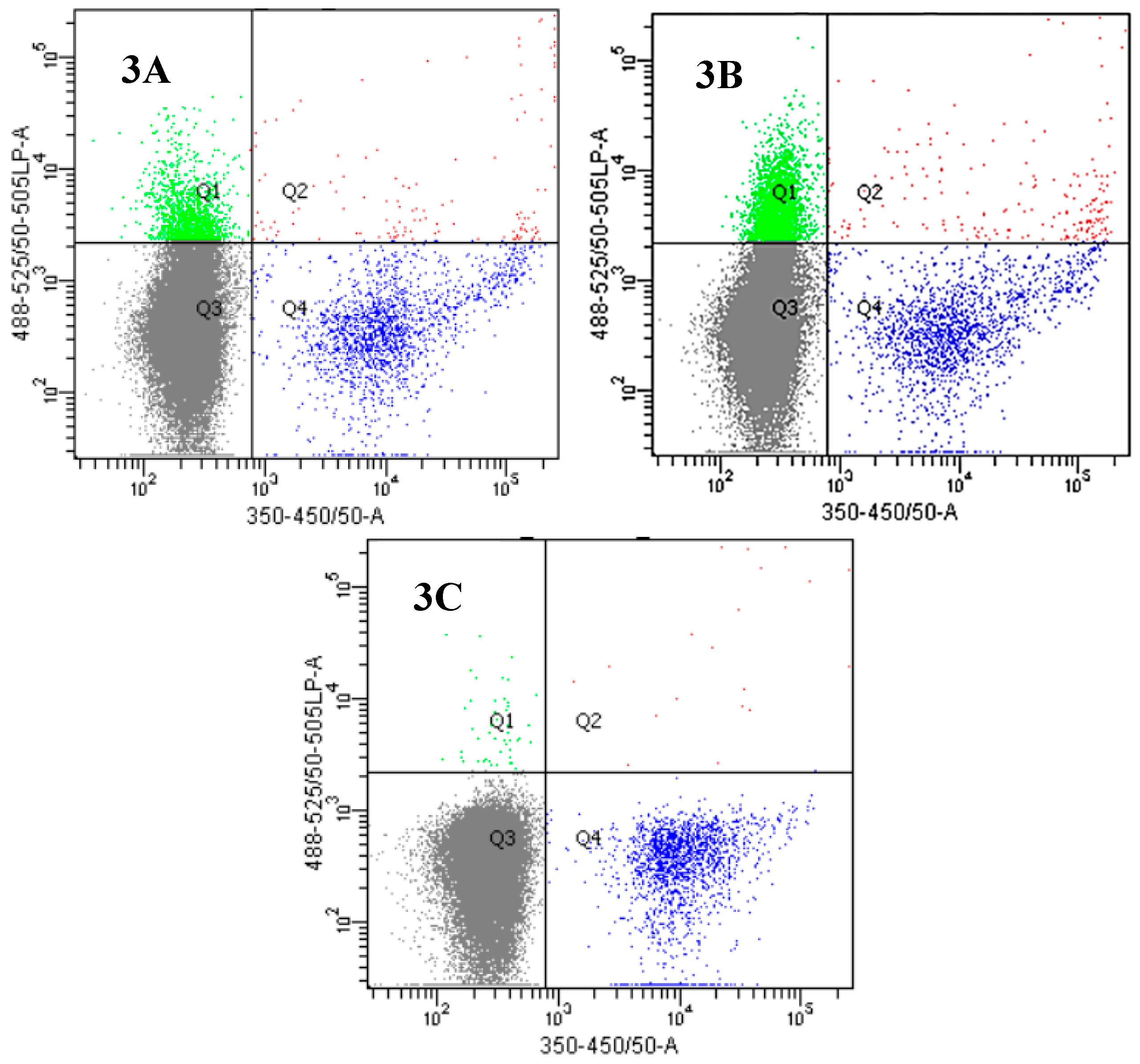
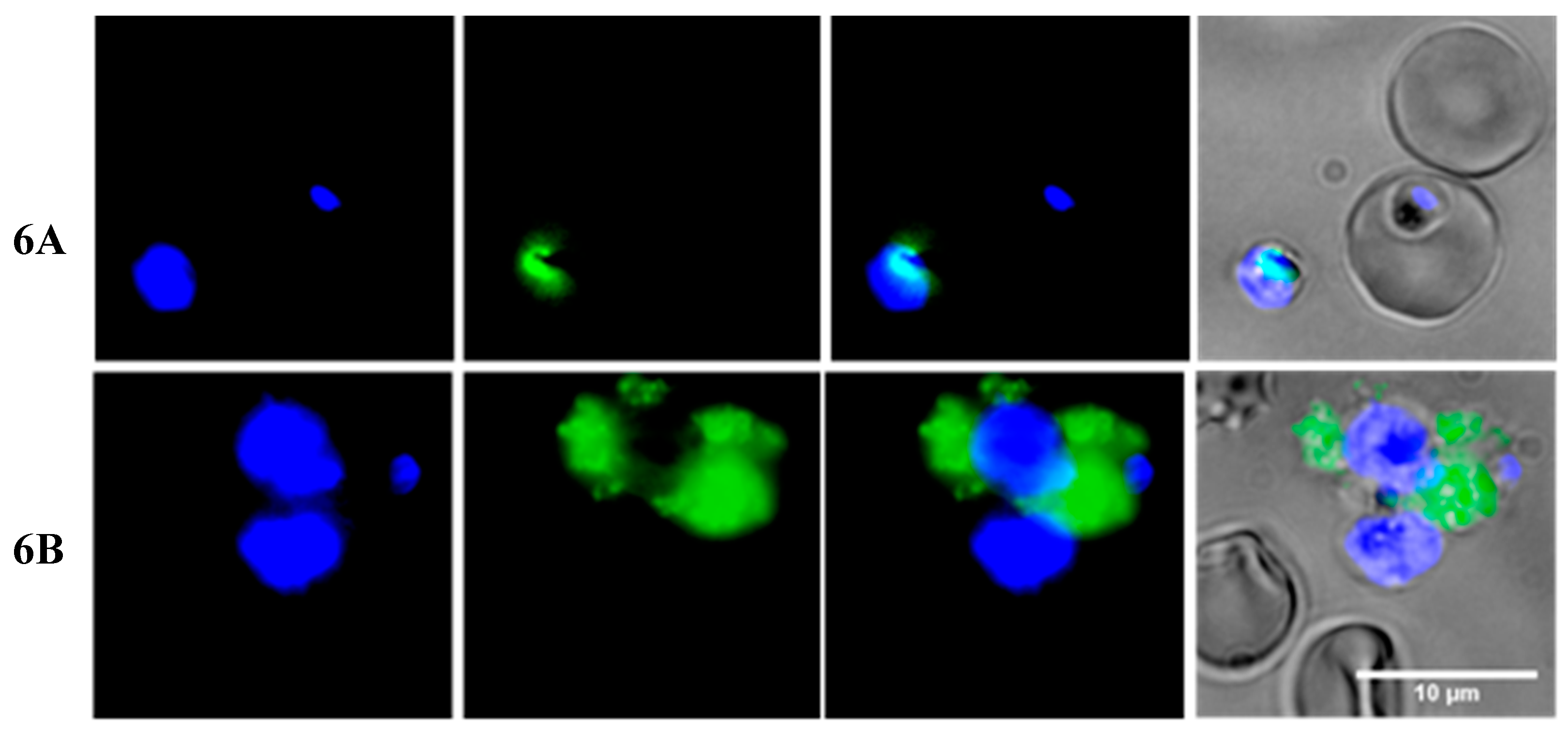
2.3. Insights into the Interactions between Chloroquine-Peptide Conjugates and Erythrocytes
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Instrumentation and General Procedures
4.3. Solution-Phase Synthesis
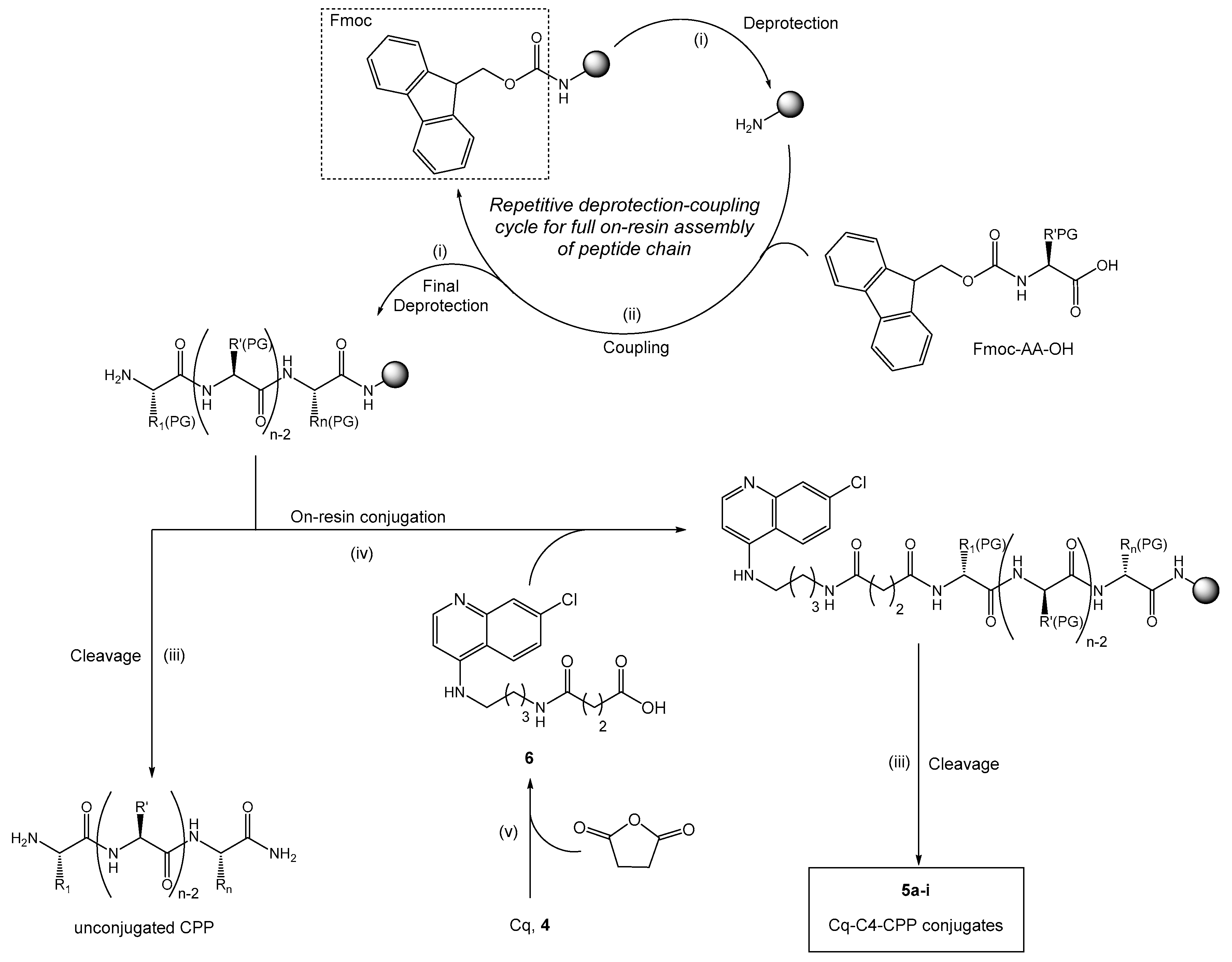
4.3.1. Synthesis of N-(7-Methylquinolin-4-yl)butane-1,4-diamine, Cq (4)
4.3.2. Synthesis of 3-({4-[(7-Methylquinolin-4-yl)amino]butyl}carbamoyl)propanoic acid, Cq-C4 (6)
4.3.3. 6-Azido-N-{4-[(7-Methylquinolin-4-yl)amino]butyl}hexanamide, Cq-C6-N3 (7)
4.3.4. Synthesis of 2-Amino-N-{4-[(7-Methylquinolin-4-yl)amino]butyl}-3-sulfanylpropanamide, Cq-Cys (8)
4.3.5. 2-Amino-N-{4-[(7-Methylquinolin-4-yl)amino]butyl}-3-(pyridin-2-yldisulfanyl)propaneimide, Cq-Cys(2-PDS) (9)
4.4. Solid-Phase Synthesis
4.4.1. General Procedures for Solid-Phase Peptide Synthesis
4.4.2. Synthesis of Cq-C4-CPP Conjugates (5a–5i)
4.4.3. Synthesis of Cq-C10-TP10 Conjugate (10)
4.4.4. Synthesis of Cq-TR-TP10 Conjugate (11)
4.4.5. Synthesis of Cq-S-S-TP10 Conjugate (12)
4.4.6. Synthesis of TP10-C4-Cq Conjugate (13)
4.4.7. Synthesis of TP10-S-S-Cq Conjugate (14)
4.4.8. Synthesis of Fluorescently-Labeled TP10-K(CF) (15) and TAT-K(CF) Peptides
4.4.9. Synthesis of Fluorescently Labeled Cq-C4-TP10-K(CF) (16) and Cq-C4-TAT-K(CF) (17) Conjugates
4.5. In Vitro Assessment of Antimalarial Activity
4.5.1. Activity of Conjugates 3a and 5a-i against P. Falciparum W2
4.5.2. Activity of Conjugates 3a, 5a, 14′ and 11–15 against P. Falciparum 3D7
4.6. In Vitro Assessment of Hemolytic Activity
4.7. Fluorescence Microscopy and Flow Cytometry Studies
4.7.1. Fluorescence Microscopy Assays
4.7.2. Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prudêncio, M.; Rodríguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Genet. 2006, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Machado, M.; Lobo, L.; Nogueira, F.; Prudêncio, M.; Teixeira, C.; Gomes, P. N-Cinnamoylation of antimalarial classics: Effects of using acyl groups other than cinnamoyl toward dual-stage antimalarials. ChemMedChem 2015, 10, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, M.J.; Bom, J.; Capela, R.; Casimiro, C.; Chambel, P.; Gomes, P.; Iley, J.; Lopes, F.; Morais, J.; Moreira, R.; et al. Imidazolidin-4-one derivatives of primaquine as novel transmission-blocking antimalarials. J. Med. Chem. 2005, 48, 888–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vale, N.; Prudêncio, M.; Marques, C.A.; Collins, M.S.; Gut, J.; Nogueira, F.; Matos, J.; Rosenthal, P.J.; Cushion, M.T.; do Rosário, E.V.; et al. Imidazoquines as antimalarial and anti-pneumocystis agents. J. Med. Chem. 2009, 52, 7800–7807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, J.; Da Cruz, F.P.; Cabrita, É.; Gut, J.; Nogueira, F.; Do Rosário, V.E.; Moreira, R.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. Novel potent metallocenes against liver stage malaria. Antimicrob. Agents Chemother. 2012, 56, 1564–1570. [Google Scholar] [CrossRef] [Green Version]
- Perez, B.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. PRIMACINS, N-Cinnamoyl-Primaquine conjugates, with improved liver-stage antimalarial activity. MedChemComm 2012, 3, 1170–1172. [Google Scholar] [CrossRef] [Green Version]
- Pérez, B.C.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Prudêncio, M.; Gomes, P. N-Cinnamoylated chloroquine analogues as dual-stage antimalarial leads. J. Med. Chem. 2013, 56, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.; Perez, B.; Albuquerque, I.S.; Machado, M.; Prudêncio, M.; Nogueira, F.; Teixeira, C.; Gomes, P. N-Cinnamoylation of antimalarial classics: Quinacrine analogues with decreased toxicity and dual-stage activity. ChemMedChem 2013, 9, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, R.; Noronha, J.; Ferreira, F.I.H.M.; Nogueira, F.; Machado, M.; Prudêncio, M.; Parapini, S.; D’Alessandro, S.; Teixeira, C.; Gomes, A.; et al. Primaquine-based ionic liquids as a novel class of antimalarial hits. RSC Adv. 2016, 6, 56134–56138. [Google Scholar] [CrossRef]
- Moles, E.; Galiano, S.; Gomes, A.; Quiliano, M.; Teixeira, C.; Aldana, I.; Gomes, P.; Fernàndez-Busquets, X. ImmunoPEGliposomes for the targeted delivery of novel lipophilic drugs to red blood cells in a falciparum malaria murine model. Biomaterials 2017, 145, 178–191. [Google Scholar] [CrossRef] [Green Version]
- Bechara, C.; Sagan, S. Cell-Penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Svensen, N.; Walton, J.G.; Bradley, M. Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 2012, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.A.; Denny, W.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2011, 1816, 232–246. [Google Scholar] [CrossRef]
- Sparr, C.; Purkayastha, N.; Kolesinska, B.; Gengenbacher, M.; Amulic, B.; Matuschewski, K.; Seebach, D.; Kamena, F. Improved efficacy of fosmidomycin against Plasmodium and Mycobacterium species by combination with the cell-penetrating peptide octaarginine. Antimicrob. Agents Chemother. 2013, 57, 4689–4698. [Google Scholar] [CrossRef] [Green Version]
- Arrighi, R.B.G.; Ebikeme, C.; Jiang, Y.; Ranford-Cartwright, L.; Barrett, M.P.; Langel, Ü.; Faye, I. Cell-penetrating peptide TP10 shows broad-spectrum activity against both Plasmodium falciparum and Trypanosoma brucei brucei. Antimicrob. Agents Chemother. 2008, 52, 3414–3417. [Google Scholar] [CrossRef] [Green Version]
- Guergnon, J.; Dessauge, F.; Dominguez, V.; Viallet, J.; Bonnefoy, S.; Yuste, V.J.; Mercereau-Puijalon, O.; Cayla, X.; Rebollo, A.; Susin, S.A.; et al. Use of penetrating peptides interacting with PP1/PP2A proteins as a general approach for a drug phosphatase technology. Mol. Pharmacol. 2006, 69, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Mansour, S.C.; De La Fuente-Núñez, C.; Hancock, R.E.W. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef]
- Aguiar, L.; Machado, M.; Sanches-Vaz, M.; Prudêncio, M.; Vale, N.; Gomes, P. Coupling the cell-penetrating peptides transportan and transportan 10 to primaquine enhances its activity against liver-stage malaria parasites. MedChemComm 2019, 10, 221–226. [Google Scholar] [CrossRef]
- Wellems, T.E.; Plowe, C.V. Chloroquine-Resistant malaria. J. Infect. Dis. 2001, 184, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Pradines, B.; Barbe, J.; Fusai, T.; Papa, V.; Houdoin, C.; Mosnier, J.; Rogier, C.; Santelli-Rouvier, C.; Alibert-Franco, S.; Parzy, D. In vitro reversal of chloroquine resistance in Plasmodium falciparum with dihydroethanoanthracene derivatives. Am. J. Trop. Med. Hyg. 2002, 66, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumai, J.; Hozumi, K.; Yamada, Y.; Katagiri, F.; Kikkawa, Y.; Nomizu, M. Effect of spacer length and type on the biological activity of peptide-polysaccharide matrices. Pept. Sci. 2016, 106, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Mbatha, L.; Chakravorty, S.; Koning, C.; Otterlo, W.; Arbuthnot, P.; Ariatti, M.; Singh, M. Spacer Length: A Determining Factor in the Design of Galactosyl Ligands for Hepatoma Cell-Specific Liposomal Gene Delivery. Curr. Drug Deliv. 2016, 13, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Agarwal, D.; Kumar, S.; Dixit, A.; Gupta, R.D.; Awasthi, S.K. In vitro antiplasmodial efficacy of synthetic coumarin-triazole analogs. Eur. J. Med. Chem. 2018, 145, 735–745. [Google Scholar] [CrossRef]
- Boechat, N.; Ferreira, M.D.L.G.; Pinheiro, L.C.S.; Jesus, A.M.L.; Leite, M.M.M.; Júnior, C.C.S.; Aguiar, A.C.C.; Andrade, I.M.; Krettli, A.U.; De Andrade, I.M. New compounds hybrids 1H-1,2,3-triazole-quinoline against Plasmodium falciparum. Chem. Boil. Drug Des. 2014, 84, 325–332. [Google Scholar] [CrossRef]
- Singh, P.; Sachdeva, S.; Raj, R.; Kumar, V.; Mahajan, M.P.; Nasser, S.; Vivas, L.; Gut, J.; Rosenthal, P.J.; Feng, T.-S.; et al. Antiplasmodial and cytotoxicity evaluation of 3-functionalized 2-azetidinone derivatives. Bioorganic Med. Chem. Lett. 2011, 21, 4561–4563. [Google Scholar] [CrossRef]
- Bakunov, S.A.; Bakunova, S.M.; Wenzler, T.; Ghebru, M.; Werbovetz, K.A.; Brun, R.; Tidwell, R.R. Synthesis and Antiprotozoal activity of cationic 1,4-diphenyl-1H-1,2,3-triazoles. J. Med. Chem. 2010, 53, 254–272. [Google Scholar] [CrossRef] [Green Version]
- Rathod, P.K.; McErlean, T.; Lee, P.-C. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1997, 94, 9389–9393. [Google Scholar] [CrossRef] [Green Version]
- Ecker, A.; Lehane, A.M.; Clain, J.; Fidock, D.A. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012, 28, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Chinappi, M.; Via, A.; Marcatili, P.; Tramontano, A. On the mechanism of chloroquine resistance in Plasmodium falciparum. PLoS ONE 2010, 5, e14064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursos, L.M.; Roepe, P.D. Chloroquine resistance in the malarial parasite, Plasmodium falciparum. Med. Res. Rev. 2002, 22, 465–491. [Google Scholar] [CrossRef] [PubMed]
- Pulcini, S.; Staines, H.M.; Lee, A.H.; Shafik, S.H.; Bouyer, G.; Moore, C.M.; Daley, D.A.; Hoke, M.J.; Altenhofen, L.M.; Painter, H.J.; et al. Mutations in the Plasmodium falciparum chloroquine resistance transporter, PfCRT, enlarge the parasite’s food vacuole and alter drug sensitivities. Sci. Rep. 2015, 5, 14552. [Google Scholar] [CrossRef] [PubMed]
- Boudhar, A.; Ng, X.W.; Loh, C.Y.; Ni Chia, W.; Tan, Z.M.; Nosten, F.; Dymock, B.W.; Tan, K.S.W. Overcoming chloroquine resistance in malaria: Design, synthesis, and structure-activity relationships of novel hybrid compounds. Antimicrob. Agents Chemother. 2016, 60, 3076–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, S.H.; Miller, S.E.; Keim, A.I.; Gorka, A.P.; Schnermann, M.J.; Schneider, J.P. An intrinsically disordered peptide facilitates non-endosomal cell entry. Angew. Chem. Int. Ed. 2016, 55, 3369–3372. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.S.; Geörg, M.; Sjölinder, H.; Sillard, R.; Lindberg, S.; Langel, Ü.; Jonsson, A.-B. Identification of cell-penetrating peptides that are bactericidal to Neisseria meningitidis and prevent inflammatory responses upon infection. Antimicrob. Agents Chemother. 2013, 57, 3704–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, R.M.; Brun, R.; Grieder, A. Microscopic and flow cytophotometric analysis of parasitemia in cultures of Plasmodium falciparum vitally stained with Hoechst 33342—application to studies of antimalarial agents. Zeitschrift für Parasitenkunde 1986, 72, 201–212. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Ciudad, S.; Guiu, M.; Arranz-Gibert, P.; Garcia, C.; Gomis, R.R.; Cecchelli, R.; García, J.; Giralt, E.; et al. MiniAp-4: A venom-inspired peptidomimetic for brain delivery. Angew. Chemie Int. Ed. 2016, 55, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Hale, V.L.; Watermeyer, J.M.; Hackett, F.; Vizcay-Barrena, G.; Van Ooij, C.; Thomas, J.A.; Spink, M.C.; Harkiolaki, M.; Duke, E.; Fleck, R.A.; et al. Parasitophorous vacuole poration precedes its rupture and rapid host erythrocyte cytoskeleton collapse in Plasmodium falciparum egress. Proc. Natl. Acad. Sci. USA 2017, 114, 3439–3444. [Google Scholar] [CrossRef] [Green Version]
- Lux, S.E. Anatomy of the red cell membrane skeleton: Unanswered questions. Blood 2016, 127, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Yue, T.; Tian, F.; Liu, Z.; Zhang, X. Erythrocyte membrane skeleton inhibits nanoparticle endocytosis. AIP Adv. 2017, 7, 065303. [Google Scholar] [CrossRef] [Green Version]
- Minetti, G.; Achilli, C.; Perotti, C.; Ciana, A. continuous change in membrane and membrane-skeleton organization during development from proerythroblast to senescent red blood cell. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Mó, M.; Alfranca, A.; Cabanas, C.; Marazuela, M.; Tejedor, R.; Ursa, M.A.; Ashman, L.K.; De Landázuri, M.O.; Sánchez-Madrid, F. Regulation of endothelial cell motility by complexes of tetraspan molecules CD81/TAPA-1 and CD151/PETA-3 with alpha 3beta 1 integrin localized at endothelial lateral junctions. J. Cell Boil. 1998, 141, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Zuccala, E.; Baum, J. Cytoskeletal and membrane remodelling during malaria parasite invasion of the human erythrocyte. Br. J. Haematol. 2011, 154, 680–689. [Google Scholar] [CrossRef]
- Callan-Jones, A.; Arriagada, O.E.A.; Massiera, G.; Lorman, V.; Abkarian, M. Red blood cell membrane dynamics during malaria parasite egress. Biophys. J. 2012, 103, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, F.; Diez, A.; Radfar, A.; Pérez-Benavente, S.; Rosario, V.E.D.; Puyet, A.; Bautista, J.M. Early transcriptional response to chloroquine of the Plasmodium falciparum antioxidant defence in sensitive and resistant clones. Acta Trop. 2010, 114, 109–115. [Google Scholar] [CrossRef]
- Palasek, S.A.; Cox, Z.J.; Collins, J.M. Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. J. Pept. Sci. 2007, 13, 143–148. [Google Scholar] [CrossRef]
- Turner, R.A.; Oliver, A.G.; Lokey, R.S. Click chemistry as a macrocyclization tool in the solid-phase synthesis of small cyclic peptides. Org. Lett. 2007, 9, 5011–5014. [Google Scholar] [CrossRef]
- Jain, N.; Friedman, S.H. A tetra-orthogonal strategy for the efficient synthesis of scaffolds based on cyclic peptides. Int. J. Pept. Res. Ther. 2018, 24, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Elbert, D.L. The kinetics of the removal of the N-methyltrityl (Mtt) group during the synthesis of branched peptides. J. Pept. Res. 2002, 60, 300–303. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Böhmová, E.; Machová, D.; Pechar, M.; Pola, R.; Venclíková, K.; Janoušková, O.; Etrych, T. Cell-Penetrating peptides: A useful tool for the delivery of various cargoes into cells. Physiol. Res. 2018, 67, S267–S279. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Lin, L. Cell penetrating peptides: A promising tool for the cellular uptake of macromolecular drugs. Protein Pept. Sci. 2018, 19, 211–220. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Freshly synthesized samples of all compounds can be made available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPP | Amino Acid Sequence (Respective MW/Da) | Cq-C4-CPP Conjugate (Respective MW/Da) |
|---|---|---|
| TP10 | AGYLLGKINLKALAALAKKIL (2182) | 5a (2512) |
| Transportan | GWTLNSAGYLLGKINLKALAALAKKIL (2840) | 5b (3170) |
| DPT-sh1 | VKKKKIKREIKI (1510) | 5c (1841) |
| DPT-sh2 | RQKRLIRQKRLIRQKRLI (2402) | 5d (2732) |
| IDR-1018 | VRLIVAVRIWRR (1535) | 5e (1867) |
| TAT | GRKKRRQRRRPPQ (1718) | 5f (2049) |
| PasTAT | FFLIPKGGRKKRRQRRRPPQ (2521) | 5g (2852) |
| R9 | RRRRRRRRR (1423) | 5h (1753) |
| Penetratin | RQIKIWFQNRRMKWKK (2246) | 5i (2576) |
| CPP | IC50/µM | SD (n = 3) | Cq-C4-CPP | IC50/µM | SD (n = 3) |
|---|---|---|---|---|---|
| TP10 | 5.5 | 0.2 | Cq-C4-TP10, 5a | 1.5 | 0.1 |
| Transportan | 3.1 | 0.8 | Cq-C4-Transportan, 5b | 5.2 | 0.2 |
| DPT-sh1 | >10 | - | Cq-C4-DPT-sh1, 5c | >10 | - |
| DPT-sh2 | >10 | - | Cq-C4-DPT-sh2, 5d | >10 | - |
| IDR-1018 | >10 | - | Cq-C4-IDR-1018, 5e | >10 | - |
| TAT | >10 | - | Cq-C4-TAT, 5f | >10 | - |
| PasTAT | >10 | - | Cq-C4-PasTAT, 5g | 8.5 | 0.2 |
| R9 | >10 | - | Cq-C4-R9, 5h | >10 | - |
| Penetratin | >10 | - | Cq-C4-Penetratin, 5i | >10 | - |
| CQ (1) | 0.699 a |
| Test Compound | MW/Da | IC50 ± SD (n = 3)/µM | % hemolysis at 10 µM ± SD (n = 3) | |
|---|---|---|---|---|
| hRBC | PiRBC | |||
| PQ-C4-TP10, 3a | 2525 | - a | 89 ± 8 | 49 ± 7 |
| Cq-C4-TP10, 5a | 2514 | 0.8 ± 0.1 | 23 ± 1 | 6.5 ± 0.4 |
| Cq-C10-TP10, 10 | 2626 | 0.8 ± 0.3 | 38 ± 2 | 19 ± 1 |
| Cq-TR-TP10, 11 | 2665 | 1.2 ± 0.2 | 23 ± 1 | 10.7 ± 0.1 |
| Cq-S-S-TP10, 12 | 2636 | 1.0 ± 0.3 | 8.5 ± 0.5 | 2.0 ± 0.3 |
| TP10-C4-Cq, 13 | 2514 | - a | 50 ± 2 | 41 ± 2 |
| TP10-S-S-Cq, 14 | 2636 | 2.3 ± 0.4 | 18.2 ± 0.8 | 7 ± 1 |
| TP10-S-S-PQ, 14′ | 2647 | - a | 99.7 ± 0.4 | 72 ± 9 |
| TP10 | 2182 | 1.9 ± 0.5 | 3.7 ± 0.1 | 3.4 ± 0.2 |
| CQ, 1 | 319.9 | 0.021b | - c | - c |
| Cq, 4 | 249.7 | 0.07 ± 0.02 | - c | - c |
| Cq + TP10 1:1 | n.a. | 0.024d | 4.4 ± 0.6 | 1.8 ± 0.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguiar, L.; Biosca, A.; Lantero, E.; Gut, J.; Vale, N.; Rosenthal, P.J.; Nogueira, F.; Andreu, D.; Fernàndez-Busquets, X.; Gomes, P. Coupling the Antimalarial Cell Penetrating Peptide TP10 to Classical Antimalarial Drugs Primaquine and Chloroquine Produces Strongly Hemolytic Conjugates. Molecules 2019, 24, 4559. https://doi.org/10.3390/molecules24244559
Aguiar L, Biosca A, Lantero E, Gut J, Vale N, Rosenthal PJ, Nogueira F, Andreu D, Fernàndez-Busquets X, Gomes P. Coupling the Antimalarial Cell Penetrating Peptide TP10 to Classical Antimalarial Drugs Primaquine and Chloroquine Produces Strongly Hemolytic Conjugates. Molecules. 2019; 24(24):4559. https://doi.org/10.3390/molecules24244559
Chicago/Turabian StyleAguiar, Luísa, Arnau Biosca, Elena Lantero, Jiri Gut, Nuno Vale, Philip J. Rosenthal, Fátima Nogueira, David Andreu, Xavier Fernàndez-Busquets, and Paula Gomes. 2019. "Coupling the Antimalarial Cell Penetrating Peptide TP10 to Classical Antimalarial Drugs Primaquine and Chloroquine Produces Strongly Hemolytic Conjugates" Molecules 24, no. 24: 4559. https://doi.org/10.3390/molecules24244559
APA StyleAguiar, L., Biosca, A., Lantero, E., Gut, J., Vale, N., Rosenthal, P. J., Nogueira, F., Andreu, D., Fernàndez-Busquets, X., & Gomes, P. (2019). Coupling the Antimalarial Cell Penetrating Peptide TP10 to Classical Antimalarial Drugs Primaquine and Chloroquine Produces Strongly Hemolytic Conjugates. Molecules, 24(24), 4559. https://doi.org/10.3390/molecules24244559