3. Materials and Methods
Melting points were measured using a Reichert microscope (Gallenkamp hot stage apparatus; Mettler-Toledo Ltd, Sydney, Australia) and are uncorrected. Infrared spectra were recorded with a Thermo Nicolet 370 FTIR spectrometer with the sample prepared as a KBr pellet. NMR data were recorded using a Bruker DPX300 instrument (
1H 300 MHz,
13C 75.6 MHz) (Bruker Pty Ltd, Preston, Victoria, Australia) at 25 °C and reported as chemical shift (δ) relative to SiMe
4. High resolution mass spectrometric analysis was carried out at the Biomedical Mass Spectrometry Facility, UNSW, and the spectra were recorded on Q-TOF Ultima API (Micromass; Waters, Rydalmere, NSW, Australia). Gravity column chromatography was carried out using Merck 230–400 mesh ASTM silica gel.
Supplementary Materials contains NMR spectroscopic data.
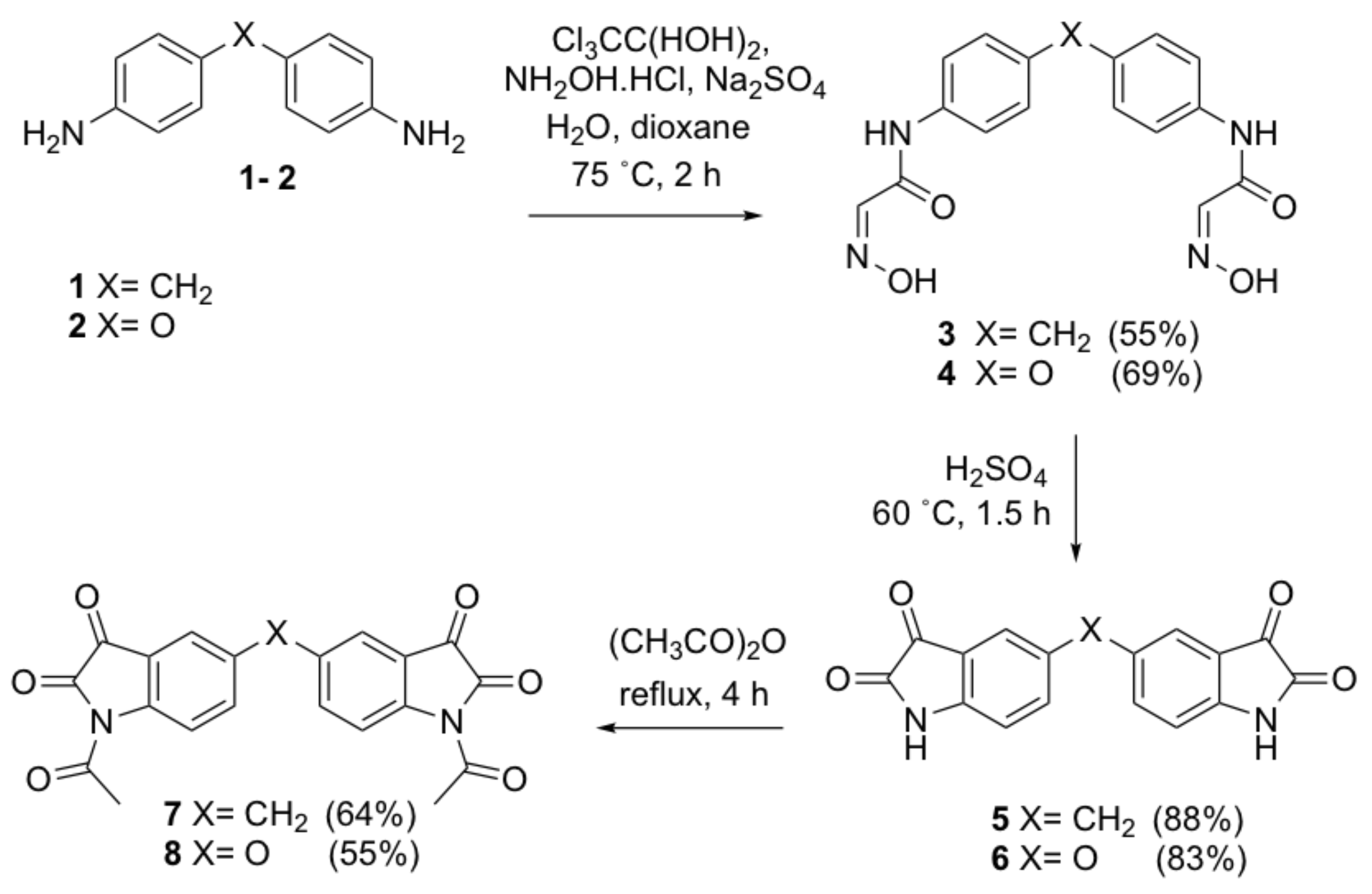
N,N’-[4,4’-bis(4,1-phenylene)] bis [2-(hydroxyimino)acetamide]methylene (3). Concentrated sodium sulfate aqueous solution (175 mL) and a solution of 4,4′-methylenedianiline 1 (10 g, 50.4 mmol) in dioxane (15 mL) was added to a stirred solution of chloral hydrate (18.5 g, 126.1 mmol) in water (175 mL). The reaction mixture was heated slowly to 75 °C until a yellow precipitate was formed. A solution of hydroxylamine hydrochloride (8.8 g, 126.7 mmol) in water (55 mL) was then added into the resulting suspension. The temperature was increased to 75 °C and the reaction mixture was stirred for a further 2 h. The reaction mixture was cooled to room temperature and left to stand overnight. The crude product was collected by filtration and recrystallized from ethyl acetate to give the title compound as yellow crystals (9.44 g, 55%); mp 272–274 °C; IR (KBr): υmax 3194, 2919, 2613, 1675, 1606, 1547, 1510, 1443, 1412, 1252, 1100, 1023, 999, 820, 765, 622, 510 cm−1; 1H NMR (DMSO-d6, 300 MHz): δ 12.13 (bs, 2H, 2 x OH) (disappears on D2O exchange), 10.14 (bs, 2H, 2 x NH) (disappears on D2O exchange), 7.65 (s, 2H, 2 x CHNOH), 7.59 (d, J = 8.5 Hz, 4H, ArH), 7.16 (d, J = 8.7 Hz, 4H, ArH), 3.85 (s, 2H, ArCH2Ar); 13C NMR (DMSO-d6, 75.6 MHz): δ 160.4 (CO), 144.4 (CHNOH), 137.2 (ArC), 136.8 (ArC), 129.2 (ArC), 120.3 (ArC), 40.2 (ArCH2Ar); HRMS (+ESI) m/z [M + Na]+ calcd for C17H16N4NaO4: 363.1069; found: 363.1055.
N,N’-[4,4’-bis(4,1-phenylene)]bis [2-(hydroxyimino)acetamide]oxide (4). This compound was prepared by the same method as compound 3 from 4,4′-oxydianiline 2 (10 g, 50.0 mmol) as orange needles (11.8 g, 69%); mp 221–223 °C; IR (KBr): υmax 3185, 2931, 2622, 1669, 1596, 1521, 1510, 1433, 1410, 1232, 1096, 1021, 1000, 822, 764, 619 cm−1; 1H NMR (DMSO-d6, 300 MHz): δ 12.22 (bs, 2H, 2 x OH) (disappears on D2O exchange), 10.22 (bs, 2H, 2 x NH) (disappears on D2O exchange), 7.69 (d, J = 9.0 Hz, 4H, ArH), 7.67 (s, 2H, 2 x CHNOH), 6.99 (d, J = 9.0 Hz, 4H, ArH); 13C NMR (DMSO-d6, 75.6 MHz): δ 153.2 (ArC), 160.4 (CO), 144.4 (CHNOH), 137.2 (ArC), 121.9 (ArC), 119.1 (ArC); HRMS (+ESI) m/z [M + Na]+ calcd for C16H14N4NaO5: 365.0862; found: 365.0851.
5,5’-Methylenediindoline-2,3-dione (5). Compound 3 (5 g, 14.7 mmol) in small portions at 60 °C was added to concentrated sulfuric acid (20 mL). The deep red reaction mixture was stirred at 60 °C for a further 30 min. Chilled water (100 mL) was added to quench the reaction. The title compound was collected by filtration as a red solid (3.96 g, 88%); mp > 300 °C; IR (KBr): υmax 3450, 3109, 1622, 1472, 1271, 1200, 1141, 906, 833, 745, 731, 711, 660, 622 cm−1; 1H NMR (DMSO-d6, 300 MHz): δ 10.99 (bs, 2H, 2 x NH) (disappears on D2O exchange), 7.51 (dd, J = 1.8, 8.1 Hz, 2H, ArH), 7.42 (s, 2H, ArH), 6.86 (d, J = 8.2 Hz, 2H, ArH), 3.89 (s, 2H, ArCH2Ar); 13C NMR (DMSO-d6, 75.6 MHz): δ 184.8 (COCONH), 159.8 (COCONH), 149.4 (ArC), 138.9 (ArC), 136.2 (ArC), 124.9 (ArC), 118.3 (ArC), 112.7 (ArC), 39.3 (ArCH2Ar); HRMS (+ESI) m/z [M + Na]+ calcd for C17H10N2NaO4: 329.0538; found: 329.0541.
5,5’-Oxydiindoline-2,3-dione (6). This compound was prepared by the same method as compound 8 from compound 4 (5.0 g, 14.6 mmol) as a red solid (4.02 g, 83%); mp > 300 °C; IR (KBr): υmax 3457, 3111, 1743, 1621, 1472, 1199, 1145, 907, 839, 744, 712, 660, 623 cm−1; 1H NMR (DMSO-d6, 300 MHz): δ 11.02 (bs, 2H, 2 x NH) (disappears on D2O exchange), 7.30 (dd, J = 2.6, 2H, 8.5 Hz, ArH), 7.09 (s, 2H, ArH), 6.93 (d, J = 8.5 Hz, 2H, ArH); 13C NMR (DMSO-d6, 75.6 MHz): δ 184.4 (COCONH), 159.9 (COCONH), 149.4 (ArC), 147.1 (ArC), 129.0 (ArC), 118.9 (COCONH), 114.7 (ArC), 114.0 (ArC); HRMS (+ESI) m/z [M + Na]+ calcd for C16H8N2NaO4: 331.0331; found: 331.0318.
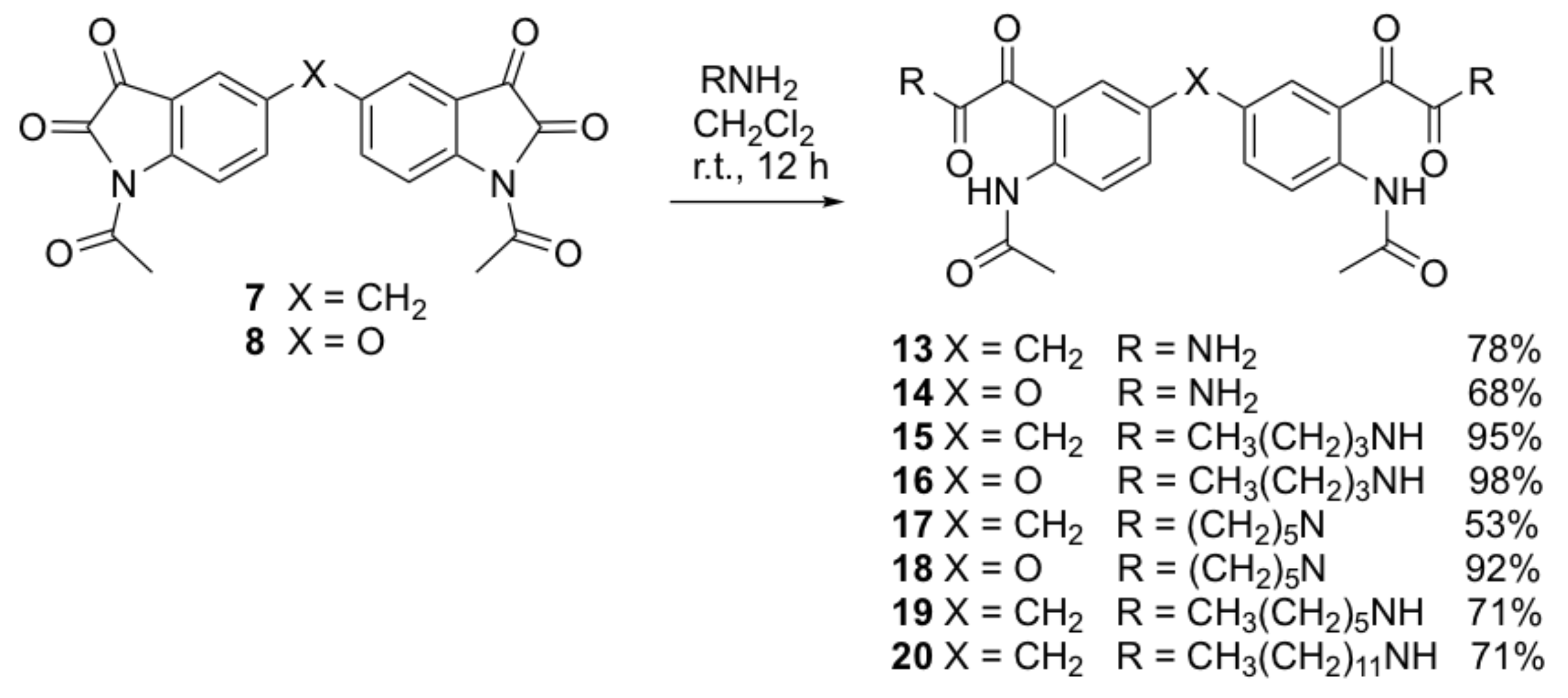
Bis(1-acetylindoline-2,3-dione)methylene (7). A suspension of compound 5 (2 g, 6.5 mmol) in acetic anhydride (30 mL) was heated to reflux for 4 h. The deep red solution was cooled to room temperature. The excess acetic anhydride was removed under vacuum. The resulting brown oily residue was redissolved in ethyl acetate (20 mL) and washed with water (2 × 20 mL) and brine (2 × 20 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was purified by flash column chromatography (dichloromethane) to yield the title compound as a bright yellow solid (1.63 g, 64%); mp 289–291 °C; IR (KBr): υmax 1785, 1712, 1756, 1617, 1588, 1481, 1443, 1371, 1346, 1306, 1294, 1250, 1227, 1201, 1168, 1124, 1042, 996, 659, 598 cm−1; 1H NMR (CDCl3, 300 MHz): δ 8.22 (d, J = 8.5 Hz, 2H, ArH), 7.73 (dd, J = 1.9, 8.5 Hz, 2H, ArH), 7.69 (s, 2H, ArH), 4.11 (s, 2H, ArCH2Ar), 2.59 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 180.5 (COCONH), 170.0 (COCH3), 158.7 (COCONH), 146.8 (ArC), 138.3 (ArC), 124.5 (ArC), 124.5 (ArC), 120.5 (ArC), 117.8 (ArC), 39.2 (ArCH2Ar), 26.3 (COCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C21H14N2NaO6: 413.0750; found: 413.0713.
5,5’-Bis(1-acetylindoline-2,3-dione)oxide (8). This compound was prepared by the same method as compound 7 from compound 6 (2.0 g, 6.5 mmol) as a bright yellow solid (1.40 g, 55%); mp > 300 °C; IR (KBr): υmax 3429, 1783, 1747, 1702, 1617, 1470, 1371, 1330, 1312, 1293, 1266, 1241, 1156, 850, 599, 468 cm−1; 1H NMR (CDCl3, 300 MHz): δ 8.31 (d, J = 9.2 Hz, 2H, ArH), 7.51 (dd, J = 2.9, 8.9 Hz, 2H, ArH), 7.31 (s, 2H, ArH), 2.57 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 179.9 (COCONH), 170.0 (COCH3), 158.6 (COCONH), 154.1 (ArC), 144.4 (ArC), 128.3 (ArC), 121.8 (ArC), 119.7 (ArC), 113.9 (ArC), 26.2 (COCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C20H12N2NaO7: 415.0542; found: 415.0526.
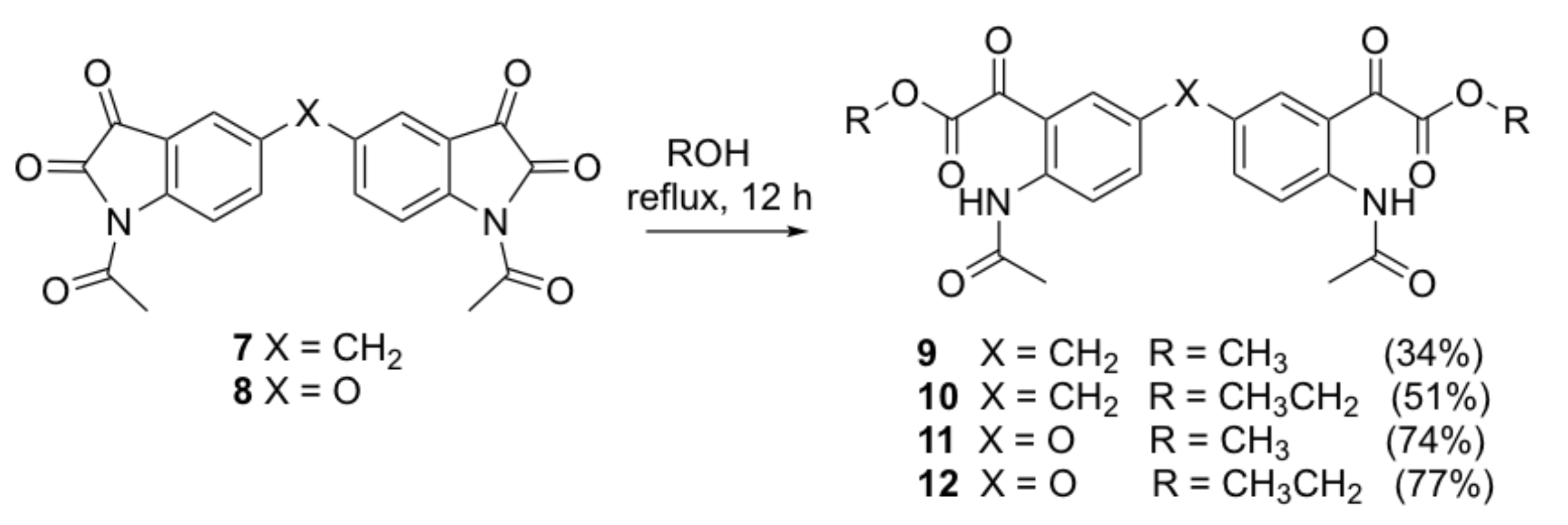
Dimethyl 2,2’-[5,5’-bis(2-acetamido-5,1-phenylene)]bis(2-oxoacetate)methylene (9). A solution of compound 7 (0.15 g, 0.38 mmol) in anhydrous methanol (20 mL) was stirred at room temperature for 24 h. A massive yellow precipitate formed. The crude compound was collected by filtration and recrystallized from n-hexane/DCM to afford the title compound as a yellow solid (0.059 g, 34%); mp 162–164 °C; IR (KBr): υmax 3303, 1747, 1733, 1702, 1652, 1595, 1528, 1420, 1340, 1295, 1250, 1226, 1160, 1013 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.91 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.66 (d, J = 8.7 Hz, 2H, ArH), 7.37 (s, 2H, ArH), 7.35 (d, J = 2.1 Hz, 2H, ArH), 3.90 (s, 2H, ArCH2Ar), 3.89 (s, 6H, 2 x COOCH3), 2.18 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 190.4 (COCH3), 169.8 (COCOO), 164.1 (COCOO), 141.7 (ArC), 138.0 (ArC), 134.5 (ArC), 133.6 (ArC), 121.6 (ArC), 117.6 (ArC), 53.4 (COOCH3), 40.2 (ArCH2Ar), 25.9 (COCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C23H22N2NaO8: 477.1274; found: 477.1243.
Diethyl 2,2’-[5,5’-bis(2-acetamido-5,1-phenylene)]bis(2-oxoacetate)methylene (10). A solution of compound 7 (0.15 g, 0.38 mmol) in absolute ethanol (20 mL) was heated to reflux for 12 h. The reaction mixture was allowed to cool to room temperature and a massive yellow precipitate formed. The crude compound was collected by filtration and recrystallized from n-hexane/DCM to afford the title compound as a yellow solid (0.095 g, 51%); mp 162–164 °C; IR (KBr): υmax 3317, 1749, 1738, 1705, 1648, 1617, 1590, 1493, 1399, 1367, 1295, 1243, 1211, 1089, 1007 cm−1; 1H NMR (CDCl3, 300 MHz): δ 11.02 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.73 (d, J = 9.0 Hz, 2H, ArH), 7.44 (s, 2H, ArH), 7.42 (dd, J = 2.2, 9.0 Hz, 2H, ArH), 4.41 (q, J = 7.2 Hz, 4H, 2 x CH2CH3), 3.97 (s, 2H, ArCH2Ar), 2.24 (s, 6H, 2 x COCH3), 1.35 (t, J = 7.1 Hz, 6H, 2 x CH2CH3); 13C NMR (CDCl3, 75.6 MHz): δ 190.2 (COCH3), 169.3 (COCOO), 163.3 (COCOO), 141.2 (ArC), 137.4 (ArC), 134.0 (ArC), 133.1 (ArC), 121.1 (ArC), 117.1 (ArC), 62.6 (CH2CH3), 39.7 (ArCH2Ar), 25.4 (OCH3), 13.9 (CH2CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C25H26N2NaO8: 505.1587; found: 505.1572.
Dimethyl 2,2’-[5,5’-bis(2-acetamido-5,1-phenylene)]bis(2-oxoacetate)oxide (11). Compound 11 was prepared by the same method as compound 9 from compound 8 (0.15 g, 0.38 mmol) as a white solid (0.129 g, 74%); mp 159–161 °C; IR (KBr): υmax 3456, 1750, 1737, 1704, 1655, 1587, 1520, 1488, 1410, 1286, 1257, 1237, 1217, 1154, 1016, 970, 783 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.82 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.72 (d, J = 9.1 Hz, 2H, ArH), 7.32 (d, J = 2.7 Hz, 2H, ArH), 7.22 (dd, J = 2.9, 9.2 Hz, 2H, ArH), 3.89 (s, 6H, 2 x COOCH3), 2.19 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 189.6 (COCH3), 169.7 (COCOO), 163.7 (COCOO), 151.3 (ArC), 139.2 (ArC), 127.7 (ArC), 123.2 (ArC), 122.9 (ArC), 118.6 (ArC), 53.6 (COOCH3), 25.9 (COCH3); HRMS (+ESI) m/z [M + H]+ calcd for C22H21N2O9: 457.1247; found: 457.1230.
Diethyl 2,2’-[5,5’-bis(2-acetamido-5,1-phenylene)]bis(2-oxoacetate)oxide (12). Compound 12 was prepared by the same method as compound 10 from compound 8 (0.15 g, 0.38 mmol) as a yellow solid (0.143 g, 77%); mp 104–106 °C; IR (KBr): υmax 3307, 2965, 1745, 1655, 1589, 1520, 1437, 1411, 1372, 1267, 1217, 1282, 1160, 1011, 842 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.92 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.79 (d, J = 9.3 Hz, 2H, ArH), 7.37 (d, J = 2.7 Hz, 2H, ArH), 7.35 (dd, J = 2.9, 9.3 Hz, 2H, ArH), 4.41 (q, J = 7.2 Hz, 4H, 2 x CH2CH3), 2.25 (s, 6H, 2 x COCH3), 1.36 (t, J = 7.1 Hz, 6H, 2 x CH2CH3); 13C NMR (CDCl3, 75.6 MHz): δ 189.6 (COCH3), 169.3 (COCOO), 162.9 (COCOO), 151.0 (ArC), 138.8 (ArC), 127.3 (ArC), 122.8 (ArC), 122.4 (ArC), 118.2 (ArC), 62.9 (CH2CH3), 25.4 (OCH3), 14.0 (CH2CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C24H24N2NaO9: 507.1380; found: 507.1360.
N,N’-{4,4’-bis-[2-(2-acetamidophenyl)-2-oxoacetamide]}methylene (13). To a solution of compound 7 (0.15 g, 0.38 mmol) in dichloromethane (20 mL) was added concentrated ammonia solution (10 mL). The reaction mixture was stirred at room temperature for 30 min. A massive white precipitate formed. The crude product was collected by filtration and purified by recrystallization from methanol to yield the title compound 13 as a white solid (0.127 g, 78%); mp 229–232 °C; IR (KBr): υmax 3310, 2933, 1692, 1641, 1588, 1522, 1466, 1411, 1359, 1310, 1279, 1201, 1177, 1132, 987, 900, 826, 735, 591 cm−1; 1H NMR (DMSO-d6, 300 MHz): δ 10.50 (bs, 2H, 2 x NH) (disappears on D2O exchange), 7.79 (s, 4H, 2 x NH2) (disappears on D2O exchange), 8.01 (s, 2H, ArH), 7.50 (d, J = 1.9 Hz, 2H, ArH), 7.44 (dd, J = 2.1, 8.5 Hz, 2H, ArH), 3.99 (s, 2H, ArCH2Ar), 2.04 (s, 6H, 2 x COCH3); 13C NMR (DMSO-d6, 75.6 MHz): δ 169.2 (COCONH2), 192.6 (COCH3), 166.3 (COCONH2), 137.4 (ArC), 136.4 (ArC), 134.8 (ArC), 131.8 (ArC), 124.3 (ArC), 122.3 (ArC), 40.0 (ArCH2Ar), 24.5 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C21H20N4NaO6: 447.1281; found: 447.1252.
N,N’-{4,4’-bis-[2-(2-acetamidophenyl)-2-oxoacetamide]}oxide (14). Compound 14 was prepared by the same method as compound 13 from compound 8 (0.15 g, 0.38 mmol) as a white solid (0.108 g, 66%); mp 183–186 °C; IR (KBr): υmax 3309, 2940, 2859, 1701, 1639, 1588, 1521, 1463, 1416, 1368, 1326, 1285, 1237, 1215, 1181, 1165, 1142, 991, 942, 842, 753, 637 cm−1; 1H NMR (DMSO-d6, 300 MHz): δ 10.36 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.00 (s, 2H, ArH), 7.74 (s, 4H, 2 x NH2) (disappears on D2O exchange), 7.50 (d, J = 1.7 Hz, 2H, ArH), 7.12 (dd, J = 1.7, 8.0 Hz, 2H, ArH), 2.14 (s, 6H, 2 x COCH3); 13C NMR (DMSO-d6, 75.6 MHz): δ 193.4 (COCH3), 169.5 (COCONH2), 166.2 (COCONH2), 137.1 (ArC), 136.3 (ArC), 130.9 (ArC), 128.9 (ArC), 120.5 (ArC), 113.2 (ArC), 26.1 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C20H18N4NaO7: 449.1073; found: 447.1059.
N,N’-{4,4’-bis-[2-(2-acetamidophenyl)-N-butyl-2-oxoacetamide]}methylene (15). A solution of n-butylamine (0.20 mL, 2.0 mmol) in anhydrous dichloromethane (20 mL)was added to a stirred solution of compound 7 (0.15 g, 0.38 mmol) in anhydrous dichloromethane (30 mL). The reaction mixture was stirred at room temperature for 12 h. The organic layer was diluted with dichloromethane (30 mL) and extracted in aqueous hydrochloric acid (0.5 M, 2 × 40 mL) and water (3 x 40 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum. The crude compound was purified by gravity column chromatography (dichloromethane-ethyl acetate/3:1) to afford the title compound 15 as a yellow solid (0.196 g, 95%); mp 144–148 °C; IR (KBr): υmax 3309, 2960, 1681, 1650, 1582, 1507, 1402, 1371, 1320, 1288, 1253, 1222, 1180, 1134, 1006, 833, 700 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.79 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.50 (d, J = 8.7 Hz, 2H, ArH), 8.07 (d, J = 2.0 Hz, 2H, ArH), 7.33 (dd, J = 2.0, 8.6 Hz, 2H, ArH), 6.81 (bs, 2H, 2 x NH butylamine) (disappears on D2O exchange), 3.91 (s, 2H, ArCH2Ar), 3.31 (dd, J = 6.8, 13.2 Hz, 4H, 2 x CH2CH2), 2.14 (s, 6H, 2 x COCH3), 1.47–1.52 (m, 4H, 2 x CH2CH2CH2CH3), 1.28–1.35 (m, 4H, 2 x CH2CH3), 0.87 (t, J = 7.6 Hz, 6H, 2 x CH3); 13C NMR (CDCl3, 75.6 MHz): δ 192.4 (COCH3), 169.5 (COCONH), 163.2 (COCONH), 140.9 (ArC), 137.4 (ArC), 134.9 (ArC), 134.7 (ArC), 121.6 (ArC), 119.4 (ArC), 40.4 (ArCH2Ar), 39.8 (CH2CH2CH2), 31.7 (CH2CH2CH2), 25.8 (OCH3), 20.4 (CH2CH2CH3), 14.1 (CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C29H36N4NaO6: 559.2533; found: 559.2513.
N,N’-{4,4’-bis-[2-(2-acetamidophenyl)-N-butyl-2-oxoacetamide]}oxide (16). Compound 16 was prepared by the same method as compound 15 from compound 8 (0.15 g, 0.38 mmol) and n-butylamine (0.20 mL, 2.0 mmol) as a yellow solid (0.202 g, 98%); mp 188–191 °C; IR (KBr): υmax 3311, 2960, 1686, 1656, 1589, 1513, 1406, 1370, 1324, 1286, 1264, 1222, 1181, 1162, 1008, 847, 699 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.75 (bs, 2H, 2 x NH) (disappears on D2O exchange), 7.99 (d, J = 3.0 Hz, 2H, ArH), 8.58 (d, J = 9.3 Hz, 2H, ArH), 7.25 (dd, J = 3.4, 9.1 Hz, 2H, ArH), 6.97 (bs, 2H, 2 x NH butylamine) (disappears on D2O exchange), 3.34 (dd, J = 6.9, 13.2 Hz, 4H, 2 x CH2CH2), 2.21 (s, 6H, 2 x COCH3), 1.51–1.56 (m, 4H, 2 x CH2CH2CH2CH3), 1.32–1.39 (m, 4H, 2 x CH2CH3), 0.91 (t, J = 7.2 Hz, 6H, 2 x CH3); 13C NMR (CDCl3, 75.6 MHz): δ 191.3 (COCH3), 169.0 (COCONH), 162.4 (COCONH), 151.2 (ArC), 137.7 (ArC), 126.8 (ArC), 123.4 (ArC), 122.7 (ArC), 120.2 (ArC), 39.4 (CH2CH2CH2), 31.2 (CH2CH2CH2), 25.3 (OCH3), 20.0 (CH2CH2CH3), 13.7 (CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C28H34N4NaO7: 561.2325; found: 561.2300.
N,N’-[4,4’-bis-(N-{2-[2-oxo-2-(piperidin-1-yl)acetyl]phenyl}acetamide)]methylene (17). Compound 17 was prepared by the same method as compound 15 from compound 7 (0.15 g, 0.38 mmol) and piperidine (0.20 mL, 2.0 mmol) as a yellow solid (0.114 g, 53%); mp 181–183 °C; IR (KBr): υmax 2939, 2859, 1701, 1642, 1591, 1520, 1451, 1412, 1367, 1326, 1298, 1251, 1182, 1136, 988, 851, 784, 752 cm−1; 1H NMR (CDCl3, 300 MHz): δ11.15 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.66 (d, J = 8.8 Hz, 2H, ArH), 7.35 (dd, J = 1.8, 8.7 Hz, 2H, ArH), 7.28 (d, J = 1.8 Hz, 2H, ArH), 3.89 (s, 2H, ArCH2Ar), 3.55 (t, J = 5.5 Hz, 4H, 2 x NCH2), 3.13 (t, J = 5.5 Hz, 4H, 2 x NCH2), 2.18 (s, 6H, 2 x COCH3), 1.51–1.57 (m, 8H, 2 x CH2CH2CH2CH2CH2), 1.29–1.38 (m, 4H, 2 x CH2CH2CH2); 13C NMR (CDCl3, 75.6 MHz): δ 196.4 (COCH3), 169.8 (COCON), 164.5 (COCON), 141.3 (ArC), 137.6 (ArC), 135.0 (ArC), 133.6 (ArC), 121.4 (ArC), 118.5 (ArC), 47.5 (NCH2), 42.5 (NCH2), 40.1 (ArCH2Ar), 25.7 (NCH2CH2), 25.9 (OCH3), 25.7 (NCH2CH2), 24.6 (CH2CH2CH2); HRMS (+ESI) m/z [M + Na]+ calcd for C31H36N4NaO6: 583.2533; found: 583.2493.
N,N’-[4,4’-bis-(N-{2-[2-oxo-2-(piperidin-1-yl)acetyl]phenyl}acetamide)]oxide (18). Compound 18 was prepared by the same method as compound 15 from compound 8 (0.15 g, 0.38 mmol) and piperidine (0.20 mL, 2.0 mmol) as a yellow solid (0.198 g, 92%); mp 158–160 °C; IR (KBr): υmax 3444, 3288, 1682, 1557, 1489, 1415, 1371, 1335, 1303, 1289, 1253, 1126, 929, 839, 669, 617, 559 cm−1; 1H NMR (CDCl3, 300 MHz): δ 11.08 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.75 (d, J = 8.8 Hz, 2H, ArH), 7.23 (dd, J = 2.8, 9.1 Hz, 2H, ArH), 7.20 (d, J = 1.9 Hz, 2H, ArH), 3.58 (t, J = 5.3 Hz, 4H, 2 x NCH2), 3.23 (t, J = 5.3 Hz, 4H, 2 x NCH2), 2.22 (s, 6H, 2 x COCH3), 1.63–1.65 (m, 4H, 2 x CH2CH2CH2), 1.51–1.57 (m, 8H, 2 x CH2CH2CH2CH2CH2); 13C NMR (CDCl3, 75.6 MHz): δ 195.2 (COCH3), 169.3 (COCON), 163.8 (COCON), 151.3 (ArC), 138.4 (ArC), 127.0 (ArC), 122.6 (ArC), 122.4 (ArC), 119.1 (ArC), 47.1 (NCH2), 42.2 (NCH2), 26.1 (NCH2CH2), 25.4 (OCH3), 25.3 (NCH2CH2), 24.2 (CH2CH2CH2); HRMS (+ESI) m/z [M + Na]+ calcd for C30H34N4NaO7: 585.2325; found: 585.2293.
2,2′-[Methylene-bis(2-acetamido-5,1-phenylene)]bis(N-hexyl-2-oxoacetamide) (19). A mixture of compound 7 (0.24 g, 0.61 mmol) and hexylamine (0.12 g, 1.22 mmol) in dichloromethane (30 mL) was heated at reflux overnight. After cooling to room temperature, the solvent was evaporated in vacuo and the crude product was purified by column chromatography using silica gel and n-hexane/DCM as eluent. The title compound 19 was obtained as an off-white solid (0.26 g, 71%); mp 182–184 °C; IR (KBr): υmax 3305, 3053, 2928, 2855, 1694, 1650, 1585, 1518, 1465, 1411, 1369, 1293, 1223, 1176, 1006, 855, 811 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.89 (s, 2H, 2 x NHCO), 8.56 (d, J = 8.6 Hz, 2H, ArH), 8.16 (d, J = 2.0 Hz, 2H, ArH), 7.42 (dd, J = 8.7, 1.9 Hz, 2H, ArH), 6.98 (t, J = 5.5 Hz, 2H, 2 x CONH), 4.00 (s, 2H, ArCH2Ar), 3.47-3.49 (q, J = 6.7, 6.3 Hz, 4H, 2 x NHCH2CH2(CH2)3CH3), 2.25 (s, 6H, 2 x COCH3), 1.55–1.66 (m, 4H, 2 x NHCH2CH2(CH2)3CH3), 1.30–1.44 (m, 12H, 2 x NHCH2CH2(CH2)3CH3), 0.93 (t, J = 6.6 Hz, 6H, 2 x NHCH2(CH2)4CH3); 13C NMR (CDCl3, 75.6 MHz): δ 162.8, 169.1, 192.0 (6 x C=O), 121.2, 134.5, 136.9 (6 x ArCH), 119.0, 134.3, 140.5 (6 x ArC), 40.0 (2 x NHCH2), 39.7 (ArCH2Ar), 25.4 (2 x COCH3), 31.4 (2 x CH2(CH2)4CH3), 22.5, 26.6, 29.2, 14.0 (2 x CH2(CH2)4CH3); HRMS (ESI) m/z [M + H]+ calcd for C33H45N4O6: 593.3333; found: 593.3326.
2,2′-[Methylene-bis(2-acetamido-5,1-phenylene)]bis(N-dodecyl-2-oxoacetamide) (20). Compound 20 was prepared by the same method as compound 19 from compound 7 (0.24 g, 0.61 mmol) and dodecylamine (0.23 g, 1.22 mmol) as an off-white solid (0.26 g, 69%); mp 162–164 °C; IR (KBr): υmax 3344, 3293, 2919, 2850, 1694, 1650, 1586, 1519, 1467, 1412, 1370, 1294, 1224, 1177, 1014, 855, 721 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.89 (s, 2H, 2 x NHCO), 8.54 (d, J = 8.5 Hz, 2H, ArH), 8.17 (d, J = 2.1 Hz, 2H, ArH), 7.45 (dd, J = 8.6, 2.12 Hz, 2H, ArH), 6.98 (t, J = 5.9 Hz, 2H, 2 x CONH), 4.03 (s, 2H, ArCH2Ar), 3.37–3.44 (q, J = 6.7, 6.8 Hz, 4H, 2 x NHCH2CH2(CH2)9CH3), 2.28 (s, 6H, 2 x COCH3), 1.56-1.66 (m, 4H, 2 x NHCH2CH2(CH2)9CH3), 1.26–1.40 (m, 36H, 2 x CH2CH2(CH2)9CH3), 0.93 (t, J = 7.0 Hz, 6H, 2 x CH2(CH2)9CH3); 13C NMR (CDCl3, 75.6 MHz): δ 162.8, 169.9, 191.7 (6 x C = O), 121.6, 134.9, 136.9 (6 x ArCH), 120.6, 134.2, 139.9 (6 x ArC), 40.0 (2 x NHCH2), 39.6 (ArCH2Ar), 25.2 (2 x COCH3), 22.7, 26.9, 29.2, 29.2, 29.4, 29.5, 29.6, 29.6, 31.3 (2 x CH2(CH2)10CH3), 14.1 (2 x CH2(CH2)10CH3); HRMS (ESI) m/z [M + Na]+calcd for C45H68N4NaO6: 761.5211; found: 761.5207.
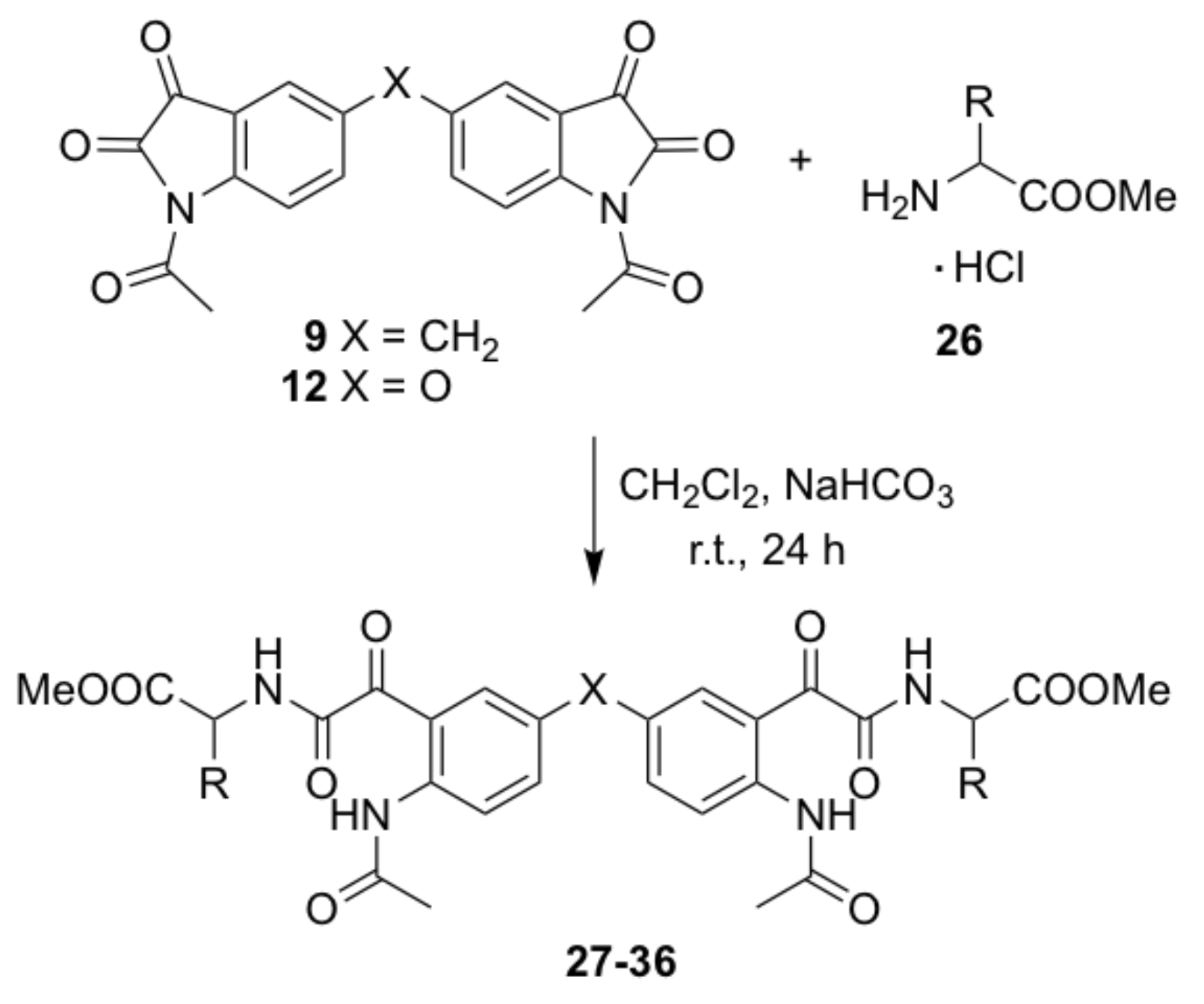
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]acetate}) methane (21). A mixture of the glycine methyl ester hydrochloride (0.24 g, 1.9 mmol) and saturated sodium hydrogen carbonate solution (3 mL) in water (7 mL) was added to a stirred solution of compound 7 (0.15 g, 0.38 mmol)) in dichloromethane (20 mL) was. The reaction mixture was stirred at room temperature for 24 h. The organic layer was diluted with dichloromethane (20 mL) and extracted in aqueous hydrochloric acid (0.5 M, 30 mL) and water (2 × 30 mL). The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under vacuum. The crude compound was purified by gravity column chromatography (dichloromethane-ethyl acetate = 4:1) to afford the title compound as a yellow solid (0.120 g, 55%); mp 204–206 °C; IR (KBr): υmax 3279, 1750, 1698, 1666, 1651, 1592, 1519, 1412, 1367, 1326, 1293, 1234, 1217, 1177, 1005, 688 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.49 (bs, 2H, 2 x NH) (disappears on D2O exchange), 9.07 (d, J = 11.2 Hz, 2H, ArH), 7.74 (s, 2H, ArH), 7.60 (d, J = 1.5 Hz, 2H, 2 x α-NH Gly) (disappears on D2O exchange), 7.49 (dd, J = 2.0, 8.5 Hz, 2H, ArH), 3.97 (d, J = 5.7 Hz, 4H, 2 x NHCH2COOCH3), 3.91 (s, 2H, ArCH2Ar), 3.67 (s, 6H, 2 x COOCH3), 2.05 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 189.8 (COCH3), 169.5 (COCONH), 168.9 (COCONH), 163.2 (COOCH3), 149.8 (ArC), 138.6 (ArC), 127.1 (ArC), 123.5 (ArC), 122.6 (ArC), 119.6 (ArC), 53.1 (COOCH3), 40.9 (NHCH2COOCH3), 38.9 (ArCH2Ar), 24.9 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C27H28N4NaO10: 591.1703; found: 591.1692.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-4-methylpentanoate})methane (22). Compound 22 was prepared by the same method as compound 21 from compound 7 (0.15 g, 0.38 mmol) and l-leucine methyl ester hydrochloride (0.30 g, 1.9 mmol) as a yellow solid (0.149 g, 57%); mp 184–186 °C; IR (KBr): υmax 3328, 2958, 1747, 1648, 1592, 1522, 1438, 1415, 1370, 1298, 1253, 1231, 1176 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.84 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.53 (d, J = 8.8 Hz, 2H, ArH), 8.01 (d, J = 1.9 Hz, 2H, 2 x α-NH Leu) (disappears on D2O exchange), 7.35 (dd, J = 2.1, 8.7 Hz, 2H, ArH), 7.25 (d, J = 8.4 Hz, 2H, ArH), 4.57–4.64 (m, 2H, 2 x NHCHCOOCH3), 3.91 (s, 2H, ArCH2Ar), 3.66 (s, 6H, 2 x COOCH3), 2.14 (s, 6H, 2 x COCH3), 1.56-1.69 (m, 4H, 2 x CH2CH(CH3)2), 1.18–1.20 (m, 2H, 2 x CH(CH3)2), 0.89 (dd, J = 2.6, 5.9 Hz, 12H, 2 x CH(CH3)2); 13C NMR (CDCl3, 75.6 MHz): δ 191.9 (COCH3), 172.9 (COCONH), 169.5 (COCONH), 163.3 (COOCH3), 141.1 (ArC), 137.7 (ArC), 134.8 (ArC), 134.6 (ArC), 121.4 (ArC), 119.0 (ArC), 52.9 (COOCH3), 51.3 (NHCHCOOCH3), 41.7 (CH2CH(CH3)2), 40.3 (ArCH2Ar), 25.8 (OCH3), 25.3 (CH(CH3)2), 23.0 (CH3), 22.2 (CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C35H44N4NaO10: 703.2955; found: 703.2948.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-4-(methylthio)butanoate})methane (23). Compound 23 was prepared by the same method as compound 21 from compound 7 (0.15 g, 0.38 mmol) and l-methionine methyl ester hydrochloride (0.38 g, 1.9 mmol) as a yellow solid (0.140 g, 51%); mp 172–174 °C; IR (KBr): υmax 3292, 1746, 1695, 1663, 1642, 1589, 1523, 1433, 1412, 1294, 1250, 1219, 1173 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.80 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.52 (d, J = 8.5 Hz, 2H, ArH), 8.01 (d, J = 1.9 Hz, 2H, 2 x α-NH Met) (disappears on D2O exchange), 7.49 (d, J = 8.1Hz, 2H, ArH), 7.35 (dd, J = 2.0, 8.7 Hz, 2H, ArH), 4.68–4.77 (m, 2H, 2 x NHCHCOOCH3), 3.91 (s, 2H, ArCH2Ar), 3.70 (s, 6H, 2 x COOCH3), 2.47 (t, J = 7.2 Hz, 4H, 2 x CH2CH2S), 2.15 (s, 6H, 2 x COCH3), 2.03 (s, 6H, 2 x SCH3), 1.05 (t, J = 7.2 Hz, 4H, 2 x CH2CH2S); 13C NMR (CDCl3, 75.6 MHz): δ 191.6 (COCH3), 171.9 (COCONH), 169.6 (COCONH), 163.3 (COOCH3), 141.1 (ArC), 137.7 (ArC), 134.8 (ArC), 134.5 (ArC), 121.5 (ArC), 118.9 (ArC), 53.2 (COOCH3), 52.0 (NHCHCOOCH3), 40.3 (ArCH2Ar), 31.6 (CH2CH2S), 30.3 (CH2CH2S), 25.8 (OCH3), 15.9 (SCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C33H40N4O10NaS2: 739.2084; found: 739.2071.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-3-methylbutanoate})methane (24). Compound 24 was prepared by the same method as compound 21 from compound 7 (0.15 g, 0.38 mmol) and l-valine methyl ester hydrochloride (0.32 g, 1.9 mmol) as a yellow solid (0.201 g, 80%); mp 204–207 °C; IR (KBr): υmax 3251, 2962, 1740, 1696, 1666, 1644, 1588, 1521, 1469, 1438, 1413, 1369, 1295, 1245, 1225, 1211, 1176, 1141 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.87 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.51 (d, J = 8.6 Hz, 2H, ArH), 7.93 (d, J = 1.9 Hz, 2H, 2 x α-NH Val) (disappears on D2O exchange), 7.39 (d, J = 8.8 Hz, 2H, ArH′), 7.32 (dd, J = 2.0, 8.8 Hz, 2H, ArH), 4.54 (dd, J = 4.9, 9.0 Hz, 2H, 2 x NHCHCOOCH3), 3.89 (s, 2H, ArCH2Ar), 3.67 (s, 6H, 2 x COOCH3), 2.18–2.22 (m, 2H, 2 x CH(CH3)2), 2.14 (s, 6H, 2 x COCH3), 0.89 (dd, J = 6.9, 12.7 Hz, 12H, 2 x CH(CH3)2); 13C NMR (CDCl3, 75.6 MHz): δ 192.3 (COCH3), 172.1 (COCONH), 169.6 (COCONH), 163.7 (COOCH3), 141.1 (ArC), 137.7 (ArC), 134.7 (ArC), 134.5 (ArC), 121.4 (ArC), 118.9 (ArC), 57.6 (NHCHCOOCH3), 52.8 (COOCH3), 40.3 (ArCH2Ar), 31.8 (CH(CH3)2), 25.8 (OCH3), 19.4 (2 x CH3), 18.0 (CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C33H40N4NaO10: 675.2642; found: 675.2616.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-3-phenylpropanoate})methane (25). Compound 25 was prepared by the same method as compound 21 from compound 7 (0.15 g, 0.38 mmol) and d-phenylalanine methyl ester hydrochloride (0.41 g, 1.9 mmol) as a yellow solid (0.081 g, 28%); mp 200–202 °C; (c 0.1 in MeOH); IR (KBr): υmax 3294, 1749, 1693, 1644, 1590, 1521, 1412, 1293, 1268, 1217, 1173, 701 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.79 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.50 (d, J = 9.0 Hz, 2H, ArH), 7.88 (s, 2H, 2 x α-NH Phe) (disappears on D2O exchange), 7.16–7.29 (d, 12H, ArH, ArH), 7.05 (d, J = 6.4 Hz, 2H, ArH), 4.86 (dd, J = 6.6, 14.8 Hz, 2H, 2 x NHCHCOOCH3), 3.84 (s, 2H, ArCH2Ar), 3.66 (s, 6H, 2 x COOCH3), 2.99-3.17 (m, 4H, 2 x CH2Ph), 2.11 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 191.7 (COCH3), 171.5 (COCONH), 169.6 (COCONH), 163.0 (COOCH3), 141.1 (ArC), 137.6 (ArC), 135.7 (ArC), 134.7 (ArC), 134.5 (ArC), 129.6 (ArC), 129.1 (ArC), 127.8 (ArC), 121.4 (ArC), 118.8 (ArC), 53.7 (NHCHCOOCH3), 53.0 (COOCH3), 40.3 (ArCH2Ar), 38.3 (CH2Ph), 25.8 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C41H40N4NaO10: 771.2642; found: 771.2610.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-3-phenylpropanoate})methane (26). Compound 26 was prepared from compound 7 (1.00 g, 2.56 mmol) and l-phenylalanine methyl ester hydrochloride (1.21 g, 5.64 mmol) in the presence of Et3N (1.04g, 10.2 mmol) as a yellow solid (0.78 mg, 48%); mp 201–202 °C; (c 0.1 in MeOH); IR υmax 3291, 1742, 1690, 1642, 1594, 1523, 1418, 1289, 1272, 1212, 1171, 705 cm−1. 1H NMR (CDCl3, 400 MHz): δ 10.80 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.51 (d, J = 9.0 Hz, 2H, ArH), 7.89 (s, 2H, 2 x α-NH Phe) (disappears on D2O exchange), 7.14-7.30 (d, 12H, ArH, ArH), 7.06 (d, J = 6.4 Hz, 2H, ArH), 4.88 (dd, J = 6.6, 14.8 Hz, 2H, 2 x NHCHCOOCH3), 3.85 (s, 2H, ArCH2Ar), 3.67 (s, 6H, 2 x COOCH3), 2.98–3.18 (m, 4H, 2 x CH2Ph), 2.12 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 101 MHz): δ 191.8 (COCH3), 172.1 (COCONH), 169.6 (COCONH), 163.0 (COOCH3), 141.1 (ArC), 137.6 (ArC), 135.7 (ArC), 134.7 (ArC), 135.6 (ArC), 129.9 (ArC), 129.2 (ArC), 128.1 (ArC), 121.2 (ArC), 119.2 (ArC), 54.1 (NHCHCOOCH3), 53.1 (COOCH3), 40.2 (ArCH2Ar), 38.4 (CH2Ph), 25.9 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C41H40N4NaO10: 771.2642; found: 771.2612.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]acetate})oxide (27). Compound 27 was prepared by the same method as compound 21 from compound 8 (0.15 g, 0.38 mmol) and glycine methyl ester hydrochloride (0.24 g, 1.9 mmol) as a yellow solid (0.061 g, 28%); mp 180–182 °C; IR (KBr): υmax 3354, 3293, 2923, 1747, 1686, 1658, 1588, 1512, 1438, 1408, 1373, 1327, 1286, 1262, 1219, 1183, 1011 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.69 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.60 (d, J = 9.3 Hz, 2H, ArH), 7.93 (d, J = 3.0 Hz, 2H, 2 x α-NH Gly) (disappears on D2O exchange), 7.36 (t, J = 5.3 Hz, 2H, ArH), 7.49 (dd, J = 3.0, 9.2 Hz, 2H, ArH), 4.05 (d, J = 5.5 Hz, 4H, 2 x NHCH2COOCH3), 3.68 (s, 6H, 2 x COOCH3), 2.17 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 190.9 (COCH3), 169.8 (COCONH), 169.5 (COCONH), 163.2 (COOCH3), 151.6 (ArC), 138.5 (ArC), 127.8 (ArC), 123.5 (ArC), 123.1 (ArC), 119.9 (ArC), 53.1 (COOCH3), 41.5 (NHCH2COOCH3), 25.8 (OCH3). HRMS (+ESI) m/z [M + Na]+ calcd for C26H26N4NaO11: 593.1496; found: 593.1472.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-4-methylpentanoate})oxide (28). Compound 28 was prepared by the same method as compound 21 from compound 8 (0.15 g, 0.38 mmol) and l-leucine methyl ester hydrochloride (0.30 g, 1.9 mmol) as a yellow solid (0.068 g, 26%); mp 106–108 °C; IR (KBr): υmax 3313, 2958, 1748, 1655, 1589, 1519, 1438, 1410, 1370, 1263, 1217, 1182, 1162, 1013, 830, 698 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.82 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.64 (d, J = 9.3 Hz, 2H, ArH), 7.83 (d, J = 2.7 Hz, 2H, 2 x α-NH Leu) (disappears on D2O exchange), 7.41 (d, J = 8.5 Hz, 2H, ArH), 7.28 (dd, J = 2.9, 9.2 Hz, 2H, ArH), 4.60–4.67 (m, 2H, 2 x NHCHCOOCH3), 3.67 (s, 6H, 2 x COOCH3), 2.21 (s, 6H, 2 x COCH3), 1.57–1.72 (m, 6H, 2 x CH2CH(CH3)2), 0.93 (d, J = 5.9 Hz, 12H, CH(CH3)2); 13C NMR (CDCl3, 75.6 MHz): δ 191.3 (COCH3), 172.7 (COCONH), 169.0 (COCONH), 162.9 (COOCH3), 151.1 (ArC), 138.0 (ArC), 127.3 (ArC), 122.9 (ArC), 122.6 (ArC), 119.4 (ArC), 52.5 (COOCH3), 50.8 (NHCHCOOCH3), 41.3 (CH2CH(CH3)2), 25.3 (OCH3), 24.9 (CH(CH3)2), 22.8 (CH3), 21.8 (CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C34H42N4NaO11: 705.2748; found: 705.2720.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-4-(methylthio)butanoate})oxide (29). Compound 29 was prepared by the same method as compound 21 from compound 8 (0.15 g, 0.38 mmol) and l-methionine methyl ester hydrochloride (0.38 g, 1.9 mmol) as a yellow solid (0.206 g, 75%); mp 102–104 °C; IR (KBr): υmax 3274, 1743, 1658, 1588, 1519, 1489, 1410, 1370, 1271, 1218, 1183, 1162 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.77 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.63 (d, J = 9.3 Hz, 2H, ArH), 7.87 (d, J = 2.9 Hz, 2H, 2 x α-NH Met) (disappears on D2O exchange), 7.65 (d, J = 8.1 Hz, 2H, ArH), 7.29 (dd, J = 2.9, 9.2 Hz, 2H, ArH), 4.74 (dd, J = 8.8, 12.9 Hz, 2H, 2 x NHCHCOOCH3), 3.72 (s, 6H, 2 x COOCH3), 2.49–2.56 (m, 4H, 2 x CH2CH2S), 2.21 (s, 6H, 2 x COCH3), 2.07 (s, 6H, 2 x SCH3), 1.03 (t, J = 6.8 Hz, 4H, 2 x CH2CH2S); 13C NMR (CDCl3, 75.6 MHz): δ 190.8 (COCH3), 171.6 (COCONH), 169.1 (COCONH), 162.7 (COOCH3), 151.1 (ArC), 138.0 (ArC), 127.3(ArC), 122.9 (ArC), 122.6 (ArC), 119.5 (ArC), 52.8 (COOCH3), 51.5 (NHCHCOOCH3), 31.1 (CH2CH2S), 29.9 (CH2CH2S), 25.3 (OCH3), 15.5 (SCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C32H38N4O11NaS2: 741.1876; found: 741.1854.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-3-methylbutanoate})oxide (30). Compound 30 was prepared by the same method as compound 21 from compound 8 (0.15 g, 0.38 mmol) and l-valine methyl ester hydrochloride (0.32 g, 1.9 mmol) as a yellow solid (0.115 g, 46%); mp 99–101 °C; IR (KBr): υmax 3215, 2992, 1743, 1701, 1660, 1591, 1518, 1485, 1417, 1370, 1289, 1256, 1215, 1210, 1016, 920, 836, 741 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.80 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.61 (d, J = 9.2 Hz, 2H, ArH), 7.70 (d, J = 2.8 Hz, 2H, 2 x α-NH Val) (disappears on D2O exchange), 7.47 (d, J = 9.1 Hz, 2H, ArH), 7.24 (dd, J = 2.8, 9.2 Hz, 2H, ArH), 4.54 (dd, J = 4.8, 9.1 Hz, 2H, 2 x NHCHCOOCH3), 3.66 (s, 6H, 2 x COOCH3), 2.16 (s, 6H, 2 x COCH3), 1.18–1.20 (m, 2H, 2 x CH(CH3)2), 0.88 (dd, J = 6.9, 15.3 Hz, 12H, 2 x CH(CH3)2); 13C NMR (CDCl3, 75.6 MHz): δ 192.0 (COCH3), 172.4 (COCONH), 169.4 (COCONH), 163.7 (COOCH3), 151.5 (ArC), 138.5 (ArC), 127.8 (ArC), 123.1 (ArC), 123.0 (ArC), 119.7 (ArC), 57.4 (NHCHCOOCH3), 52.8 (COOCH3), 31.8 (CH(CH3)2), 25.7 (OCH3), 18.0 (CH3), 19.4 (CH3); HRMS (+ESI) m/z [M + Na]+ calcd for C33H40N4NaO10: 677.2435; found: 677.2419.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-3-phenylpropanoate})oxide (31). Compound 31 was prepared by the same method as compound 21 from compound 8 (0.15 g, 0.38 mmol) and d-phenylalanine methyl ester hydrochloride (0.41 g, 1.9 mmol) as a yellow solid (0.196 g, 68%); mp 200–202 °C; IR (KBr): υmax 3298, 1745, 1669, 1587, 1518, 1496, 1439, 1410, 1370, 1267, 1218, 1180, 702 cm−1; 1H NMR (CDCl3, 300 MHz): δ 10.78 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.67 (d, J = 9.3 Hz, 2H, ArH), 7.79 (d, J = 2.9 Hz, 2H, 2 x α-NH Phe) (disappears on D2O exchange), 7.22-7.31 (m, 12H, ArH), 7.11 (dd, J = 1.7, 8.0 Hz, 2H, ArH), 4.86–4.93 (m, 2H, 2 x NHCHCOOCH3), 3.70 (s, 6H, 2 x COOCH3), 3.00–3.19 (m, 4H, 2 x CH2Ph), 2.21 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 75.6 MHz): δ 190.9 (COCH3), 171.3 (COCONH), 169.0 (COCONH), 162.3 (COOCH3), 151.1 (ArC), 138.1 (ArC), 135.2 (ArC), 129.2 (ArC), 128.8 (ArC), 127.4 (ArC), 127.3 (ArC), 123.0 (ArC), 122.6 (ArC), 119.3 (ArC), 53.2 (NHCHCOOCH3), 52.6 (COOCH3), 37.9 (2 x CH2Ph), 25.4 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C40H38N4NaO11: 773.2435; found: 773.2419.
N,N’-(4,4’-bis-{methyl-2-[2-(2-acetamidophenyl)-2-oxoacetamido]-3-phenylpropanoate})oxide (32). Compound 32 was prepared by the same method as compound 21 from compound 8 (1.00 g, 2.55 mmol), l-phenylalanine methyl ester hydrochloride (1.21 g, 5.61 mmol) and Et3N (1.03 g, 10.20 mmol) as a yellow solid (1.50 g, 78%); mp 201–202 °C; [α]D -12 (c 0.1 in MeOH); IR: υmax 3292, 1743, 1671, 1582, 1520, 1494, 1440, 1411, 1371, 1265, 1211, 1178, 701 cm−1; 1H NMR (CDCl3, 400 MHz): δ 10.74 (bs, 2H, 2 x NH) (disappears on D2O exchange), 8.65 (d, J = 9.2 Hz, 2H, ArH), 7.80 (d, J = 3.0 Hz, 2H, 2 x α-NH Phe) (disappears on D2O exchange), 7.21–7.34 (m, 12H, ArH), 7.10 (dd, J = 1.8, 8.0 Hz, 2H, ArH), 4.87–4.91 (m, 2H, 2 x NHCHCOOCH3), 3.71 (s, 6H, 2 x COOCH3), 3.00–3.20 (m, 4H, 2 x CH2Ph), 2.22 (s, 6H, 2 x COCH3); 13C NMR (CDCl3, 101 MHz): δ 190.8 (COCH3), 171.2 (COCONH), 168.5 (COCONH), 162.0 (COOCH3), 151.2 (ArC), 137.8 (ArC), 135.1 (ArC), 129.1 (ArC), 128.7 (ArC), 127.3 (ArC), 127.1 (ArC), 123.0 (ArC), 122.5 (ArC), 119.0 (ArC), 53.1 (NHCHCOOCH3), 52.5 (COOCH3), 37.7 (2 x CH2Ph), 25.3 (OCH3); HRMS (+ESI) m/z [M + Na]+ calcd for C40H38N4NaO11: 773.2435; found: 773.2428.
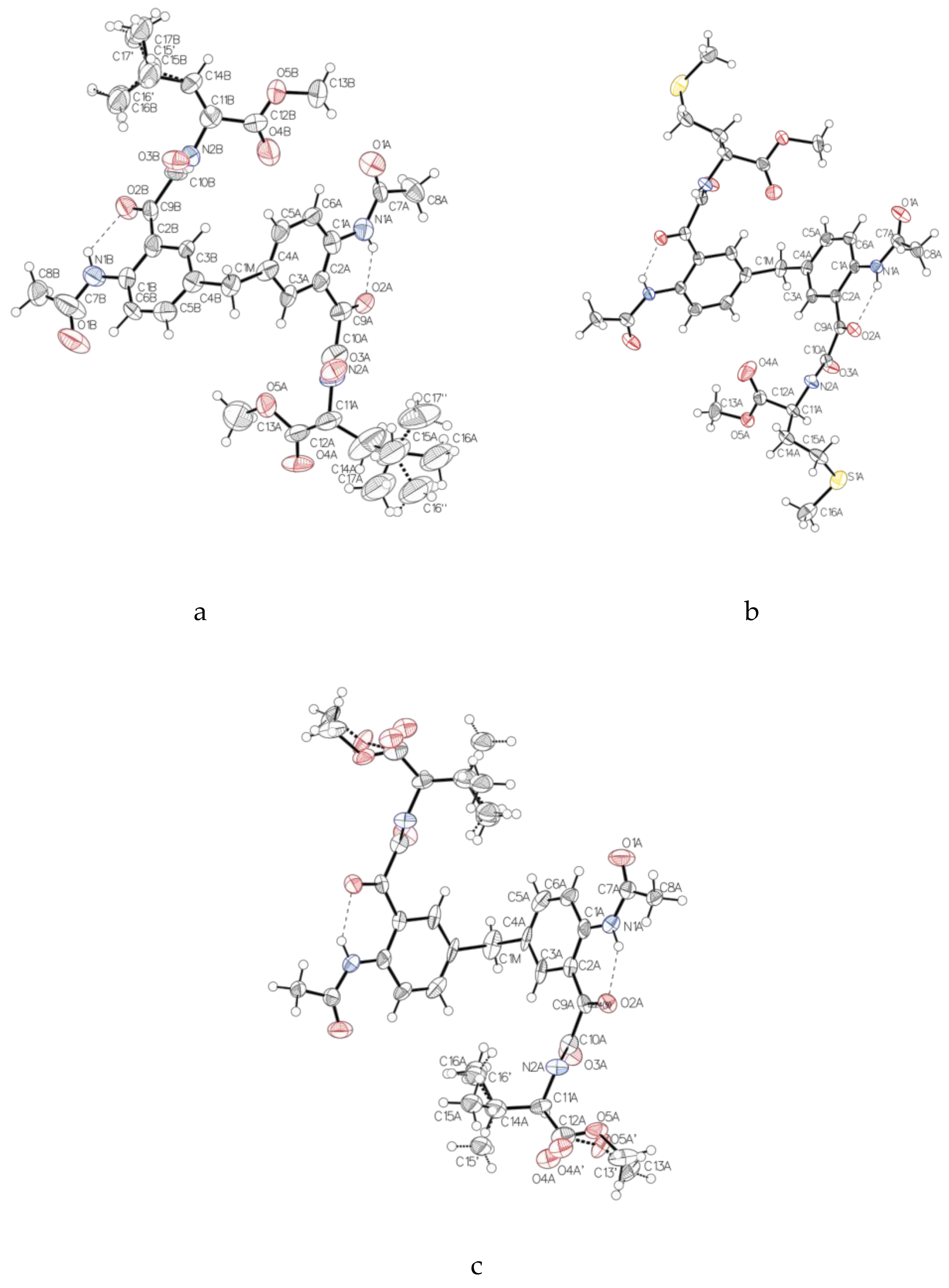
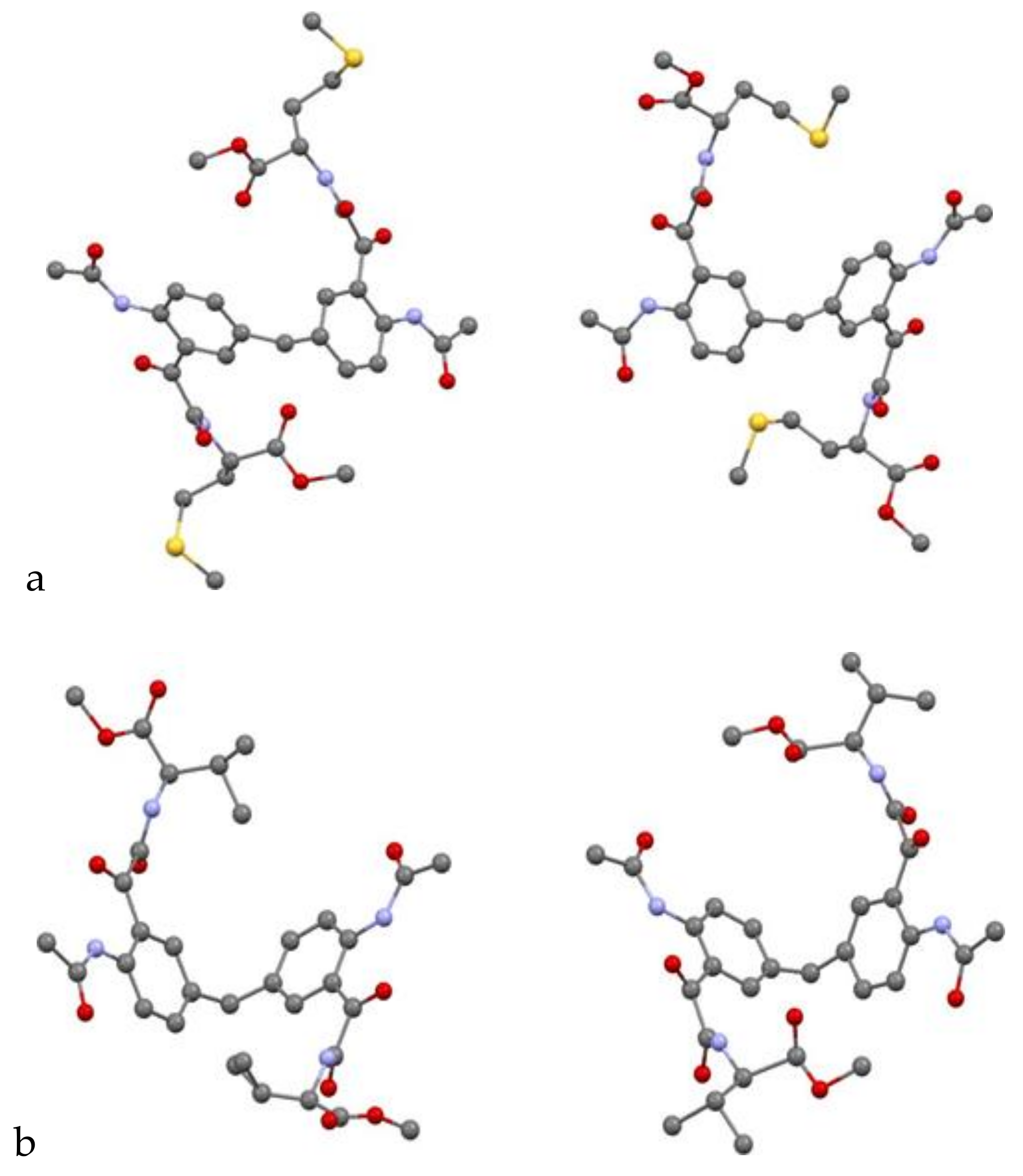
Single-Crystal X-Ray Diffraction
The X-ray diffraction measurements for compounds
22–
24 were carried out at MX1 and MX2 beamlines at the Australian Synchrotron Facility, Melbourne. The procedure for diffraction intensity measurements on both beamlines was similar. The crystal was mounted on the goniometer using a cryo loop for diffraction measurements, and it was coated with paraffin oil and then quickly transferred to the cold stream using cryo stream attachment. Data were collected using Si<111> monochromated synchrotron X-ray radiation (λ = 0.71023 Å) at 100(2) K and were corrected for Lorentz and polarization effects using the XDS software [
23]. The structure was solved by direct methods and the full-matrix least-squares refinements were carried out using SHELXL [
26]. X-ray crystallographic information files (CIF) for the structures
22–
24 are CCDC 1505262, 1,505, 263 and 1956306. A copy of the data can be obtained free of charge from CCDC, 12 Union Road, Cambridge CB2 1EZ, UK or email: deposit@ccdc.cam.ac.uk.




 and
and 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}