
Synthetic Strategies for Dinucleotides Synthesis


Abstract
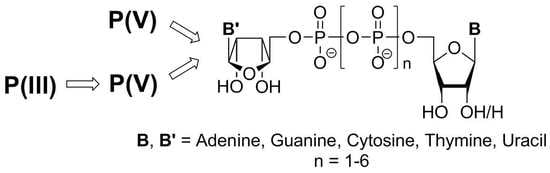
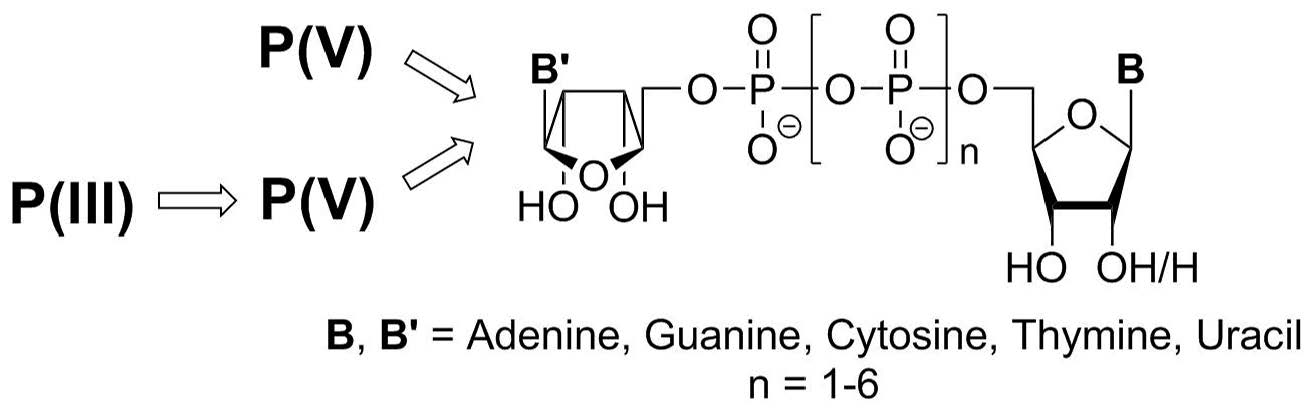
1. Introduction
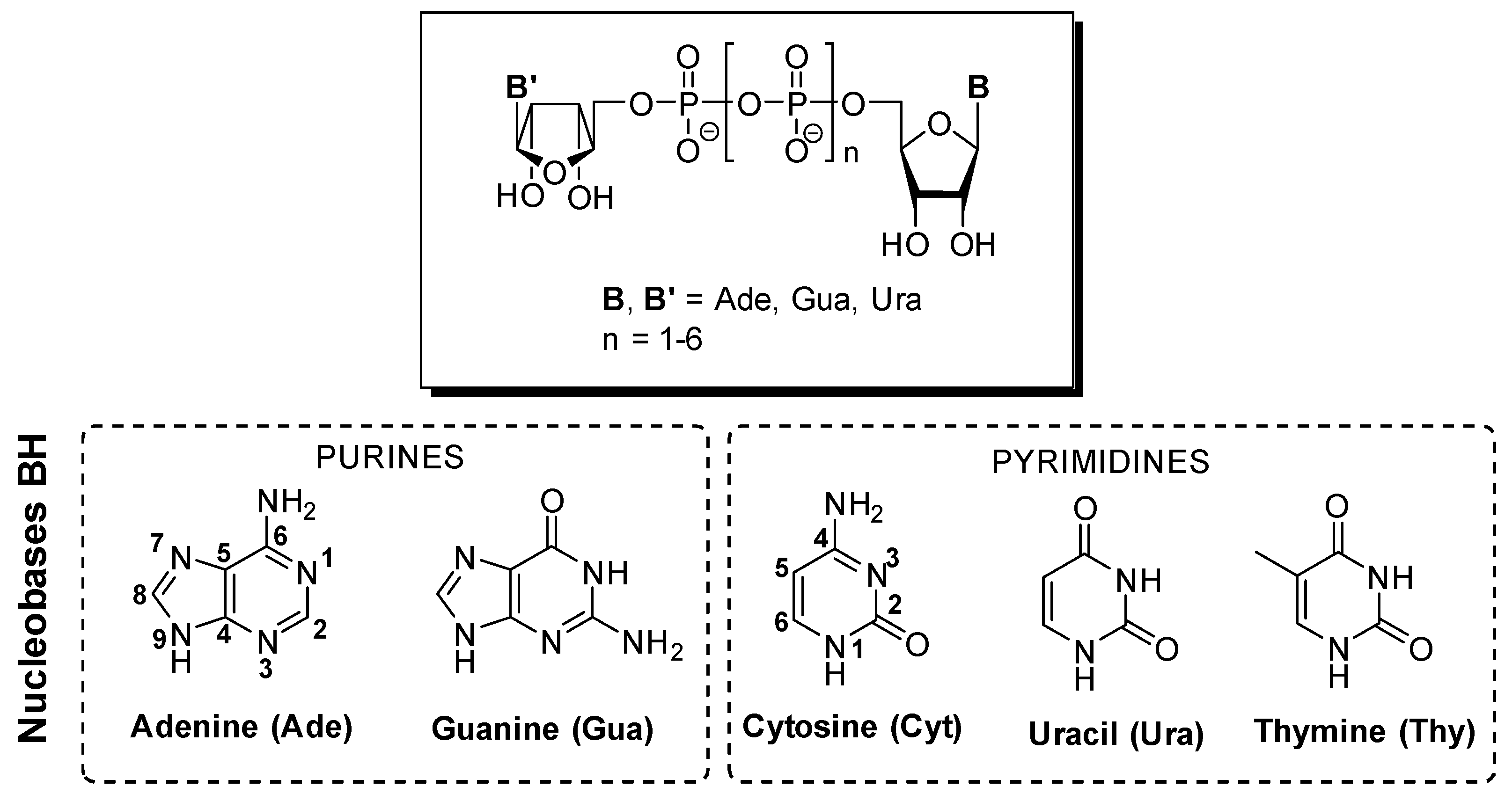
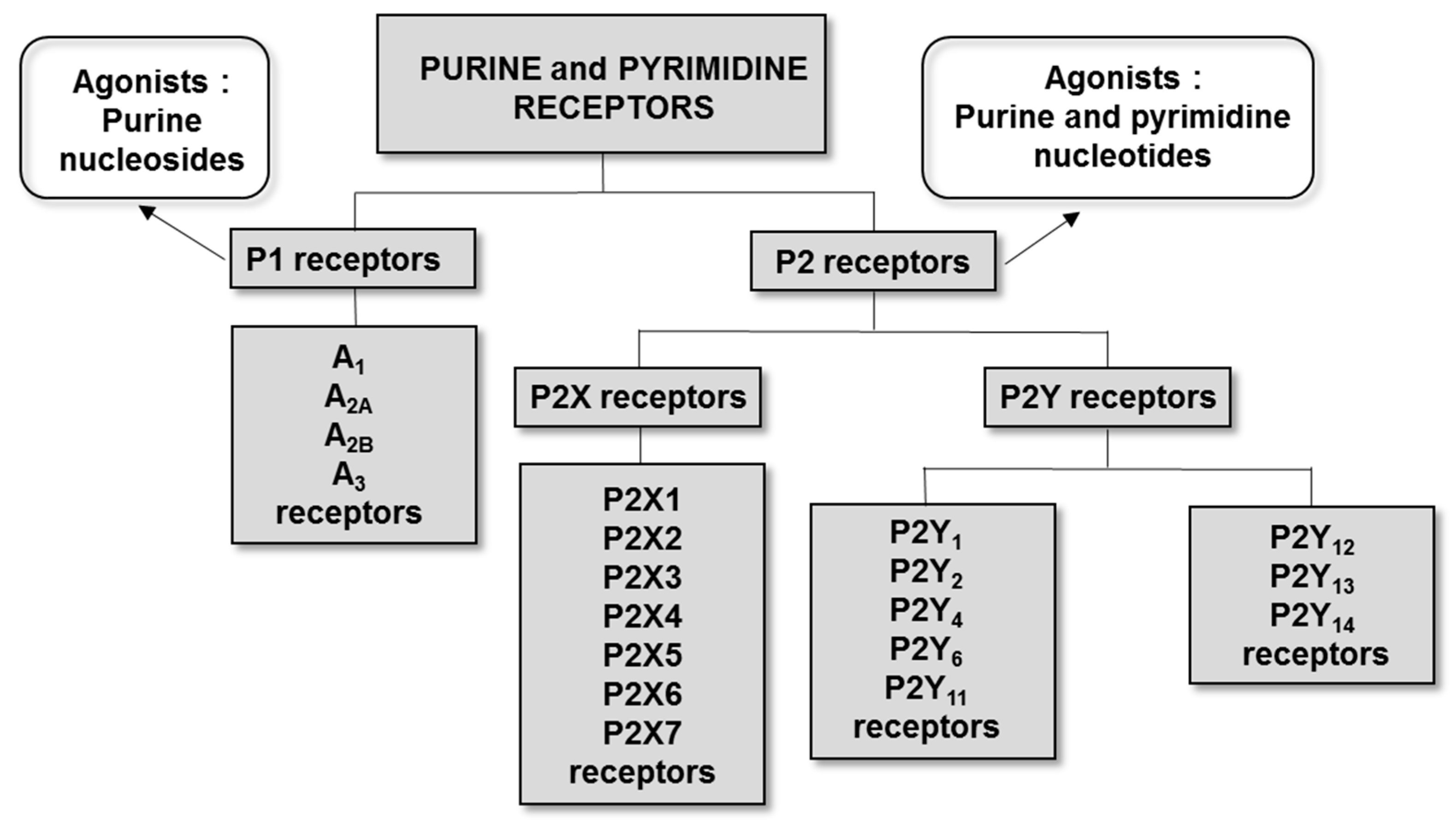
2. Interaction of DNPs with Purine and Pyrimidine Receptors
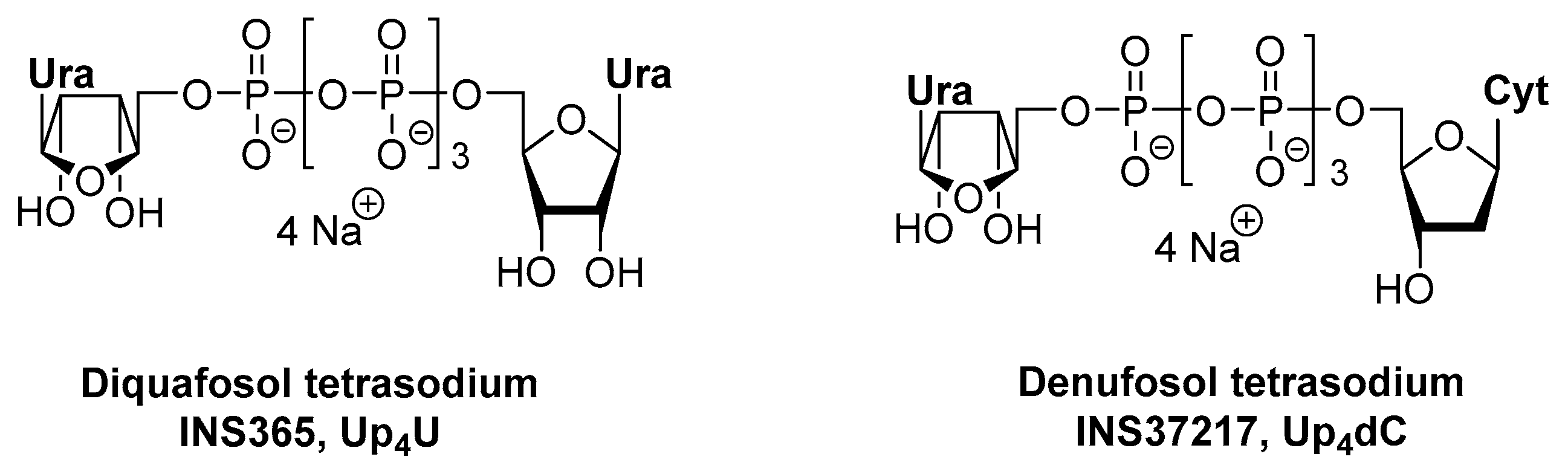
2.1. Up4U for the Treatment of Dry Eye Disease (DED)
2.2. Up4dC for the Treatment of Cystic Fibrosis
3. Synthesis Based on P(V) Chemistry
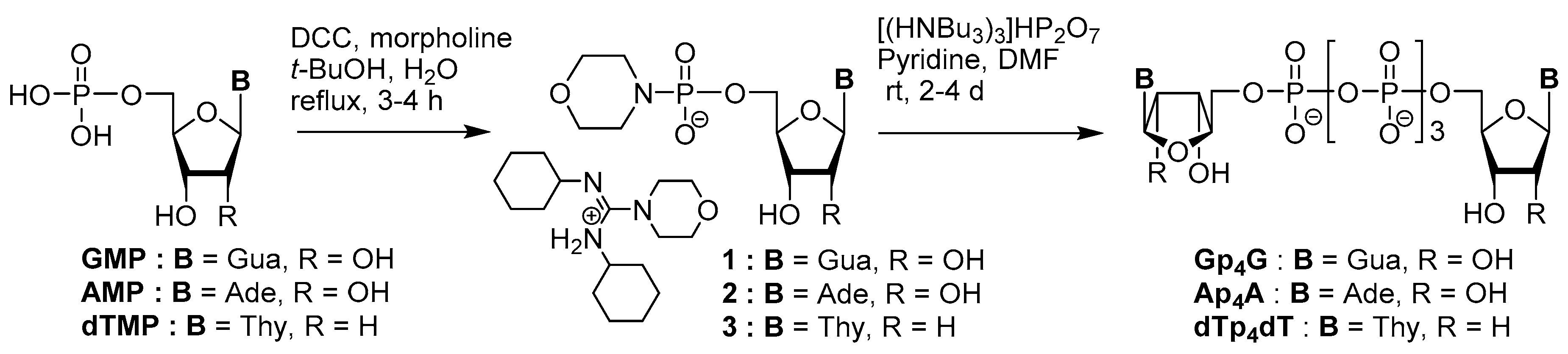
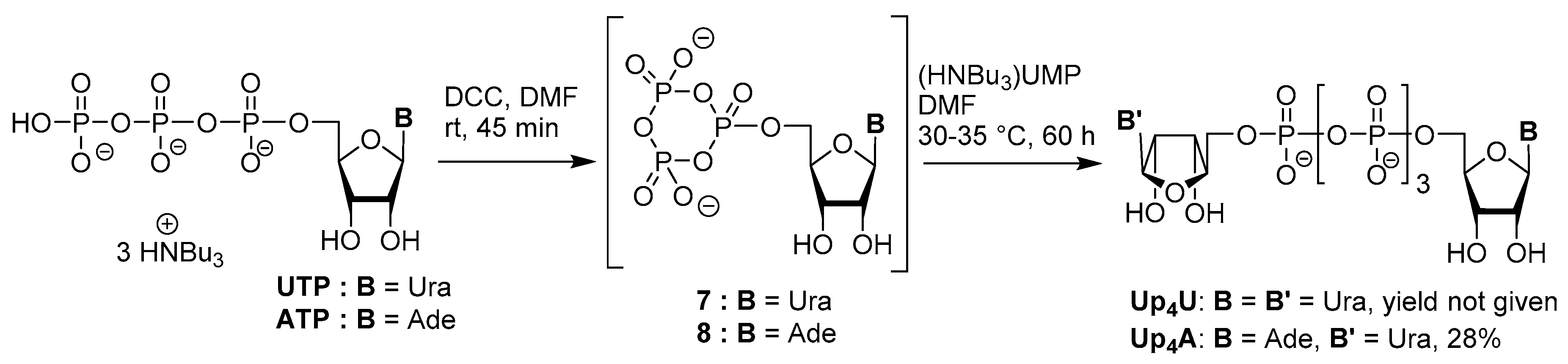
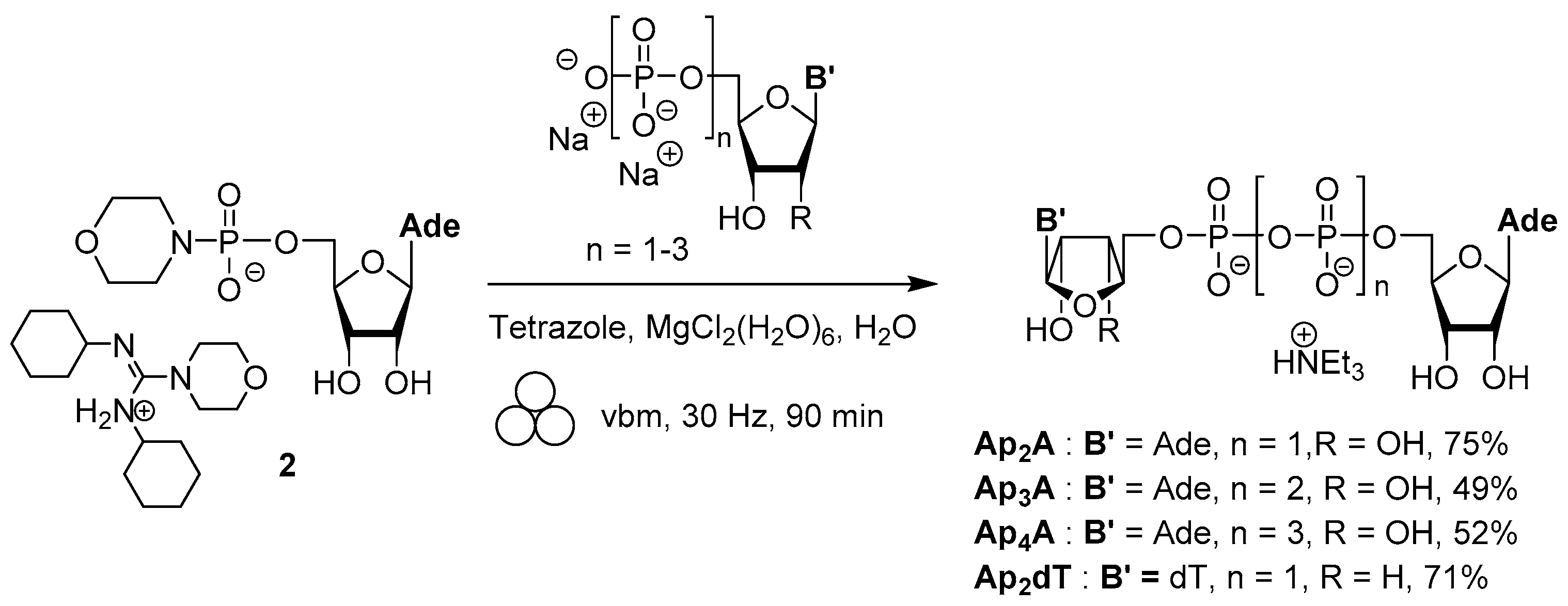
3.1. Synthesis via a Phosphoromorpholidate Intermediate
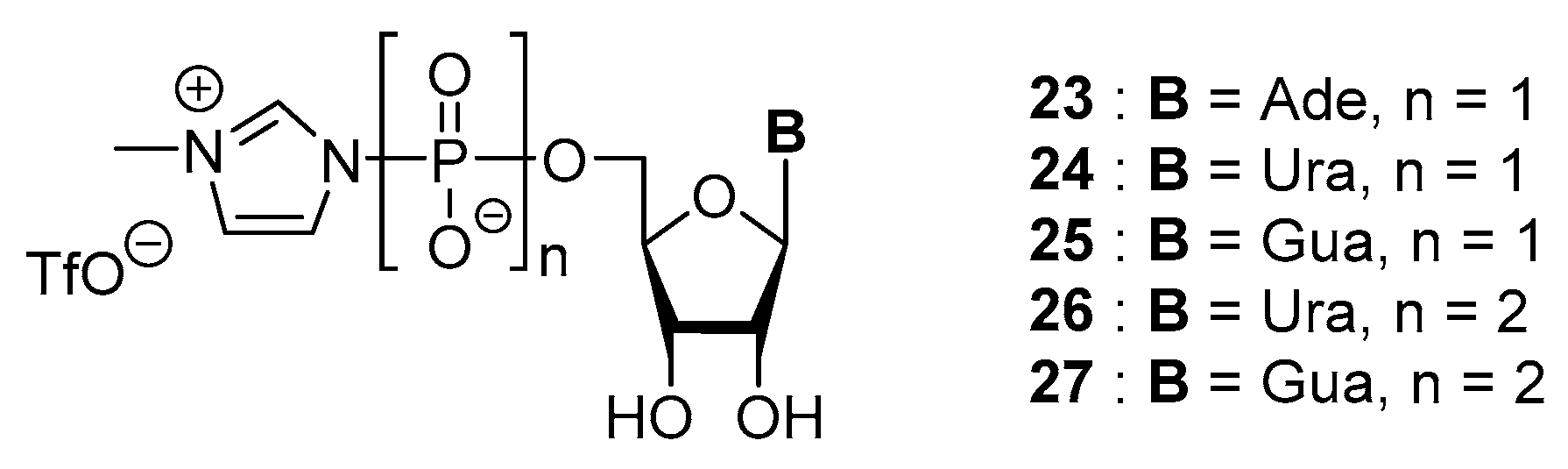
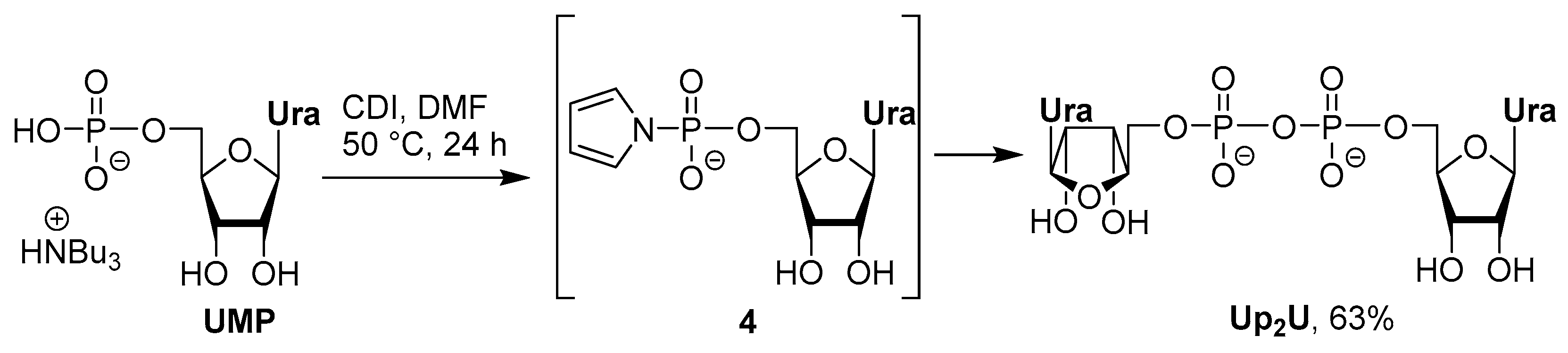
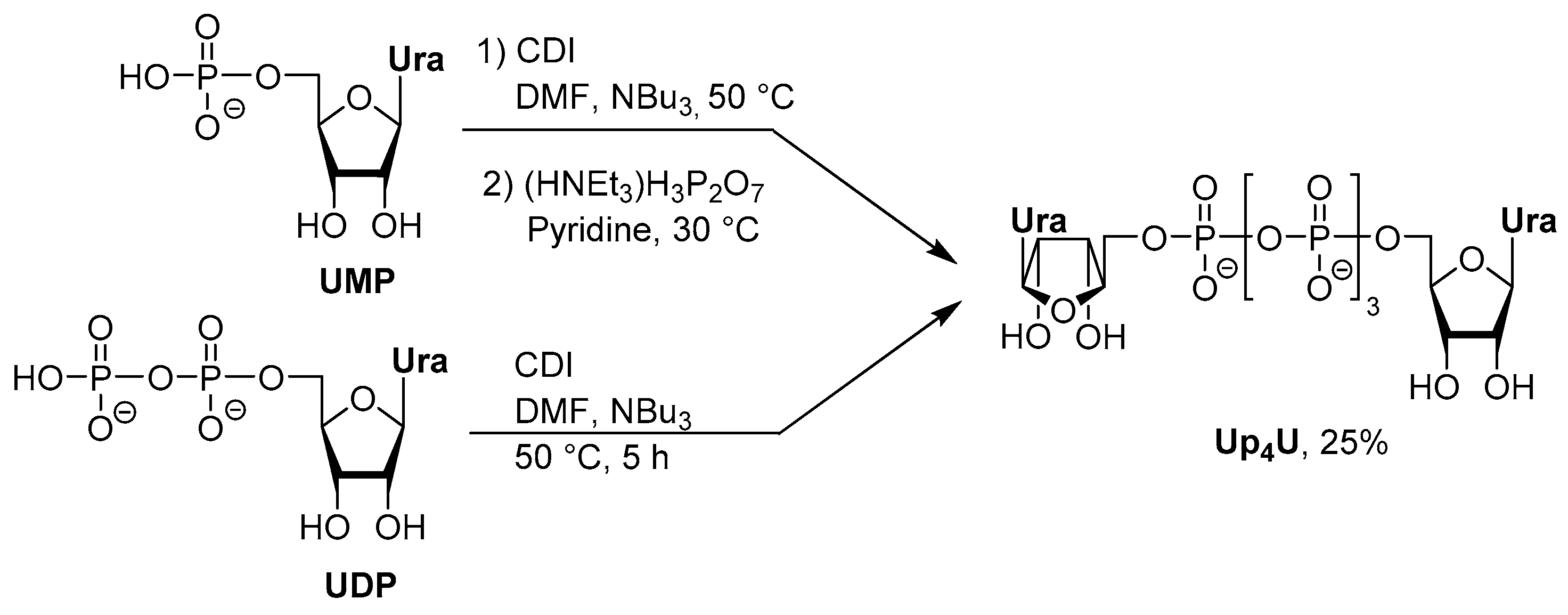
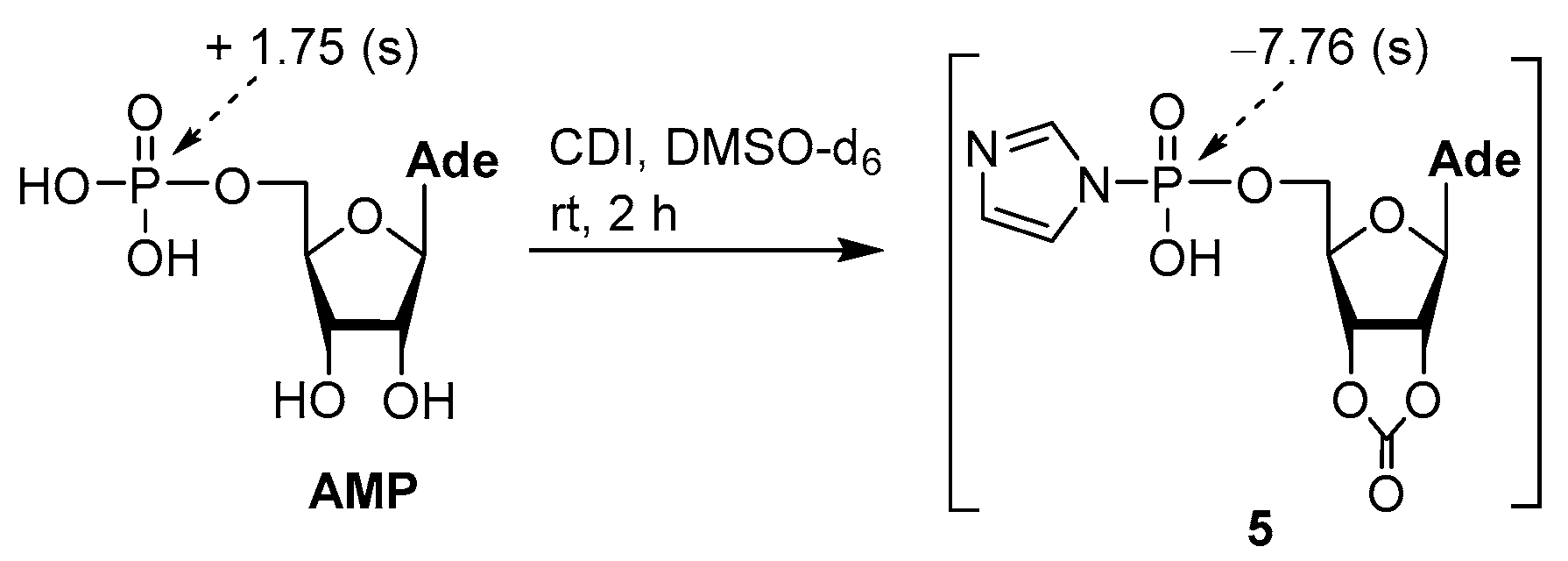
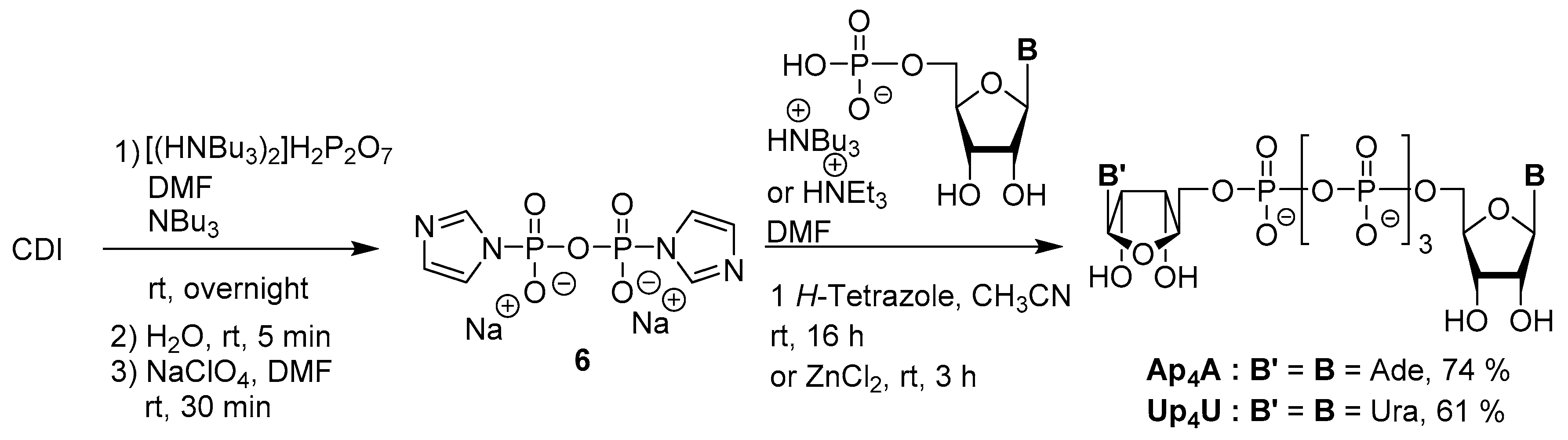
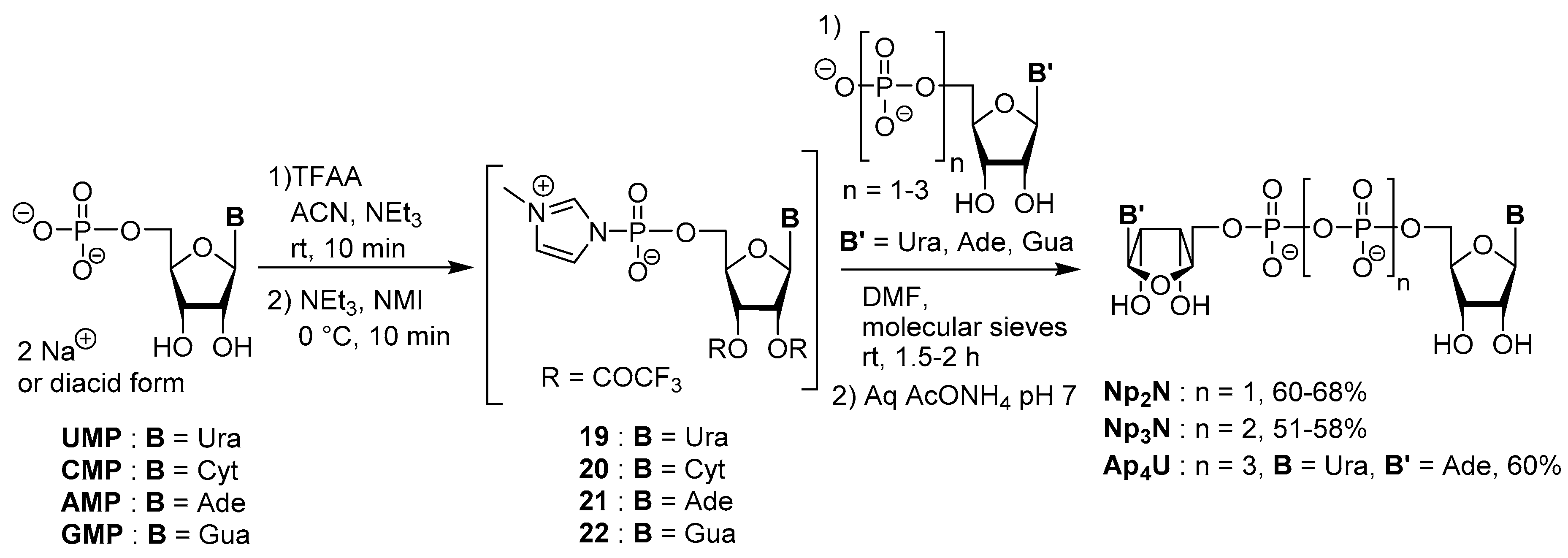
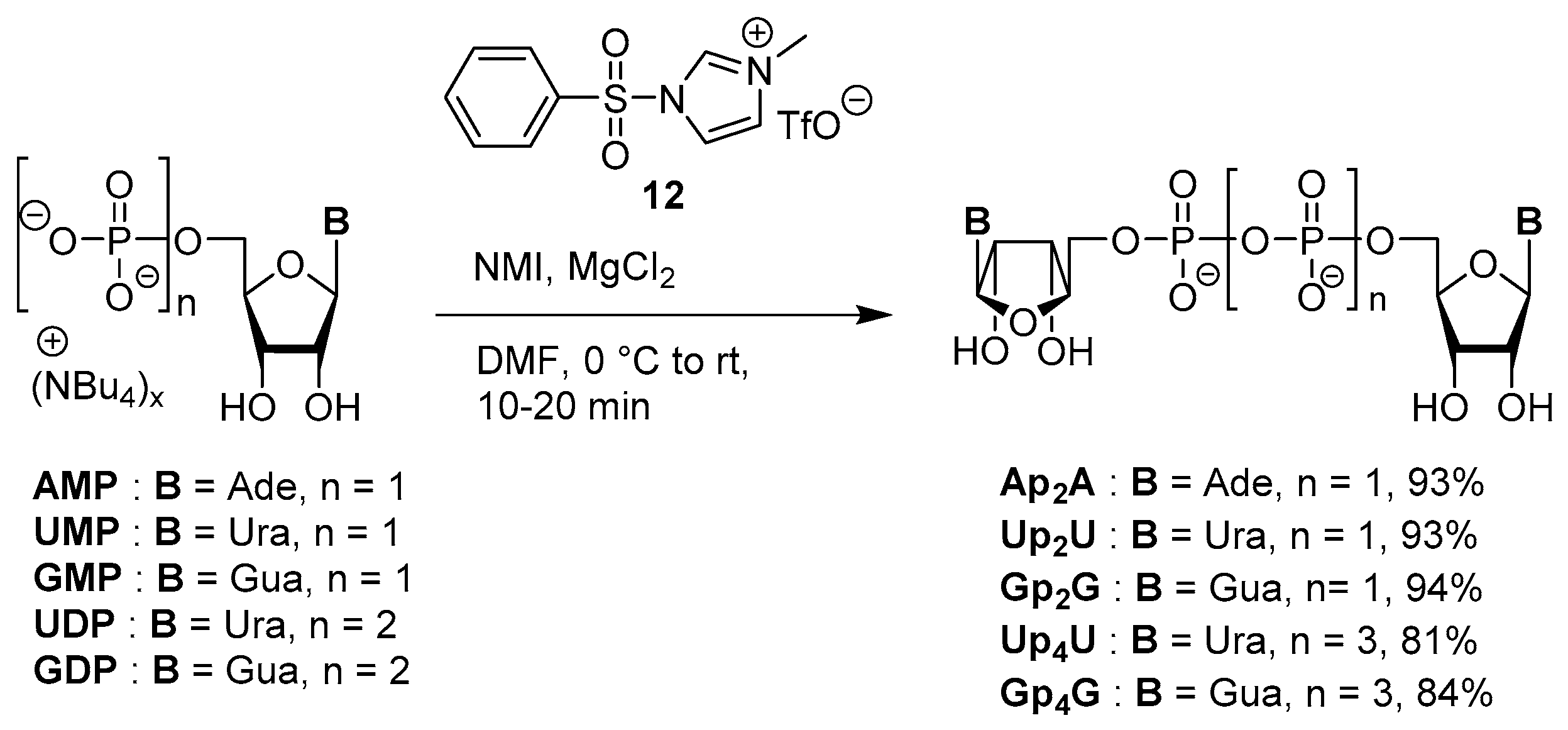
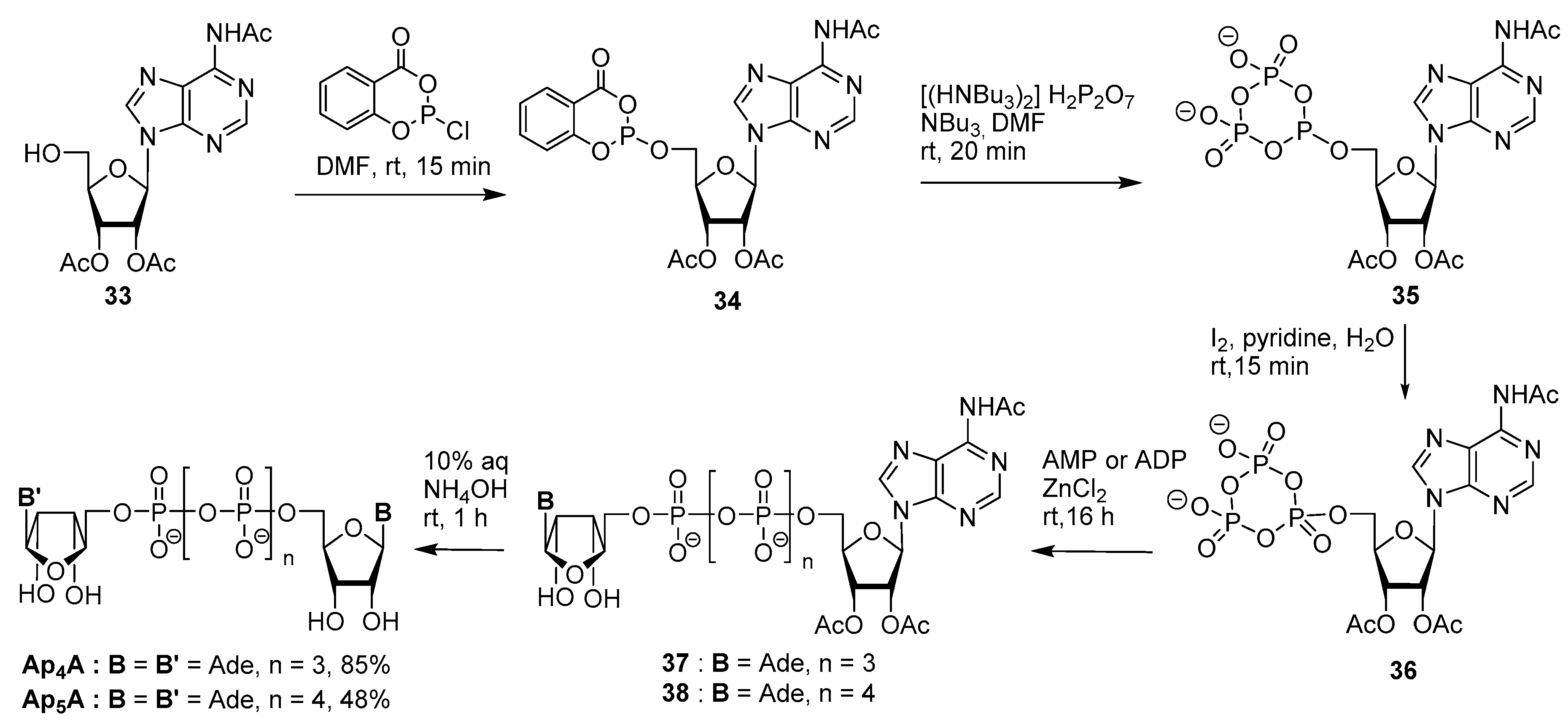
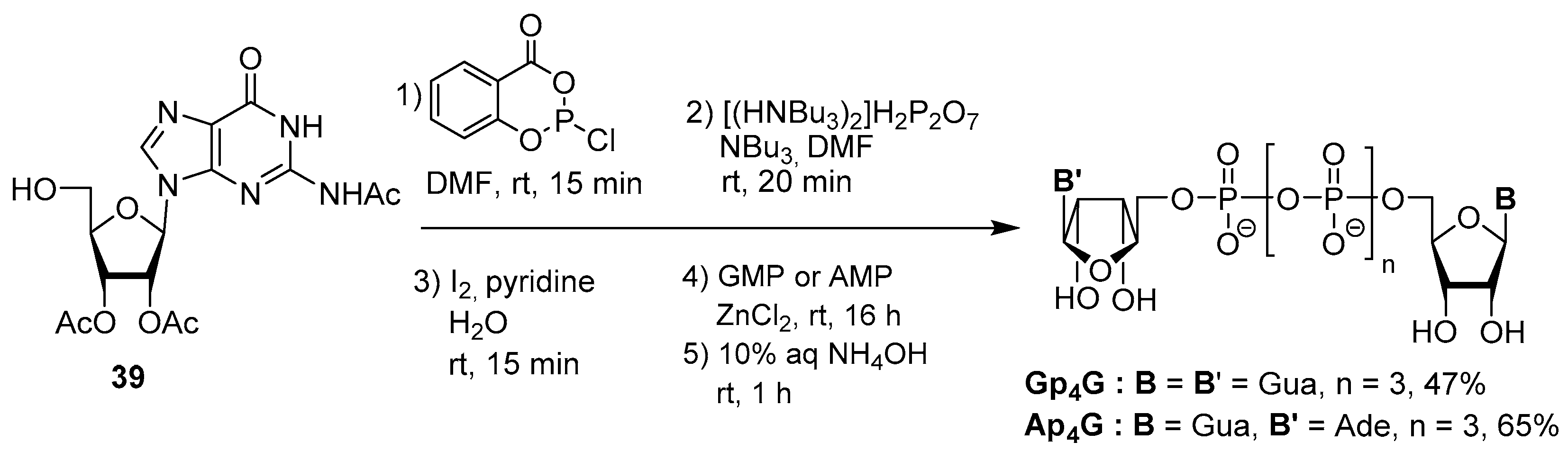
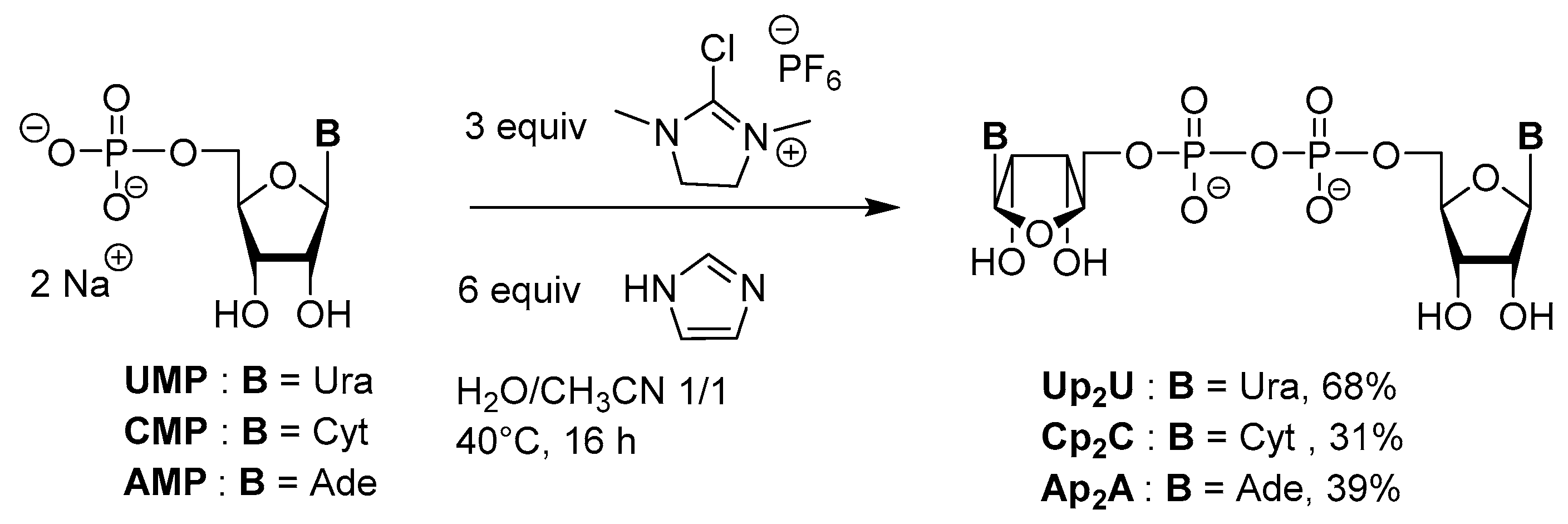
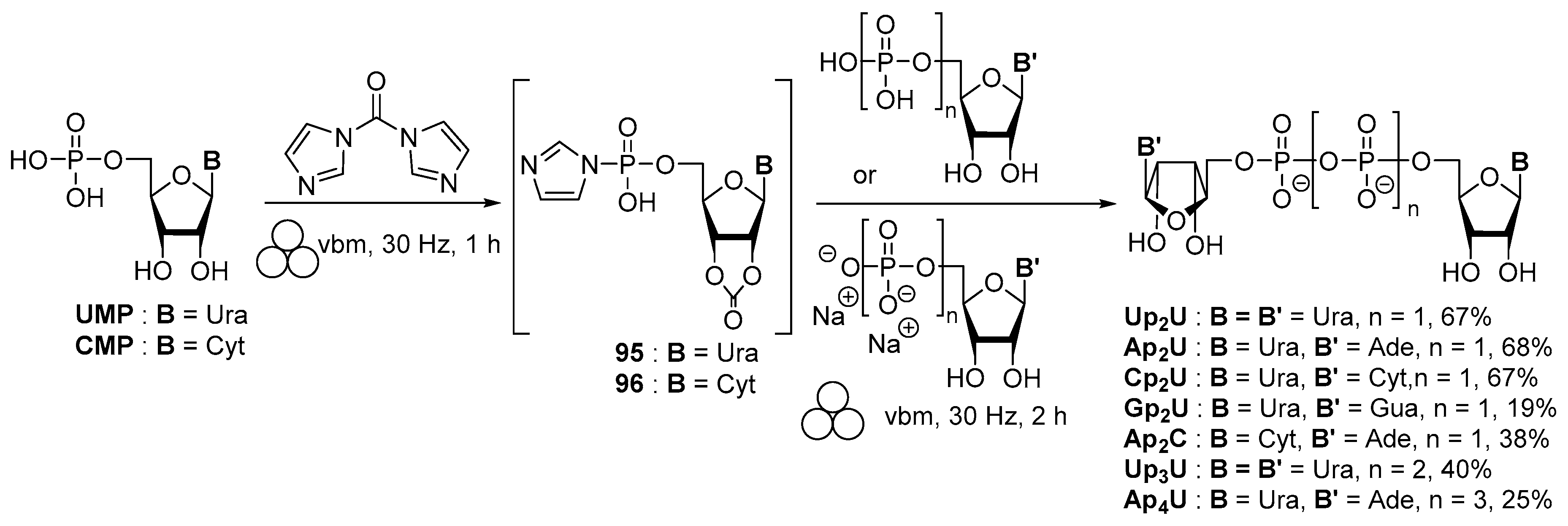
3.2. Synthesis via a Phosphorimidazolide Intermediate
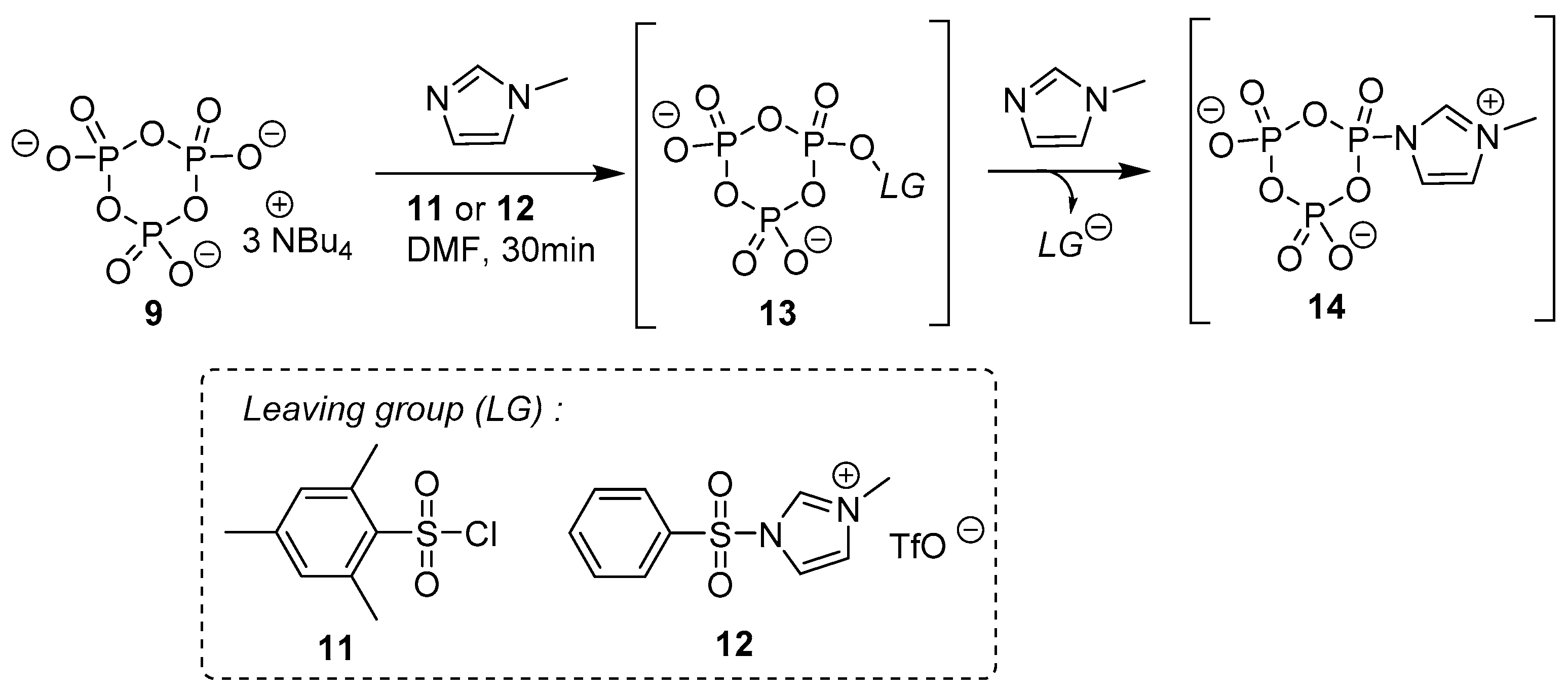
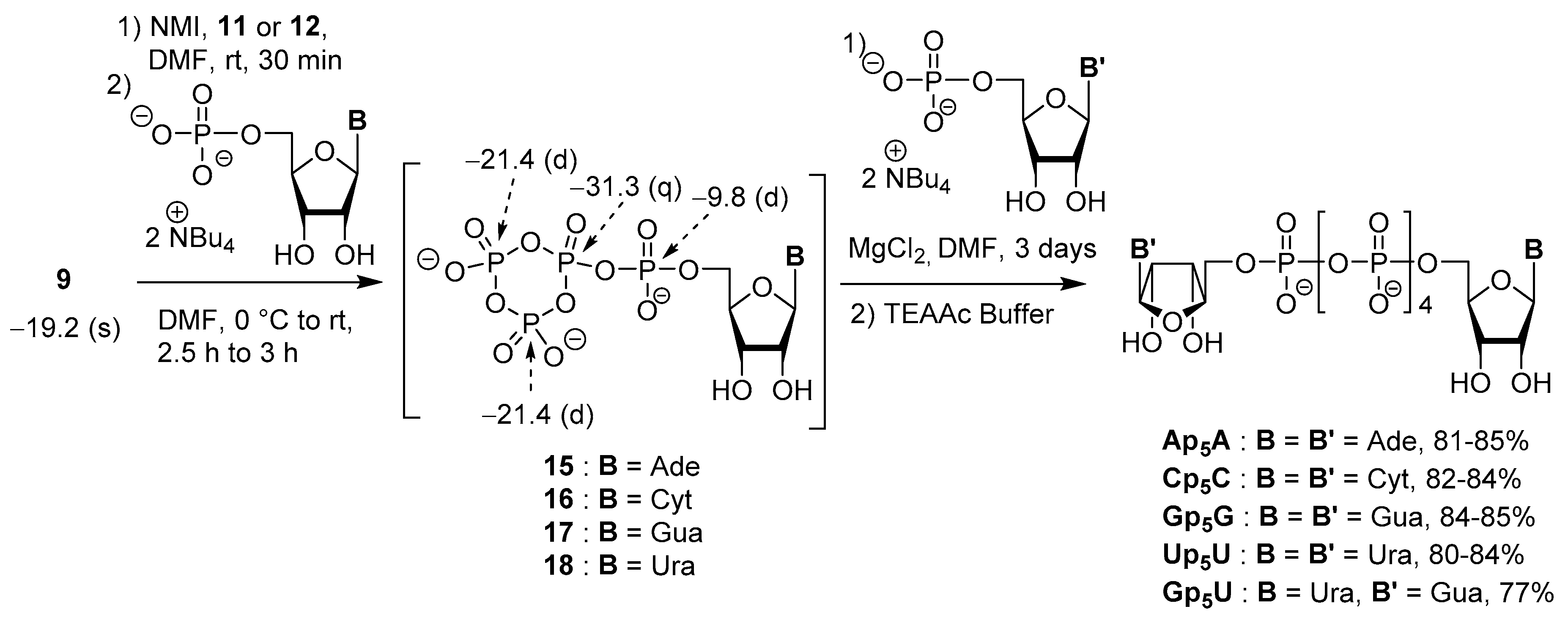
3.3. Synthesis via a Cyclic Trimetaphosphate Intermediate
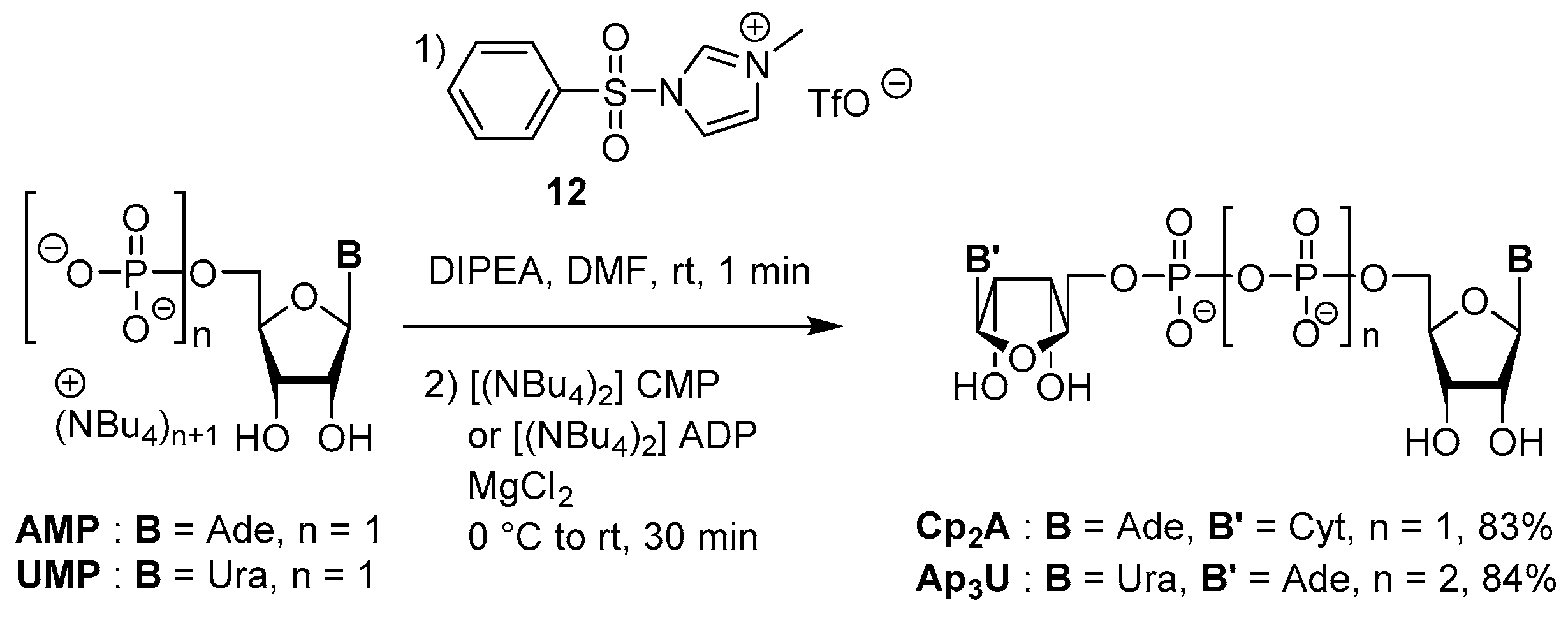
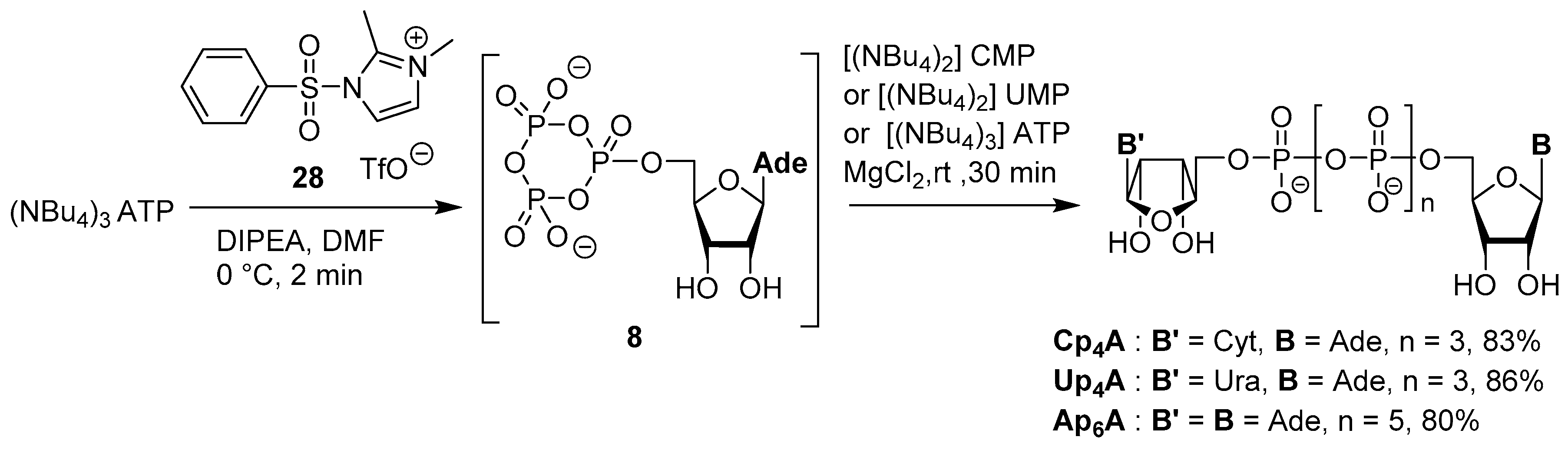
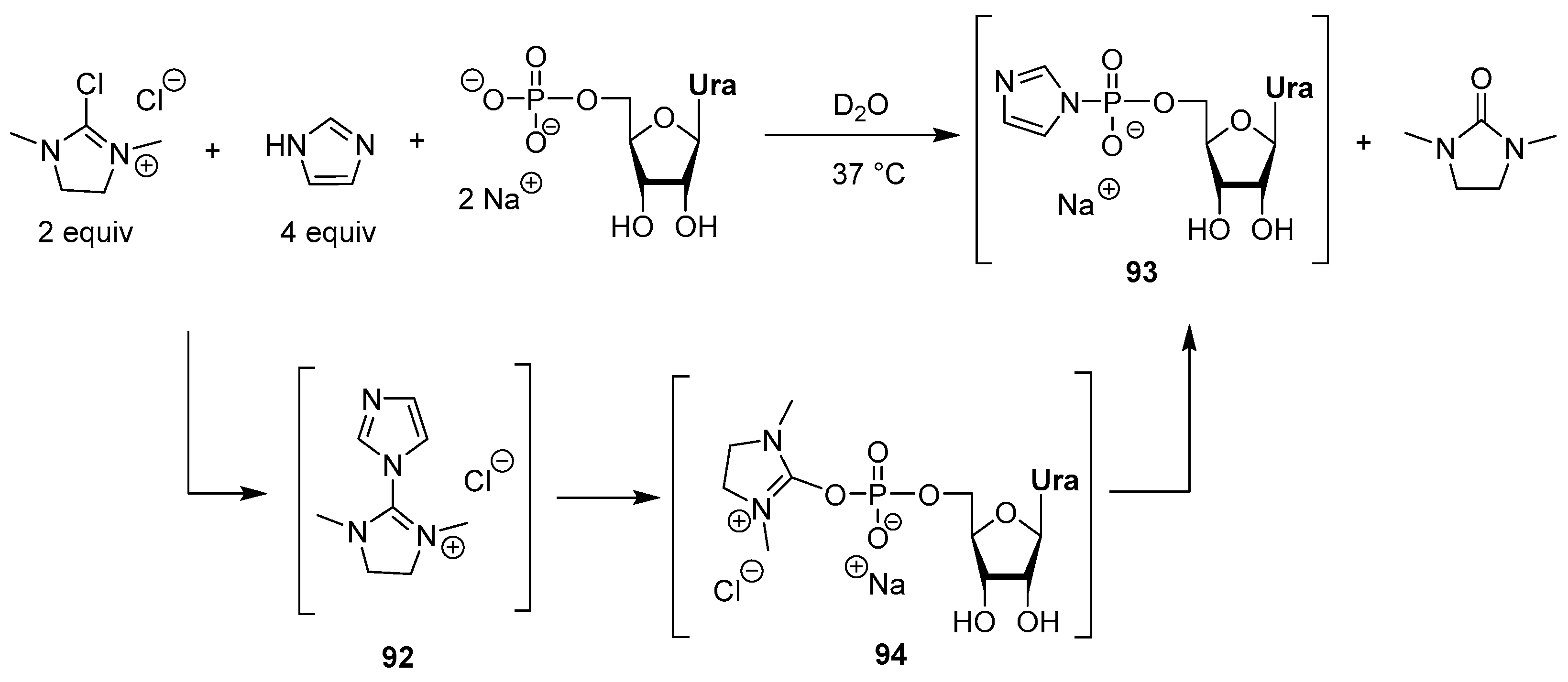
3.4. Synthesis via a Phosphoromethylimidazolium Intermediate
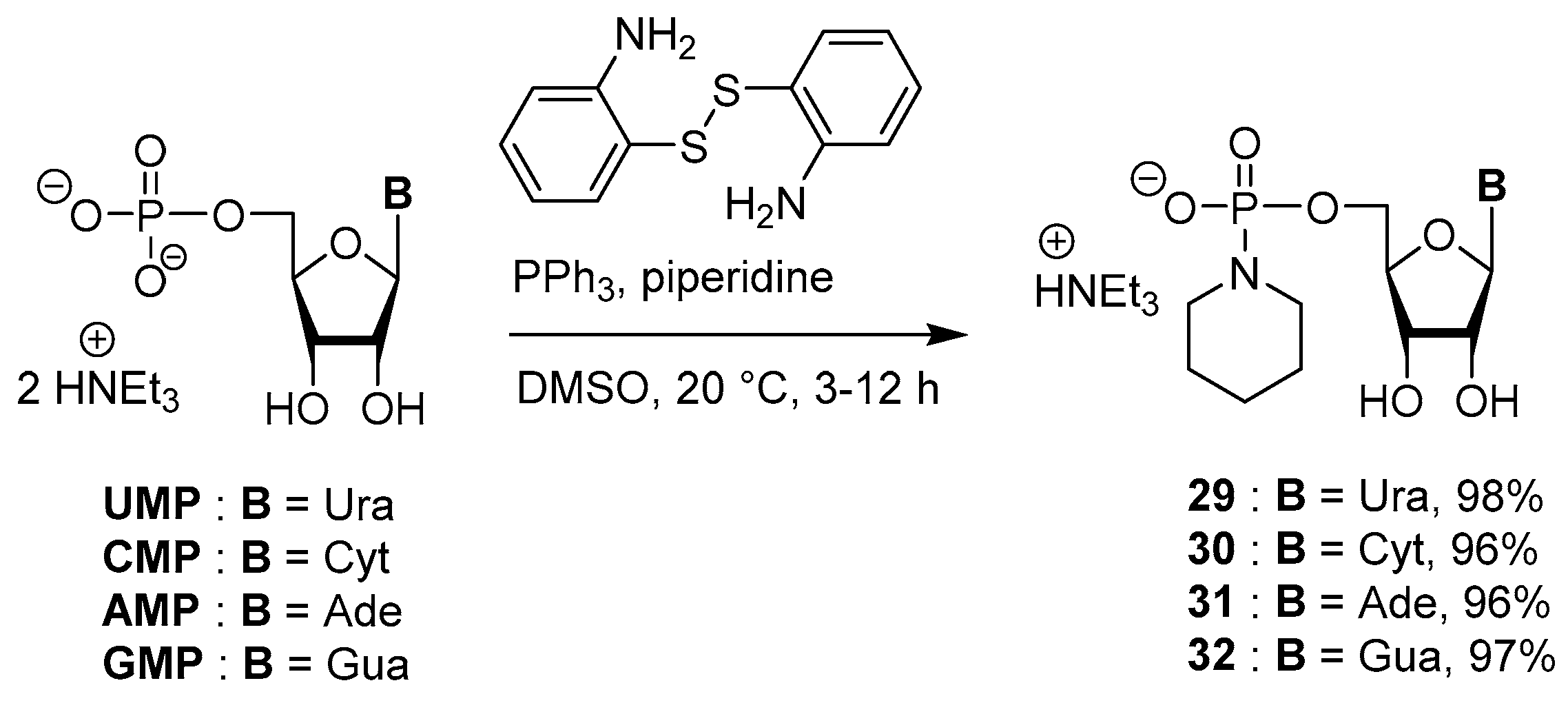
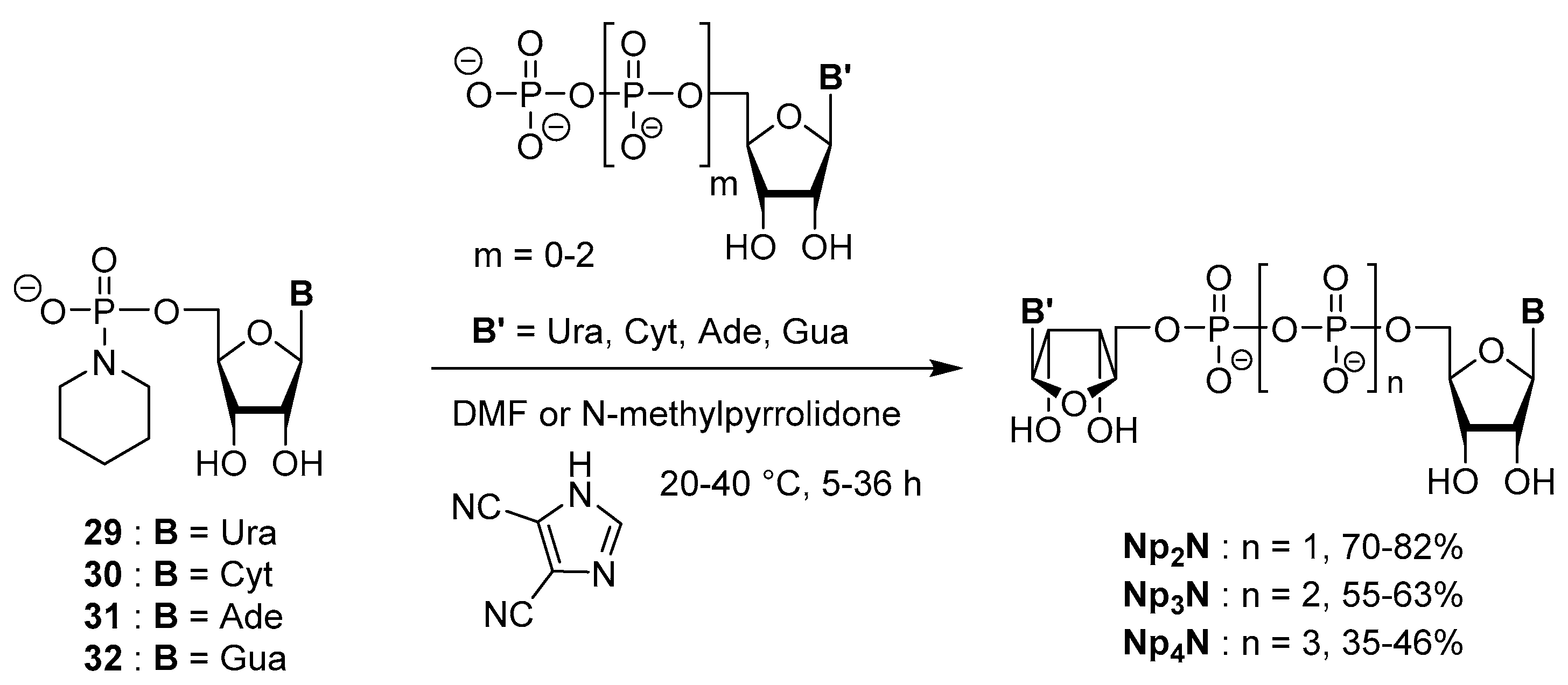
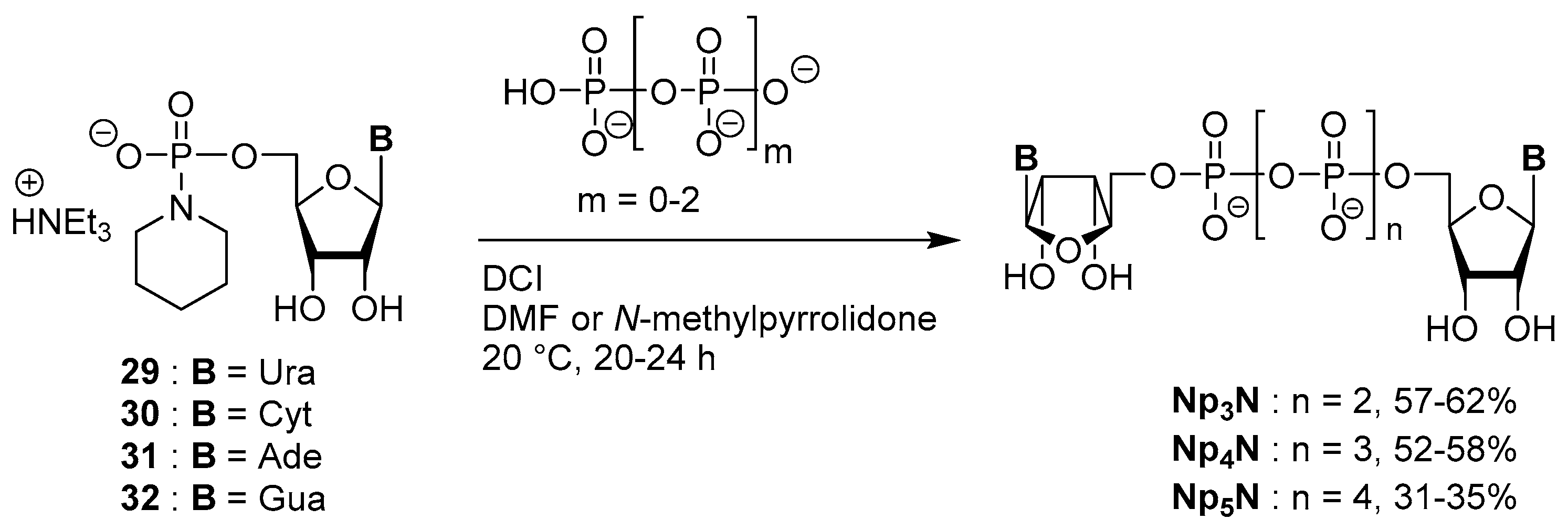
3.5. Synthesis via a Phosphoropiperidate Intermediate
4. Synthesis Based on P(III) Chemistry
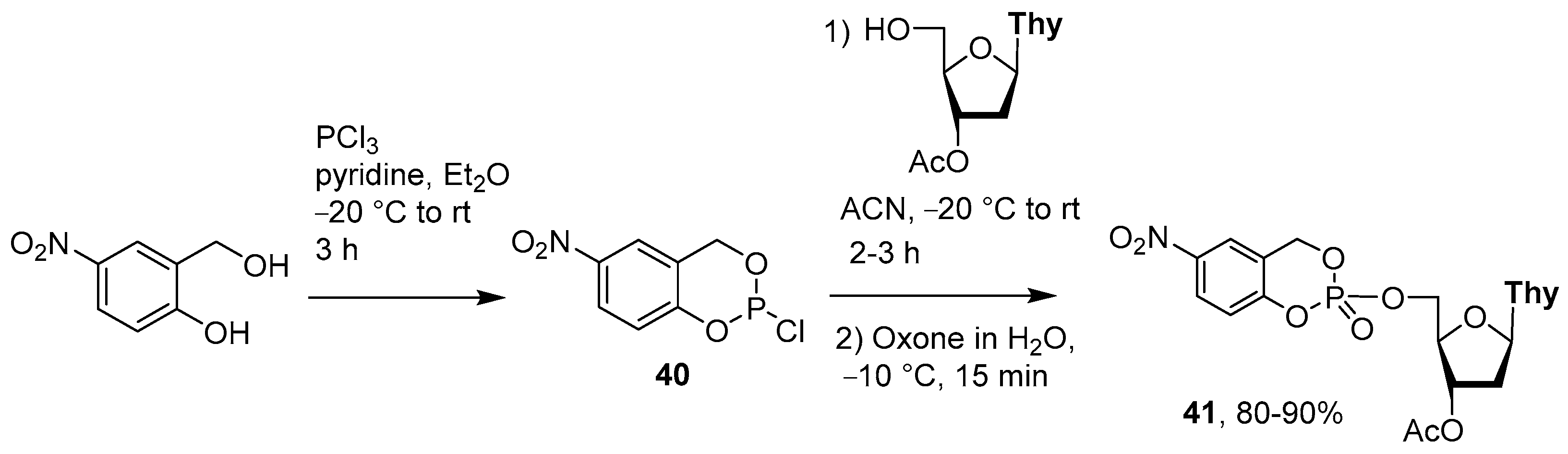
4.1. Synthesis via a Salicylphosphite Intermediate
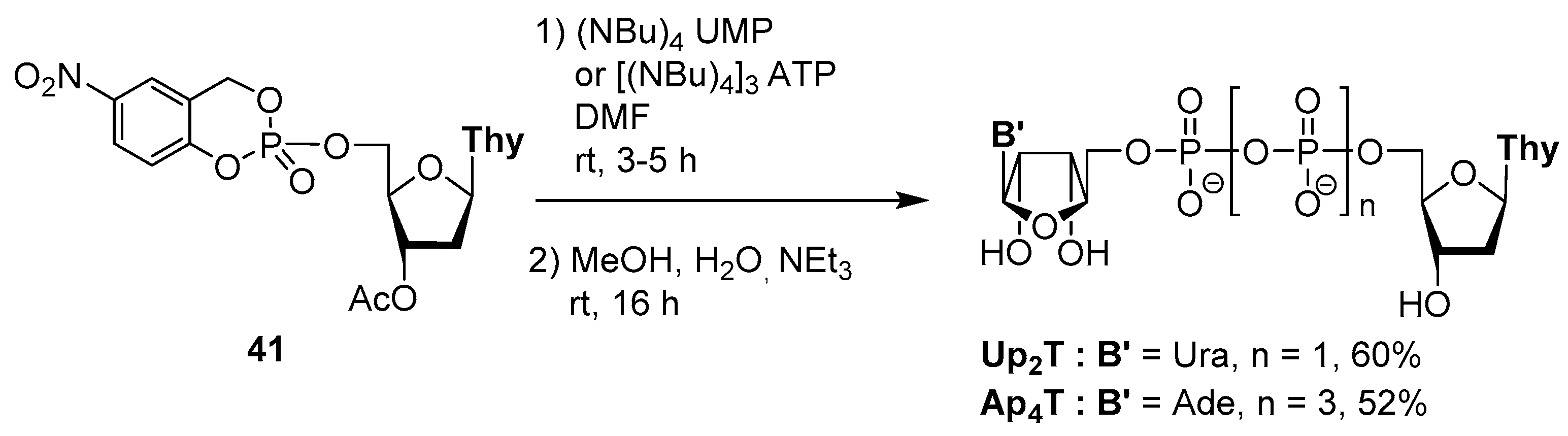
4.2. Synthesis via a Cyclosaligenyl Phosphite Intermediate
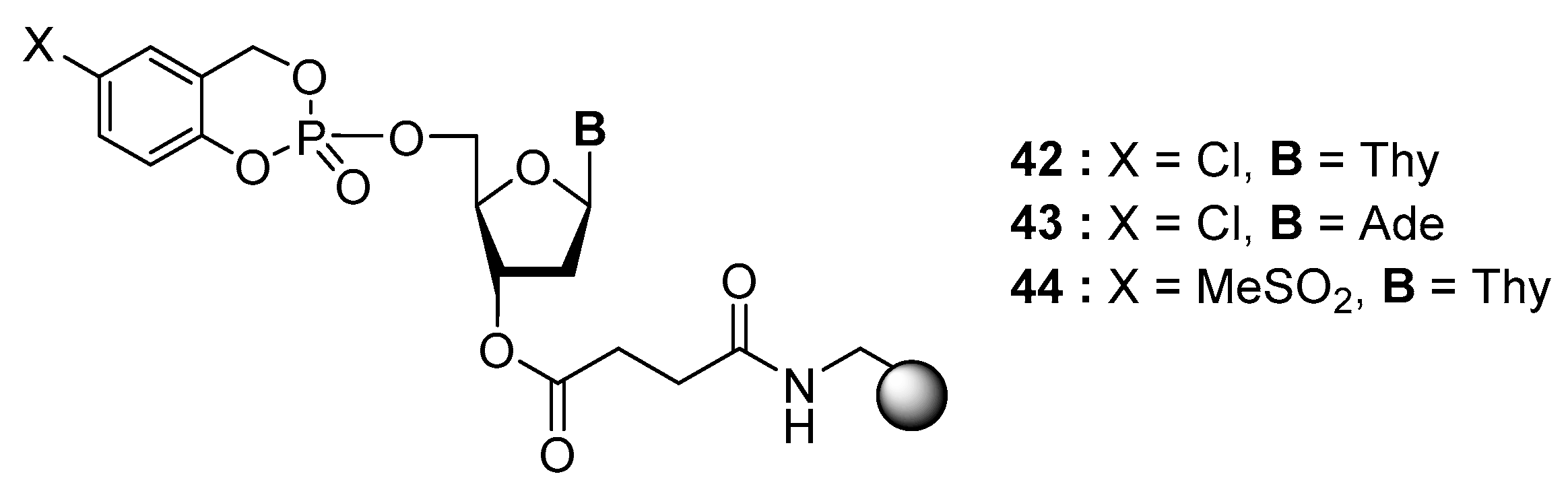
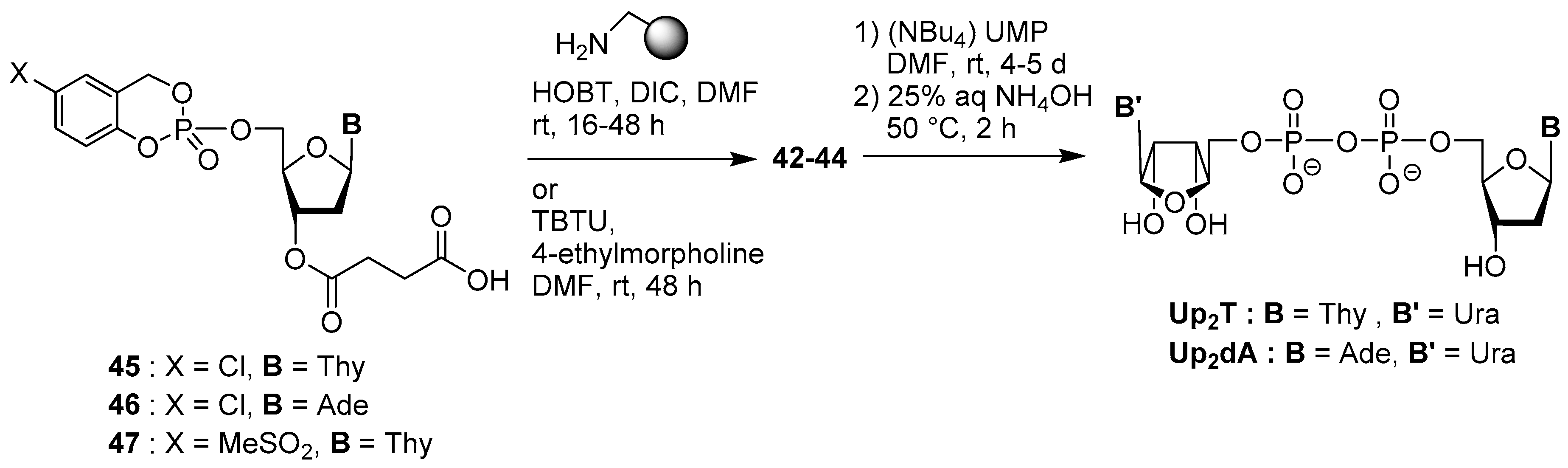
4.3. Synthesis via a Supported Phosphite Intermediate
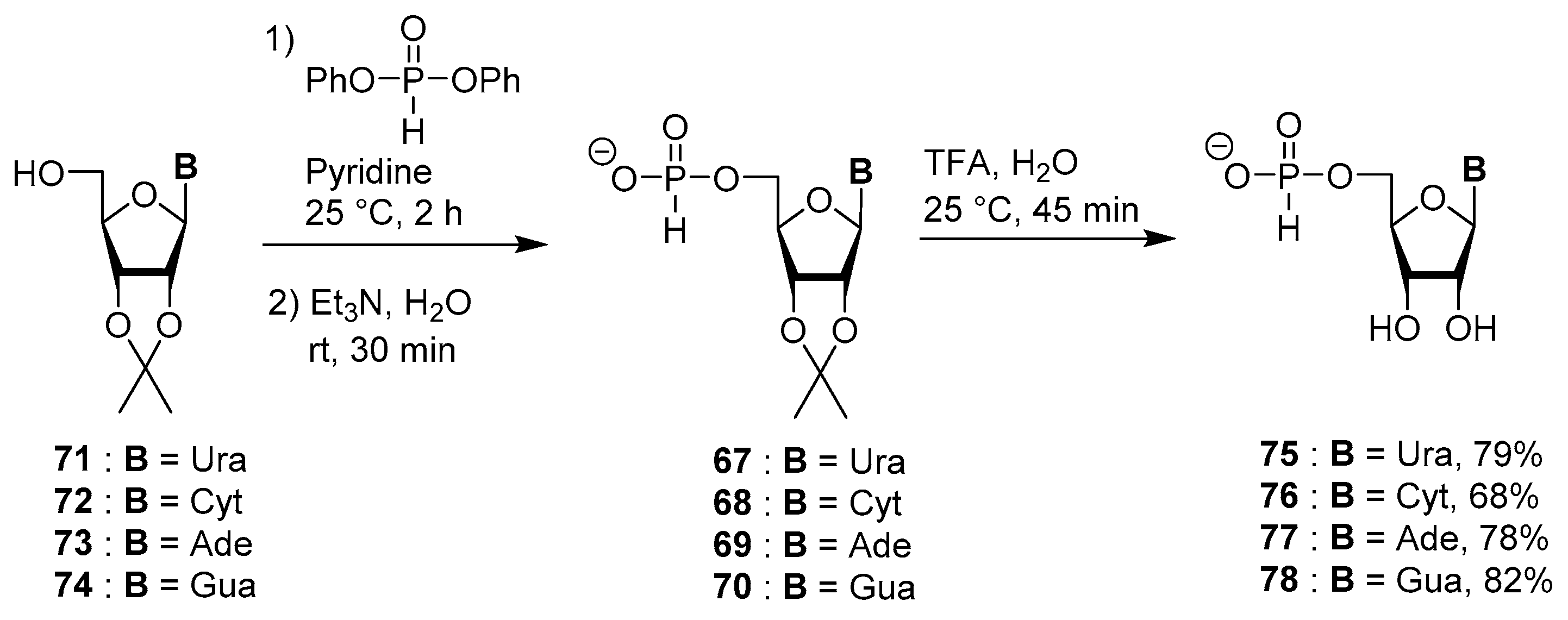
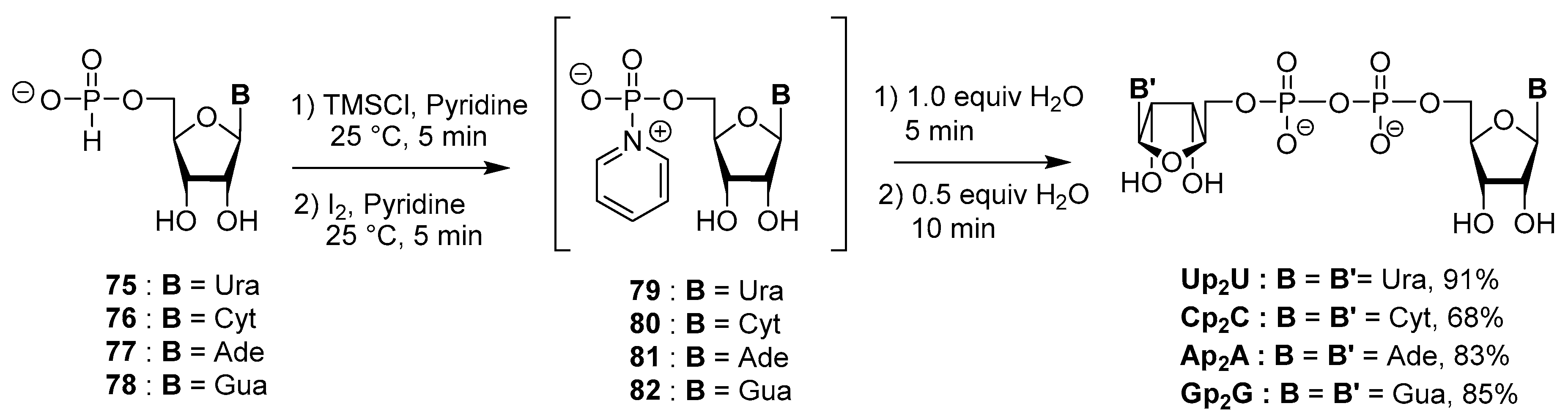
4.4. Synthesis via a 5′-H-Phosphonate Intermediate
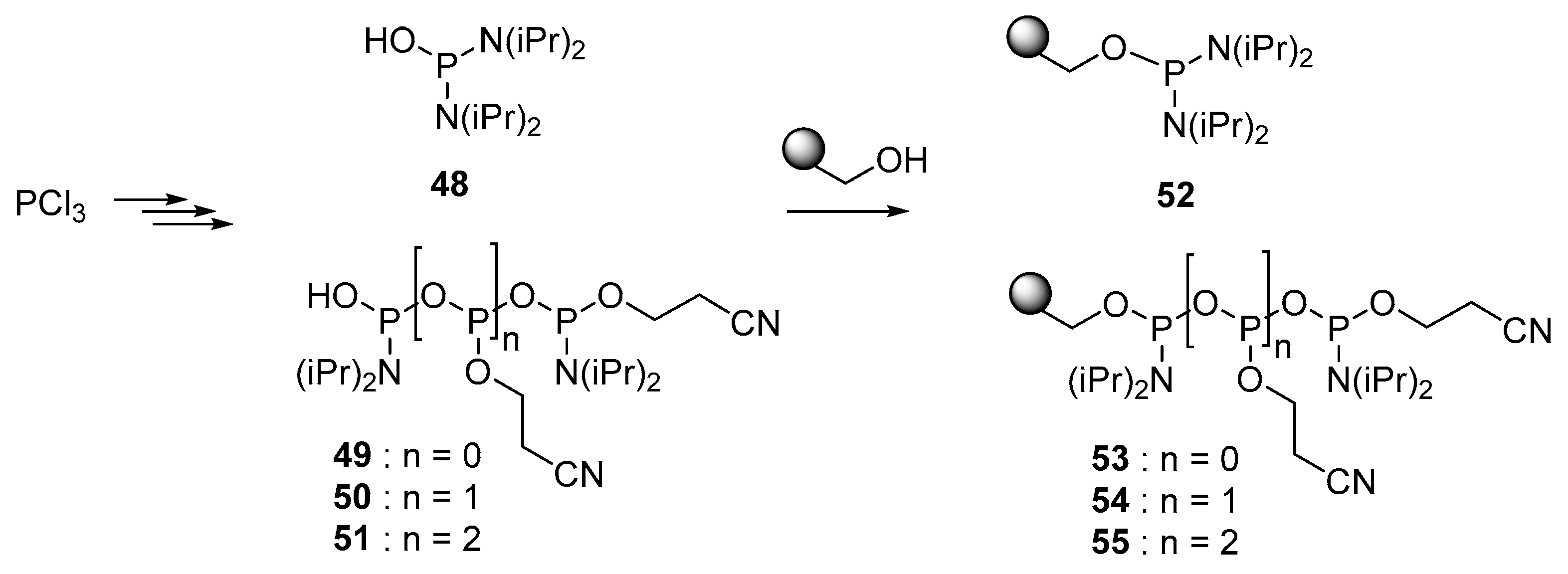
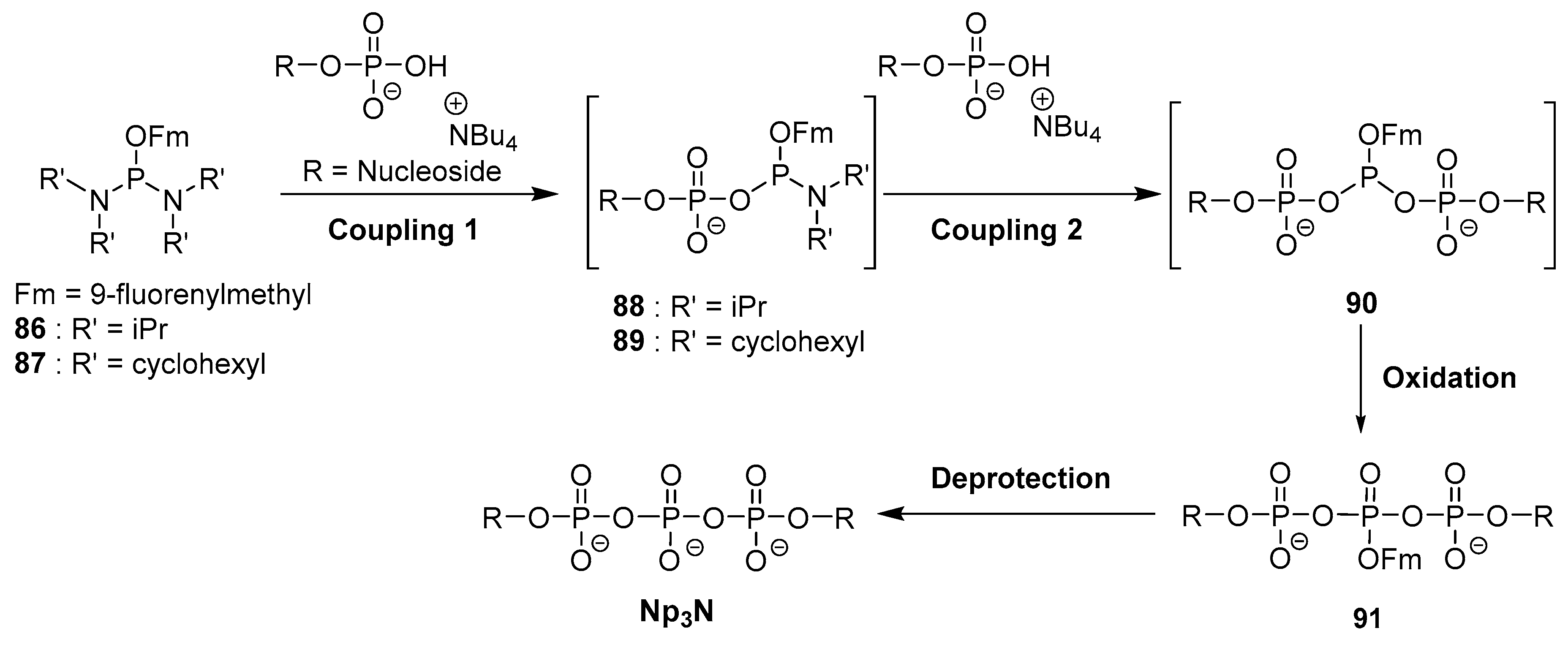
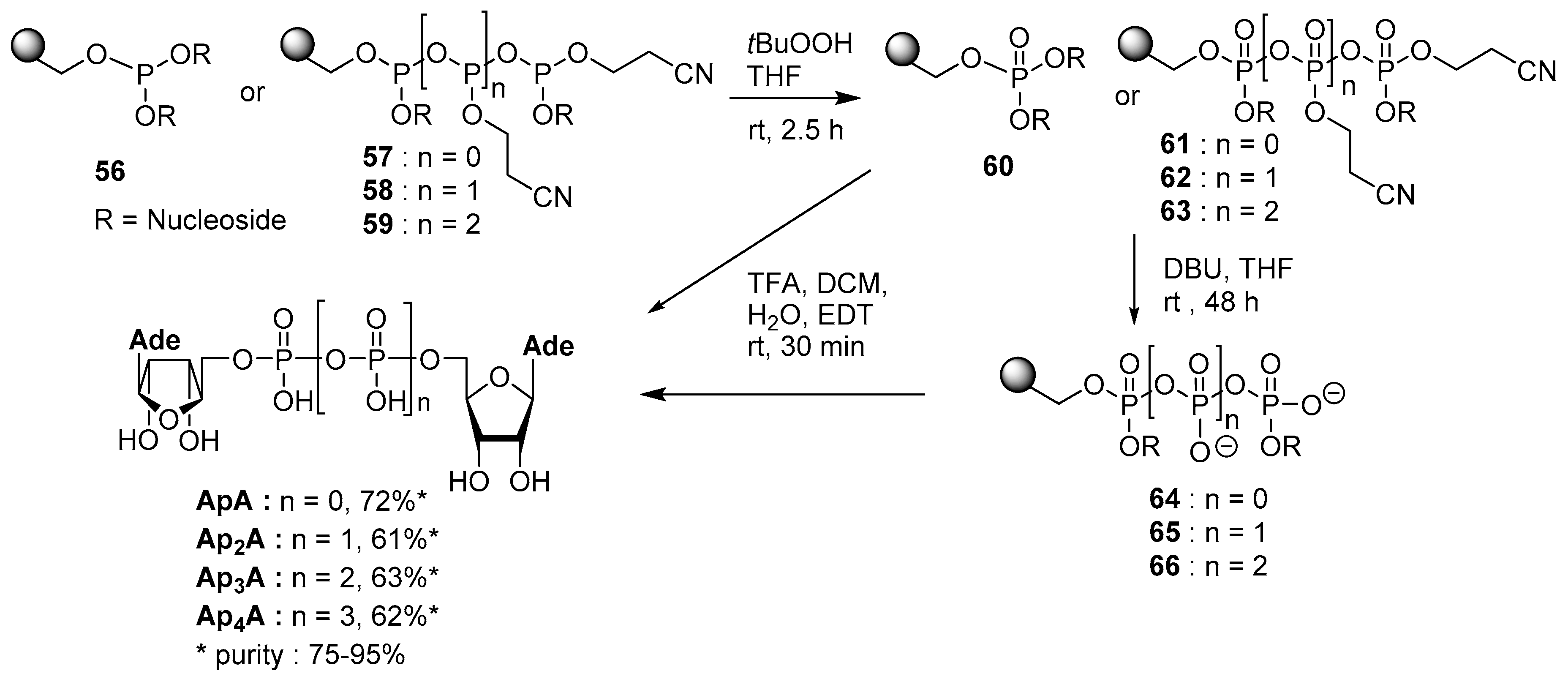
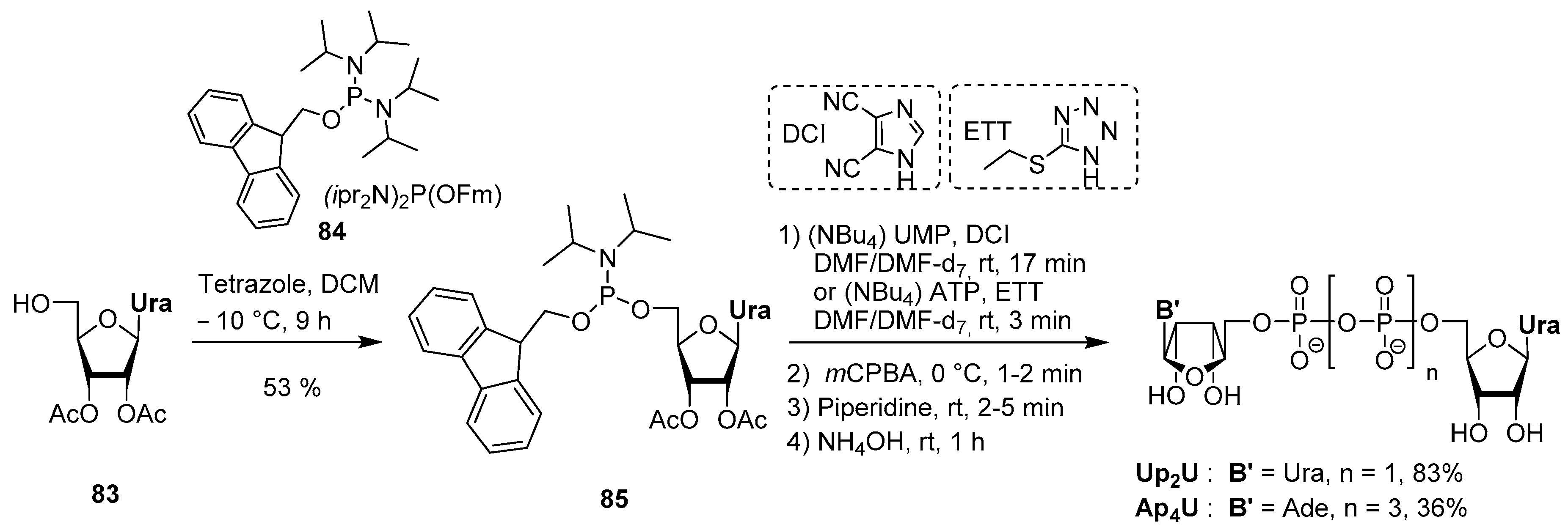
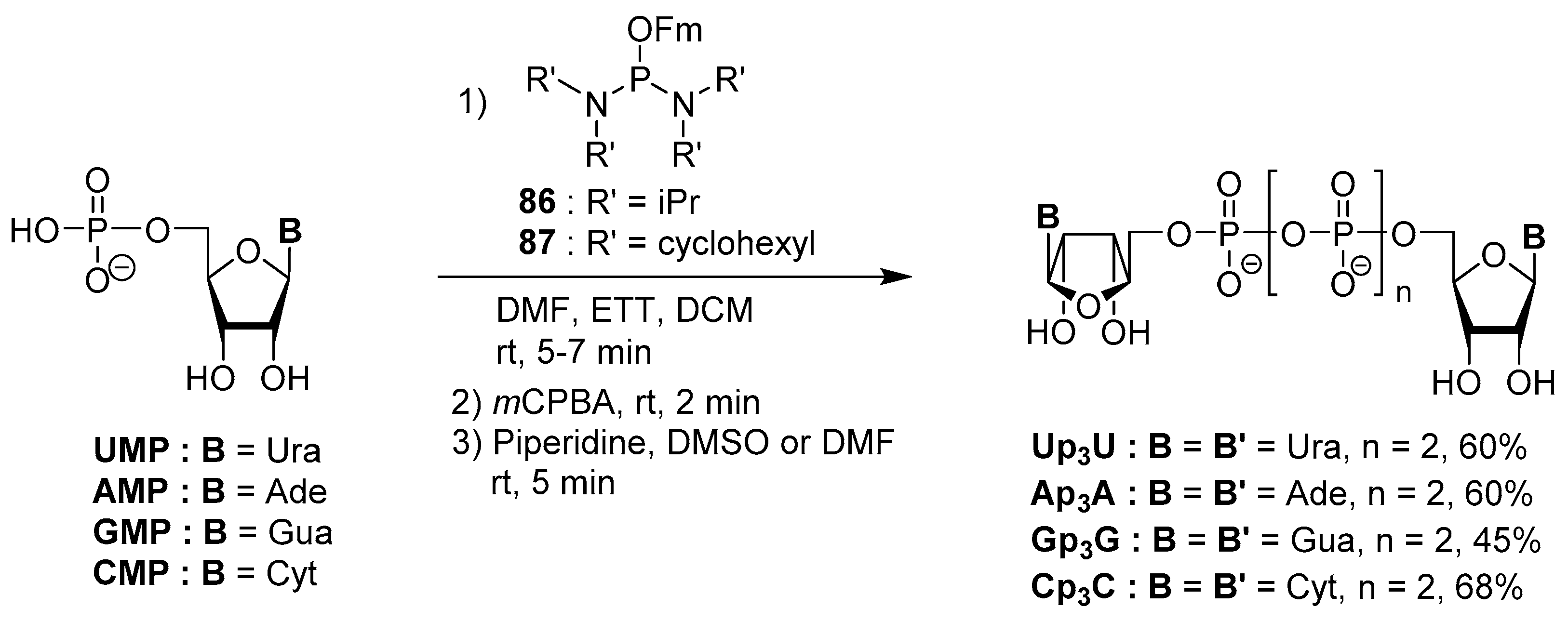
4.5. Synthesis via a Phosphoramidite Intermediate
5. Green Chemistry Approaches
6. Purification and Characterization
6.1. Purification
6.2. Physico-Chemical Properties
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roy, B.; Depaix, A.; Périgaud, C.; Peyrottes, S. Recent trends in nucleotide synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, V.; Van Der Giet, M.; Mischak, H.; Morgan, M.; Zidek, W.; Jankowski, J. Dinucleoside polyphosphates: Strong endogenous agonists of the purinergic system. Br. J. Pharmacol. 2009, 157, 1142–1153. [Google Scholar] [CrossRef]
- Christie, S.M.H.; Elmore, D.T.; Kenner, G.W.; Todd, A.R.; Weymouth, F.J. Syntheses of P1P2-diadenosine-5′ and P1P2-diuridine-5′ pyrophosphates. J. Chem. Soc. 1953, 2947–2953. [Google Scholar] [CrossRef]
- Rapaport, E.; Zamecnik, P.C. Presence of diadenosine 5′,5′’’ -P1, P4-tetraphosphate (Ap4A) in mamalian cells in levels varying widely with proliferative activity of the tissue: A possible positive “pleiotypic activator”. Proc. Natl. Acad. Sci. USA 1976, 73, 3984–3988. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, V.; Tölle, M.; Vanholder, R.; Schönfelder, G.; Van Der Giet, M.; Henning, L.; Schlüter, H.; Paul, M.; Zidek, W.; Jankowski, J. Uridine adenosine tetraphosphate: A novel endothelium- derived vasoconstrictive factor. Nat. Med. 2005, 11, 223–227. [Google Scholar] [CrossRef]
- Zhou, Z.; Matsumoto, T.; Jankowski, V.; Pernow, J.; Mustafa, S.J.; Duncker, D.J.; Merkus, D. Uridine adenosine tetraphosphate and purinergic signaling in cardiovascular system: An update. Pharmacol. Res. 2019, 141, 32–45. [Google Scholar] [CrossRef]
- Delicado, E.G.; Miras-Portugal, M.T.; Carrasquero, L.M.G.; Leon, D.; Pérez-Sen, R.; Gualix, J. Dinucleoside polyphosphates and their interaction with other nucleotide signaling pathways. Pflügers Archiv. Eur. J. Physiol. 2006, 452, 563–572. [Google Scholar] [CrossRef]
- Hampton, A.; Kappler, F.; Picker, D. Species- or isozyme-specific enzyme inhibitors. 4. Design of a two-site inhibitor of adenylate kinase with isozyme selectivity. J. Med. Chem. 1982, 25, 638–644. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Müller, C.E. Nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) and its inhibitors. MedChemComm 2017, 8, 823–840. [Google Scholar] [CrossRef]
- Götz, K.H.; Hacker, S.M.; Mayer, D.; Dürig, J.-N.; Stenger, S.; Marx, A. Inhibitors of the Diadenosine Tetraphosphate Phosphorylase Rv2613c of Mycobacterium tuberculosis. ACS Chem. Biol. 2017, 12, 2682–2689. [Google Scholar] [CrossRef]
- Hacker, S.M.; Mortensen, F.; Scheffner, M.; Marx, A. Selective monitoring of the enzymatic activity of the tumor suppressor Fhit. Angew. Chem. Int. Ed. 2014, 53, 10247–10250. [Google Scholar] [CrossRef] [PubMed]
- Guranowski, A. Specific and nonspecific enzymes involved in the catabolism of mononucleoside and dinucleoside polyphosphates. Pharmacol. Ther. 2000, 87, 117–139. [Google Scholar] [CrossRef]
- Hodgson, D.R. Physicochemical aspects of aqueous and nonaqueous approaches to the preparation of nucleosides, nucleotides and phosphate ester mimics. Adv. Phys. Org. Chem. 2017, 51, 187–219. [Google Scholar]
- Xu, Z. A review on the chemical synthesis of pyrophosphate bonds in bioactive nucleoside diphosphate analogs. Bioorganic Med. Chem. Lett. 2015, 25, 3777–3783. [Google Scholar] [CrossRef]
- Bezold, D.; Dürr, T.; Singh, J.; Jessen, H.J. Cyclotriphosphate: A brief history, recent developments, and perspectives in synthesis. Chem. Eur. J. 2019. [Google Scholar] [CrossRef]
- Jenal, U.; Reinders, A.; Lori, C. Cyclic di-GMP: Second messenger extraordinaire. Nat. Rev. Microbiol. 2017, 15, 271–284. [Google Scholar] [CrossRef]
- Mlynarska-Cieslak, A.; Depaix, A.; Grudzien-Nogalska, E.; Sikorski, P.J.; Warminski, M.; Kiledjian, M.; Jemielity, J.; Kowalska, J. Nicotinamide-containing di- and trinucleotides as chemical tools for studies of NAD-capped RNAs. Org. Lett. 2018, 20, 7650–7655. [Google Scholar] [CrossRef]
- Shanmugasundaram, M.; Senthilvelan, A.; Kore, A.R. Recent advances in synthesis and biological activity of modified cap analogs. Curr. Org. Chem. 2017, 21, 2530–2560. [Google Scholar] [CrossRef]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic mRNA capping. Beilstein J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef]
- Warminski, M.; Sikorski, P.J.; Kowalska, J.; Jemielity, J. Applications of phosphate modification and labeling to study (m)RNA caps. Top. Curr. Chem. 2017, 375, 16. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signalling: Therapeutic developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar]
- Burnstock, G. The therapeutic potential of purinergic signalling. Biochem. Pharmacol. 2018, 151, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, M.; Muller, C.E. Tools and drugs for uracil nucleotide-activated P2Y receptors. Pharmacol. Ther. 2018, 190, 24–80. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A. Structure-Based approaches to ligands for G-protein-coupled adenosine and P2Y Receptors, from small molecules to nanoconjugates. J. Med. Chem. 2013, 56, 3749–3767. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling. Br. J. Pharmacol. 2006, 147, S172–S181. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol. Rev. 2006, 58, 58–86. [Google Scholar] [CrossRef] [PubMed]
- White, N.; Burnstock, G. P2 receptors and cancer. Trends Pharmacol. Sci. 2006, 27, 211–217. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug Discov. 2008, 7, 575–590. [Google Scholar] [CrossRef]
- Erlinge, D. P2Y Receptors in health and disease. Adv. Pharmacol. 2011, 61, 417–439. [Google Scholar]
- Xu, P.; Feng, X.; Luan, H.; Wang, J.; Ge, R.; Li, Z.; Bian, J. Current knowledge on the nucleotide agonists for the P2Y2 receptor. Bioorg. Med. Chem. 2018, 26, 366–375. [Google Scholar] [CrossRef]
- Douglass, J.G.; Patel, R.I.; Yerxa, B.R.; Shaver, S.R.; Watson, P.S.; Bednarski, K.; Plourde, R.; Redick, C.C.; Brubaker, K.; Jones, A.C.; et al. Lipophilic modifications to dinucleoside polyphosphates and nucleotides that confer antagonist properties at the platelet P2Y12 receptor. J. Med. Chem. 2008, 51, 1007–1025. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.H.; Hilderman, R.H.; Pintor, J.J.; Schlüter, H.; King, B.F. Diadenosine polyphosphates as extracellular signal molecules. Drug Dev. Res. 2001, 52, 260–273. [Google Scholar] [CrossRef]
- Carracedo, G.; Crooke, A.; Guzman-Aranguez, A.; De Lara, M.J.P.; Martin-Gil, A.; Pintor, J. The role of dinucleoside polyphosphates on the ocular surface and other eye structures. Prog. Retin. Eye Res. 2016, 55, 182–205. [Google Scholar] [CrossRef]
- Pintor, J.; Díaz-Hernández, M.; Gualix, J.; Gómez-Villafuertes, R.; Hernando, F.; Miras-Portugal, M.T. Diadenosine polyphosphate receptors. from rat and guinea-pig brain to human nervous system. Pharmacol. Ther. 2000, 87, 103–115. [Google Scholar] [CrossRef]
- Lau, O.C.F.; Samarawickrama, C.; Skalicky, S.E. P2Y2 receptor agonists for the treatment of dry eye disease: A review. Clin. Ophthalmol. 2014, 8, 327–334. [Google Scholar]
- Keating, G.M. Diquafosol ophthalmic solution 3%: A review of its use in dry eye. Drugs 2015, 75, 911–922. [Google Scholar] [CrossRef]
- Kellerman, D.; Mospan, A.R.; Engels, J.; Schaberg, A.; Gorden, J.; Smiley, L. Denufosol: A review of studies with inhaled P2Y2 agonists that led to phase 3. Pulm. Pharmacol. Ther. 2008, 21, 600–607. [Google Scholar] [CrossRef]
- Accurso, F.J.; Moss, R.B.; Wilmott, R.W.; Anbar, R.D.; Schaberg, A.E.; Durham, T.A.; Ramsey, B.W. Denufosol tetrasodium in patients with cystic fibrosis and normal to mildly impaired lung function. Am. J. Respir. Crit. Care Med. 2011, 183, 627–634. [Google Scholar] [CrossRef]
- Yerxa, B.R.; Sabater, J.R.; Davis, C.W.; Stutts, M.J.; Lang-Furr, M.; Picher, M.; Jones, A.C.; Cowlen, M.; Dougherty, R.; Boyer, J.; et al. Pharmacology of INS37217 [P1-(Uridine 5′)-P4- (2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J. Pharmacol. Exp. Ther. 2002, 302, 871–880. [Google Scholar] [CrossRef]
- Deterding, R.R.; LaVange, L.M.; Engels, J.M.; Mathews, D.W.; Coquillette, S.J.; Brody, A.S.; Millard, S.P.; Ramsey, B.W. Phase 2 randomized safety and efficacy trial of nebulized denufosol tetrasodium in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 362–369. [Google Scholar] [CrossRef]
- Moss, R.B. Pitfalls of drug development: Lessons learned from trials of denufosol in cystic fibrosis. J. Pediatr. 2013, 162, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Durham, T.; Navratil, T.; Schaberg, A.; Accurso, F.J.; Wainwright, C.; Barnes, M.; Moss, R.B. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Reiss, J.R.; Moffatt, J.G. Dismutation reactions of nucleoside polyphosphates. 3. The synthesis of α,ω-Dinucleoside 5′-polyphosphates. J. Org. Chem. 1965, 30, 3381–3387. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.; Moffatt, J.G. Dismutation reactions of nucleoside polyphosphates. V. Syntheses of P1,P4-Di(guanosine-5′) tetraphosphate and P1,P3-Di(guanosine-5′) triphosphate. J. Am. Chem. Soc. 1966, 88, 838–842. [Google Scholar] [CrossRef]
- Moffatt, J.G.; Khorana, H.G. Nucleoside polyphosphates. X. synthesis and some reactions of nucleoside-5’phosphoromorpholidates and related compounds. Improved methods for the preparation of nucleoside-5’polyphosphates. J. Am. Chem. Soc. 1961, 83, 649–658. [Google Scholar] [CrossRef]
- Millo, E.; Zocchi, E.; Galatini, A.; Benatti, U.; Damonte, G. Simple Synthesis of P1P2-Diadenosine 5′-Pyrophosphate. Synth. Commun. 2008, 38, 3260–3269. [Google Scholar] [CrossRef]
- Shaver, S.R.; Rideout, J.L.; Pendergast, W.; Douglass, J.G.; Brown, E.G.; Boyer, J.L.; Patel, R.I.; Redick, C.C.; Jones, A.C.; Picher, M.; et al. Structure–Activity relationships of dinucleotides: Potent and selective agonists of P2Y receptors. Purinergic Signal. 2005, 1, 183–191. [Google Scholar] [CrossRef]
- Ko, H.; Carter, R.L.; Cosyn, L.; Petrelli, R.; De Castro, S.; Besada, P.; Zhou, Y.; Cappellacci, L.; Franchetti, P.; Grifantini, M.; et al. Synthesis and potency of novel uracil nucleotides and derivatives as P2Y2 and P2Y6 receptor agonists. Bioorganic Med. Chem. 2008, 16, 6319–6332. [Google Scholar] [CrossRef]
- Pendergast, W.; Yerxa, B.R.; Douglass, J.G.; Shaver, S.R.; Dougherty, R.W.; Redick, C.C.; Sims, I.F.; Rideout, J.L. Synthesis and P2Y receptor activity of a series of uridine dinucleoside 5′-polyphosphates. Bioorganic Med. Chem. Lett. 2001, 11, 157–160. [Google Scholar] [CrossRef]
- Stern, N.; Major, D.T.; Gottlieb, H.E.; Weizman, D.; Fischer, B. What is the conformation of physiologically-active dinucleoside polyphosphates in solution? Conformational analysis of free dinucleoside polyphosphates by NMR and molecular dynamics simulations. Org. Biomol. Chem. 2010, 8, 4637–4652. [Google Scholar] [CrossRef]
- Liu, K.K.-C.; Sakya, S.M.; O’Donnell, C.J.; Flick, A.C.; Ding, H.X. Synthetic approaches to the 2010 new drugs. Bioorganic Med. Chem. 2012, 20, 1155–1174. [Google Scholar] [CrossRef] [PubMed]
- Zatorski, A.; Goldstein, B.M.; Colby, T.D.; Jones, J.P.; Pankiewicz, K.W. Potent inhibitors of human inosine monophosphate dehydrogenase type II. Fluorine-substituted analogues of thiazole-4-carboxamide adenine dinucleotide. J. Med. Chem 1995, 38, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Rejman, D.; Bonnac, L.; Pankiewicz, K.W.; Patterson, S.E. Nucleoside-5′-phosphoimidazolides: Reagents for facile synthesis of dinucleoside pyrophosphates. Curr. Protoc. Nucleic Acid Chem. 2006, 23, 13.4.1–13.4.10. [Google Scholar] [CrossRef] [PubMed]
- Yanachkov, I.B.; Dix, E.J.; Yanachkova, M.I.; Wright, G.E. P1, P2-diimidazolyl derivatives of pyrophosphate and bis-phosphonates - synthesis, properties, and use in preparation of dinucleoside tetraphosphates and analogs. Org. Biomol. Chem. 2011, 9, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Depaix, A.; Peyrottes, S.; Roy, B. Water-Medium synthesis of nucleoside 5′-polyphosphates. Curr. Protoc. Nucleic Acid Chem. 2017, 69, 13.16.1–13.16.11. [Google Scholar]
- Depaix, A.; Peyrottes, S.; Roy, B. One-Pot synthesis of nucleotides and conjugates in aqueous medium. Eur. J. Org. Chem. 2017, 2, 241–245. [Google Scholar] [CrossRef]
- Smith, M.; Khorana, H.G. Nucleoside Polyphosphates. VI. 1 An improved and general method for the synthesis of Ribo-and Deoxyribonucleoside 5’-Triphosphates. J. Am. Chem. Soc. 1958, 80, 1141–1145. [Google Scholar] [CrossRef]
- Ng, K.-M.E.; Orgel, L.E. The action of a water-soluble carbodiimide on adenosine-5′-polyphosphates. Nucleic Acids Res. 1987, 15, 3573–3580. [Google Scholar] [CrossRef][Green Version]
- Mohamady, S.; Taylor, S.D. Synthesis of nucleoside tetraphosphates and dinucleoside pentaphosphates via activation of cyclic trimetaphosphate. Org. Lett. 2013, 15, 2612–2615. [Google Scholar] [CrossRef]
- Mohamady, S.; Desoky, A.; Taylor, S.D. Sulfonyl imidazolium salts as reagents for the rapid and efficient synthesis of nucleoside polyphosphates and their conjugates. Org. Lett. 2012, 14, 402–405. [Google Scholar] [CrossRef]
- Mohsen, M.G.; Ji, D.; Kool, E.T. Polymerase synthesis of four-base DNA from two stable dimeric nucleotides. Nucleic Acids Res. 2019, 47, 9495–9501. [Google Scholar] [CrossRef] [PubMed]
- Kanavarioti, A.; Lu, J.; Rosenbach, M.T.; Hurley, T.B. Unexpectedly Facile Synthesis of Symmetrical P1,P2-Dinucleoside-5′pyrophosphates. Tetrahedron Lett. 1991, 32, 6065–6068. [Google Scholar] [CrossRef]
- Bogachev, V.S. Synthesis of deoxynucleoside 5′-triphosphates using trifluoroacetic anhydride as an activating reagent. Bioorg. Khim 1996, 22, 699–705. [Google Scholar]
- Mohamady, S.; Taylor, S.D. General Procedure for the Synthesis of Dinucleoside Polyphosphates. J. Org. Chem. 2011, 76, 6344–6349. [Google Scholar] [CrossRef]
- Mohamady, S.; Taylor, S.D. Rapid and efficient synthesis of nucleoside polyphosphates and their conjugates using sulfonyl imidazolium salts. Curr. Protoc. Nucleic Acid Chem. 2012, 51, 13.11.1–13.11.24. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, S.S.; Sun, J.; Wang, C.J.; Liu, S.; Liu, G.D.; Ma, C. Efficient synthesis of nucleoside 5′-triphosphates and their β,γ-bridging oxygen-modified analogs from nucleoside 5′-phosphates. Tetrahedron Lett. 2014, 55, 2114–2118. [Google Scholar] [CrossRef]
- Sun, Q.; Gong, S.-S.; Liu, S.; Sun, J.; Liu, G.-D.; Ma, C. 4,5-Dicyanoimidazole-promoted synthesis of dinucleoside polyphosphates and their analogs. Tetrahedron 2014, 70, 4500–4506. [Google Scholar] [CrossRef]
- Sun, Q.; Sun, J.; Gong, S.-S.; Wang, C.-J.; Wang, X.-C. Synthesis of nucleoside tetraphosphates and dinucleoside pentaphosphates from nucleoside phosphoropiperidates via the activation of P(V)-N bond. Chin. Chem. Lett. 2015, 26, 89–92. [Google Scholar] [CrossRef]
- Sun, Q.; Sun, J.; Gong, S.-S.; Wang, C.-J.; Wang, X.-C. One-pot synthesis of symmetrical dinucleoside polyphosphates and analogs via 4,5-dicyanoimidazole-promoted tandem P–O coupling reactions. Tetrahedron Lett. 2014, 55, 5785–5788. [Google Scholar] [CrossRef]
- Ludwig, J.; Eckstein, F. Rapid and efficient synthesis of nucleoside 5′-O-(1-thiotriphosphates), 5′-triphosphates, and 2′,3′-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one. J. Org. Chem. 1989, 54, 631–635. [Google Scholar] [CrossRef]
- Han, Q.; Gaffney, B.L.; Jones, R.A. One-Flask synthesis of dinucleoside tetra- and pentaphosphates. Org. Lett. 2006, 8, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Meier, C. cycloSal-pronucleotides design of chemical trojan horses. Mini-Rev. Med. Chem. 2002, 2, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Meier, C. cycloSal Phosphates as chemical trojan horses for intracellular nucleotide and glycosylmonophosphate delivery — Chemistry meets biology. Eur. J. Org. Chem. 2006, 2006, 1081–1102. [Google Scholar] [CrossRef]
- Warnecke, S.; Meier, C. Synthesis of Nucleoside Di- and Triphosphates and Dinucleoside Polyphosphates with cycloSal-Nucleotides. J. Org. Chem. 2009, 74, 3024–3030. [Google Scholar] [CrossRef]
- Wendicke, S.; Warnecke, S.; Meier, C. Efficient Synthesis of Nucleoside Diphosphate Glycopyranoses. Angew. Chem. Int. Ed. 2008, 47, 1500–1502. [Google Scholar] [CrossRef]
- Tonn, V.C.; Meier, C. Solid-Phase synthesis of (poly)phosphorylated nucleosides and conjugates. Chem. Eur. J. 2011, 17, 9832–9842. [Google Scholar] [CrossRef]
- Ahmadibeni, Y.; Parang, K. Solid-Phase Synthesis of Symmetrical 5‘,5‘-Dinucleoside Mono-, Di-, Tri-, and Tetraphosphodiesters. Org. Lett. 2007, 9, 4483–4486. [Google Scholar] [CrossRef]
- Kistemaker, H.A.V.; Meeuwenoord, N.J.; Overkleeft, H.S.; Van Der Marel, G.A.; Filippov, D.V. On the synthesis of oligonucleotides interconnected through pyrophosphate linkages. Eur. J. Org. Chem. 2015, 2015, 6084–6091. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, S.; Sun, J.; Gong, S.; Xiao, Q.; Shen, L. One-pot synthesis of symmetrical P1,P2-dinucleoside-5′-diphosphates from nucleoside-5′-H-phosphonates: Mechanistic insights into reaction path. Tetrahedron Lett. 2013, 54, 3842–3845. [Google Scholar] [CrossRef]
- Sun, Q.; Edathil, J.P.; Wu, R.; Smidansky, E.D.; Cameron, C.E.; Peterson, B.R. One-Pot Synthesis of Nucleoside 5′-Triphosphates from Nucleoside 5′-H-Phosphonates. Org. Lett. 2008, 10, 1703–1706. [Google Scholar] [CrossRef][Green Version]
- Jessen, H.J.; Ahmed, N.; Hofer, A. Phosphate esters and anhydride—recent strategies targeting nature’s favoured modifications. Org. Biomol. Chem. 2014, 12, 3526–3530. [Google Scholar] [CrossRef] [PubMed]
- Cremosnik, G.S.; Hofer, A.; Jessen, H.J. Iterative synthesis of nucleoside oligophosphates with phosphoramidites. Angew. Chem. Int. Ed. 2014, 53, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.; Cremosnik, G.S.; Müller, A.C.; Giambruno, R.; Trefzer, C.; Superti-Furga, G.; Jessen, H.J. Modular synthesis of modified phosphoanhydrides. Chem. Eur. J. 2015, 21, 10116–10122. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.; Marques, E.; Kieliger, N.; Gatter, S.-K.N.; Jordi, S.; Ferrari, E.; Hofmann, M.; Fitzpatrick, T.B.; Hottiger, M.O.; Jessen, H.J. Chemoselective dimerization of phosphates. Org. Lett. 2016, 18, 3222–3225. [Google Scholar] [CrossRef]
- Tanaka, H.; Yoshimura, Y.; Jørgensen, M.R.; Cuesta-Seijo, J.A.; Hindsgaul, O. A simple synthesis of sugar nucleoside diphosphates by chemical coupling in water. Angew. Chem. Int. Ed. 2012, 51, 11531–11534. [Google Scholar] [CrossRef]
- Eguaogie, O.; Vyle, J.S.; Conlon, P.F.; Gîlea, M.A.; Liang, Y. Mechanochemistry of nucleosides, nucleotides and related materials. Beilstein J. Org. Chem. 2018, 14, 955–970. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef]
- Bowmaker, G.A. Solvent-assisted mechanochemistry. Chem. Commun. 2013, 49, 334–348. [Google Scholar] [CrossRef]
- Takacs, L. The historical development of mechanochemistry. Chem. Soc. Rev. 2013, 42, 7649–7659. [Google Scholar] [CrossRef]
- Wang, G.W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Friščić, T.; Mottillo, C.; Titi, H.M. Mechanochemistry for synthesis. Angew. Chem. Int. Ed. Engl. 2019, in press. [Google Scholar]
- Ravalico, F.; Messina, I.; Berberian, M.V.; James, S.L.; Migaud, M.E.; Vyle, J.S. Rapid synthesis of nucleotide pyrophosphate linkages in a ball mill. Org. Biomol. Chem. 2011, 9, 6496–6497. [Google Scholar] [CrossRef] [PubMed]
- Appy, L.; Depaix, A.; Bantreil, X.; Lamaty, F.; Peyrottes, S.; Roy, B. Straightforward ball-milling access to dinucleoside 5′,5′-polyphosphates via phosphorimidazolide intermediates. Chem. Eur. J. 2019, 25, 2477–2481. [Google Scholar] [PubMed]
- Verma, S.K.; Ghorpade, R.; Pratap, A.; Kaushik, M. Solvent free, N,N′-carbonyldiimidazole (CDI) mediated amidation. Tetrahedron Lett. 2012, 53, 2373–2376. [Google Scholar] [CrossRef]
- Metro, T.-X.; Bonnamour, J.; Reidon, T.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Mechanosynthesis of amides in the total absence of organic solvent from reaction to product recovery. Chem. Commun. 2012, 48, 11781–11793. [Google Scholar] [CrossRef]
- Métro, T.-X.; Bonnamour, J.; Reidon, T.; Duprez, A.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Comprehensive study of the organic-solvent-free CDI-Mediated acylation of various nucleophiles by mechanochemistry. Chem. Eur. J. 2015, 21, 12787–12796. [Google Scholar] [CrossRef]
- Métro, T.-X.; Martinez, J.; Lamaty, F. 1,1′-Carbonyldiimidazole and mechanochemistry: A shining green combination. ACS Sustain. Chem. Eng. 2017, 5, 9599–9602. [Google Scholar] [CrossRef]
- Banoub, J.H.; Newton, R.P.; Esmans, E.; Ewing, D.F.; Mackenzie, G. Recent developments in mass spectrometry for the characterization of nucleosides, nucleotides, oligonucleotides, and nucleic acids. Chem. Rev. 2005, 105, 1869–1915. [Google Scholar] [CrossRef]
- Watanabe, D.; Ishikawa, M.; Yamasaki, M.; Ozaki, M.; Katayama, T.; Nakajima, H. Tetrasodium P1,P4-Bis(5′-adenosyl)tetraphosphate Dodecahydrate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, 52, 338–340. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Savithri, H.S.; Murthy, A.R.N. Crystal structures of Salmonella typhimurium propionate kinase and its complex with Ap4A: Evidence for a novel Ap4A synthetic activity. Proteins 2008, 70, 1379–1388. [Google Scholar] [CrossRef]
- Baker, M.D.; Holloway, D.E.; Swaminathan, G.J.; Acharya, K.R. Crystal structures of eosinophil-derived neurotoxin (EDN) in complex with the inhibitors 5‘-ATP, Ap3A, Ap4A, and Ap5A. Biochemistry 2006, 45, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Kliche, W.; Wiesmuller, L.; Reinstein, J. Crystal structure of the complex of UMP/CMP kinase from Dictyostelium discoideum and the bisubstrate inhibitor P1-(5′-adenosyl) P5-(5′-uridyl) pentaphosphate (UP5A) and Mg2+ at 2.2 A: Implications for water-mediated specificity. Biochemistry 1996, 35, 9716–9727. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.B.; Phillips, G.N., Jr. Crystal structures of Bacillus stearothermophilus adenylate kinase with bound Ap5A, Mg2+ Ap5A, and Mn2+ Ap5A reveal an intermediate lid position and six coordinate octahedral geometry for bound Mg2+ and Mn2+. Proteins 1998, 32, 276–288. [Google Scholar] [CrossRef]
- Chen, J.; Kohler, B. Base stacking in adenosine dimers revealed by femtosecond transient absorption spectroscopy. J. Am. Chem. Soc. 2014, 136, 6362–6372. [Google Scholar] [CrossRef] [PubMed]






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
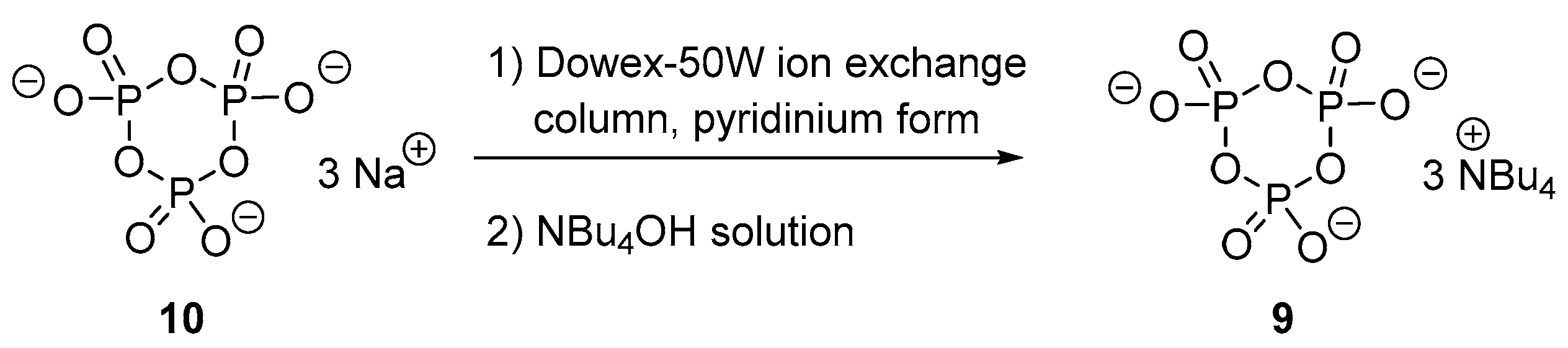{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Chromatography | Eluants | References |
|---|---|---|
| Anion-exchange | ||
| DEAE-Sephadex® | 0.02 M TEAB, pH 8; 10% ACN | [54] |
| TOYOPEARL® DEAE-650M | 0.2 M TEAB, pH 8; 10% ACN | [54] |
| DEAE-Sephadex® | Gradient of aq. NH4HCO3 | [67,69] |
| Q Sepharose® Fast Flow | Gradient of aq. NH4HCO3 | [83,84] |
| DEAE Sepharose® Fast Flow | Gradient of aq. NH4HCO3 | [83,84] |
| C-18 reverse-phase | ||
| HPLC | 0.01 M NH4HCO2, pH 4, 50% MeOH | [46] |
| Semi-preparative HPLC | TEAAc pH 7 or 9; 6–12% ACN | [59,60,64] |
| Preparative HPLC | 0.1 M NH4HCO3; ACN | [71] |
| Preparative HPLC | TEAB pH 7.8; 65% ACN | [92] |
| RP-18 silica gel column | Water | [74] |
| RP-18 silica gel column | 0.1 M TEAB, pH 7.5; 20% ACN | [55,56] |
| RP-18 silica gel column | 0.1 M TEAAc, pH 7; 5–10% ACN | [93] |
| Gel filtration | ||
| Sephadex® LH-20 | Deionized water | [79] |
| NpnN | δ Ranges | References |
|---|---|---|
| Np2N’ | 1 singlet (P1 + P2) from −9.6 to −11.5 ppm | [47,49,50,53,56,60,64,67,74,76,77,79,83,93] |
| Np3N | 1 doublet (P1 + P3) from −9.2 to −11.7 ppm 1 triplet (P2) from −21.0 to −23.4 ppm | [47,49,50,54,60,64,67,69,84,93] |
| Np4N | 1 multiplet or 1 broad singlet or 1 doublet (P1 + P4) from −8.8 to −11.5 ppm | [47,49,50,54,60,64,67,69,71,74,93] |
| 1 multiplet or 1 br singlet or 1 doublet (P2 + P3) from −20.6 to −23.1 ppm | ||
| Np5N | 1 multiplet or 1 br singlet or 1 doublet (P1 + P5) from −8.7 to −11.4 ppm | [49,50,59,69,71,84] |
| 1 multiplet or 1 br singlet or 1 doublet (P2 + P3 + P4) from −20.2 to −22.5 ppm | ||
| Np6N | 1 doublet at −10.1 ppm (P1 + P6) 1 multiplet at −21.7 ppm (P 2+ P3 + P4 + P5) | [49] |
| Np7N | 1 broad singlet or 1 doublet from −10.1 to −11.6 ppm (P1 + P7) | [49,84] |
| 1 multiplet from −21.7 to −23.5 ppm (P2 + P3 + P4 + P5 + P6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Appy, L.; Chardet, C.; Peyrottes, S.; Roy, B. Synthetic Strategies for Dinucleotides Synthesis. Molecules 2019, 24, 4334. https://doi.org/10.3390/molecules24234334
Appy L, Chardet C, Peyrottes S, Roy B. Synthetic Strategies for Dinucleotides Synthesis. Molecules. 2019; 24(23):4334. https://doi.org/10.3390/molecules24234334
Chicago/Turabian StyleAppy, Lucie, Crystalle Chardet, Suzanne Peyrottes, and Béatrice Roy. 2019. "Synthetic Strategies for Dinucleotides Synthesis" Molecules 24, no. 23: 4334. https://doi.org/10.3390/molecules24234334
APA StyleAppy, L., Chardet, C., Peyrottes, S., & Roy, B. (2019). Synthetic Strategies for Dinucleotides Synthesis. Molecules, 24(23), 4334. https://doi.org/10.3390/molecules24234334