
Energetic Butterfly: Heat-Resistant Diaminodinitro trans-Bimane

 , ,
, ,
Abstract
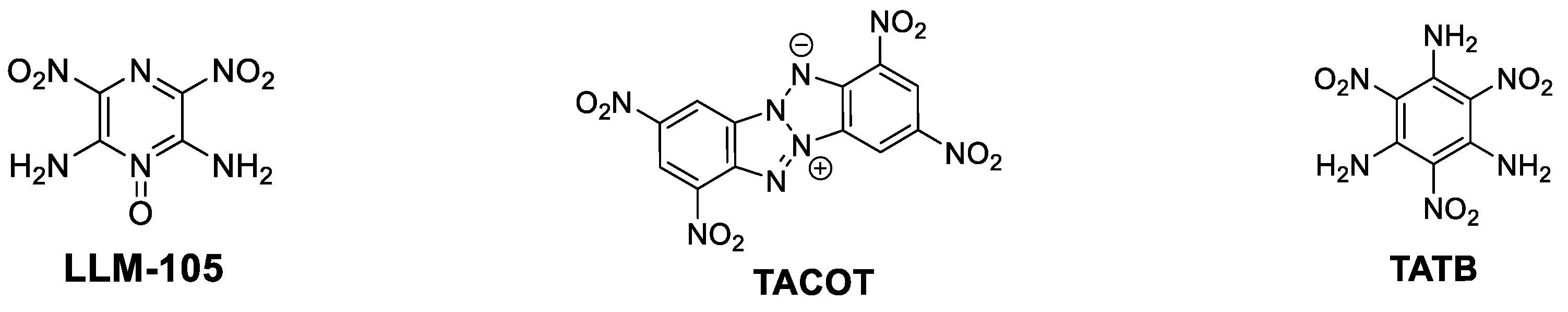
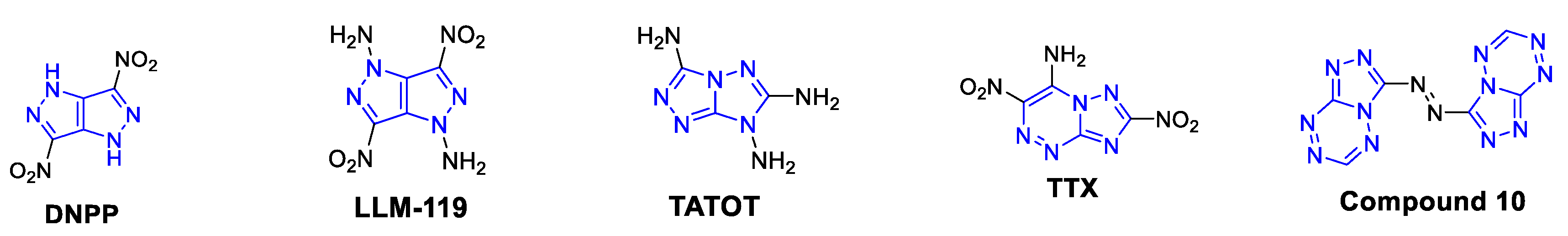
:1. Introduction
2. Results and Discussion
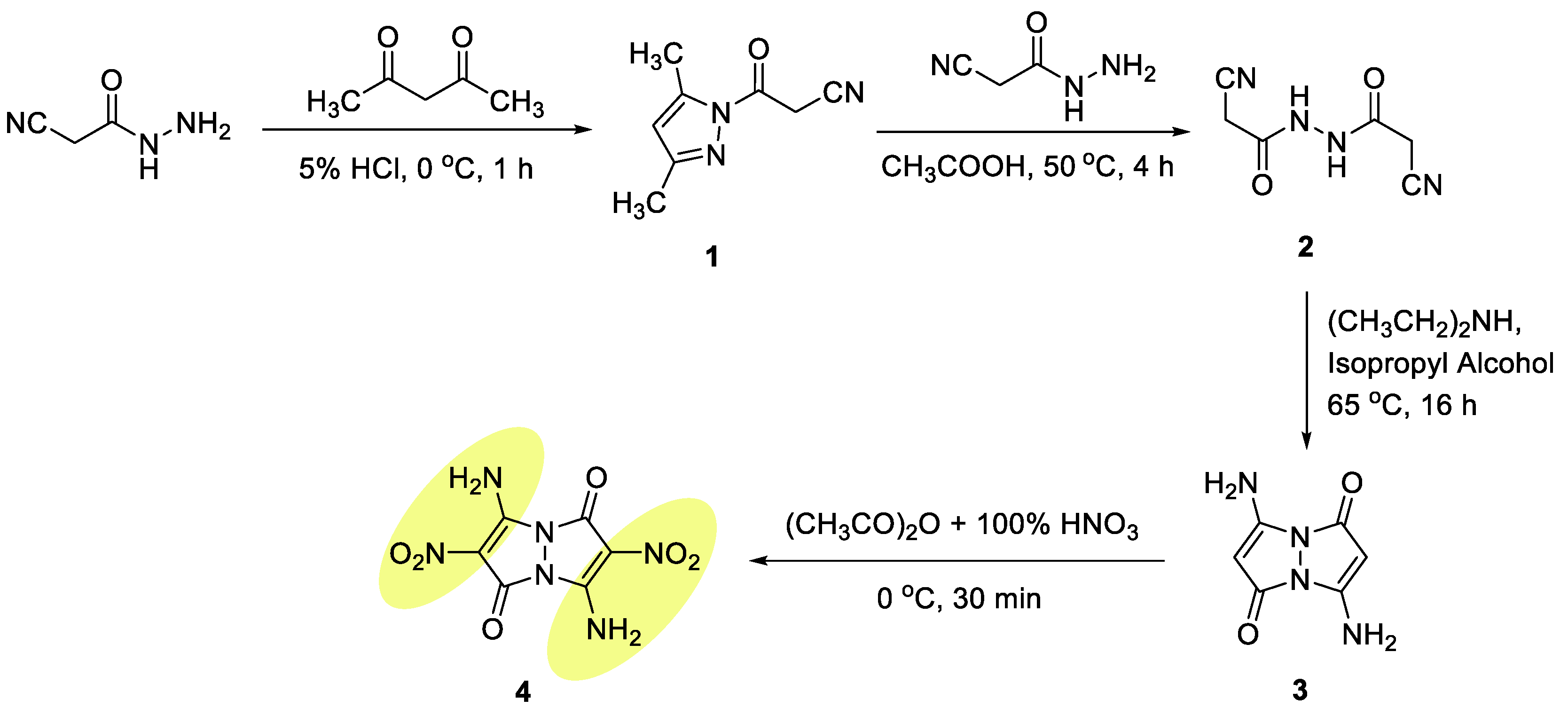
2.1. Synthesis
2.2. Spectral Studies
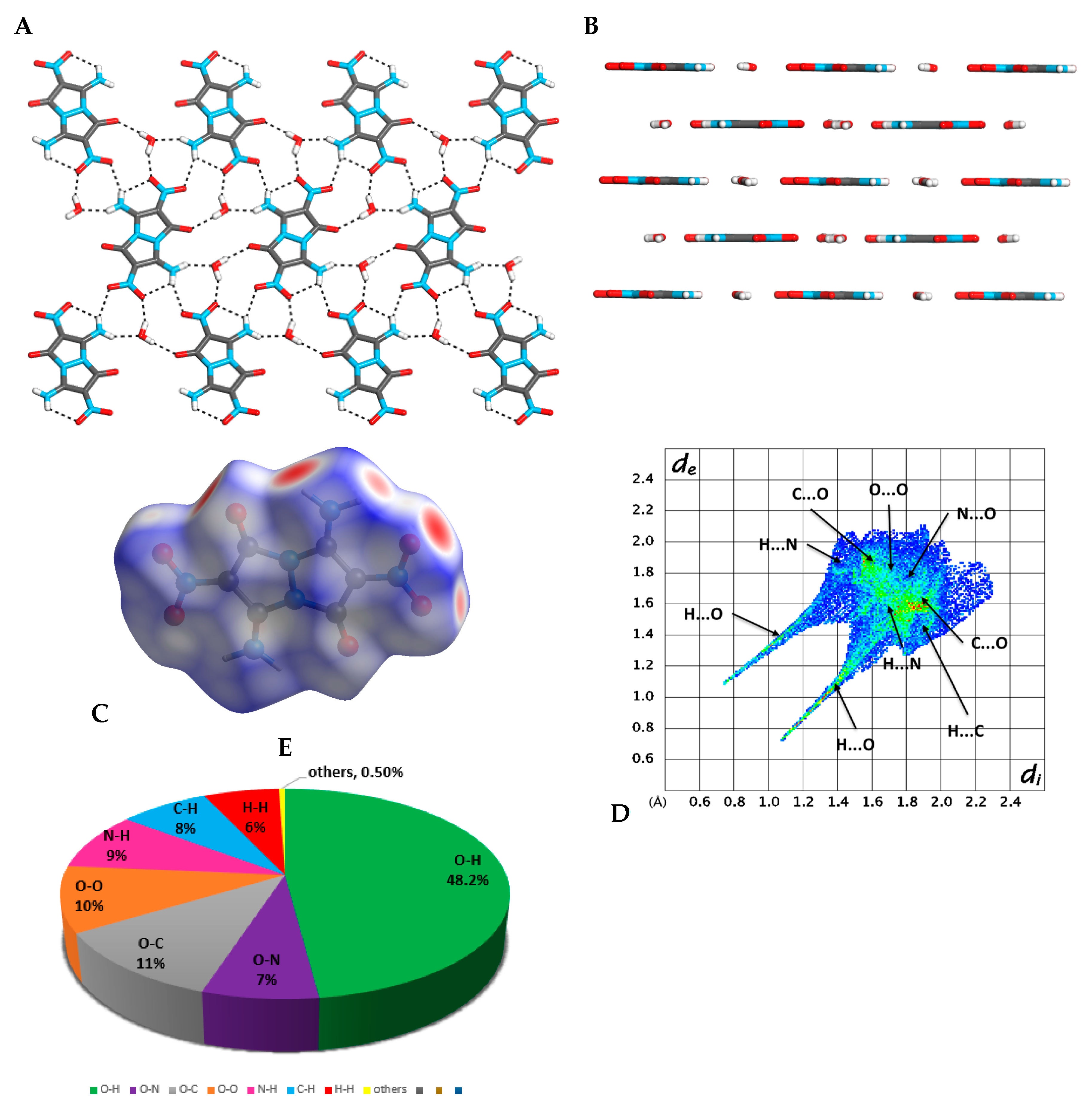
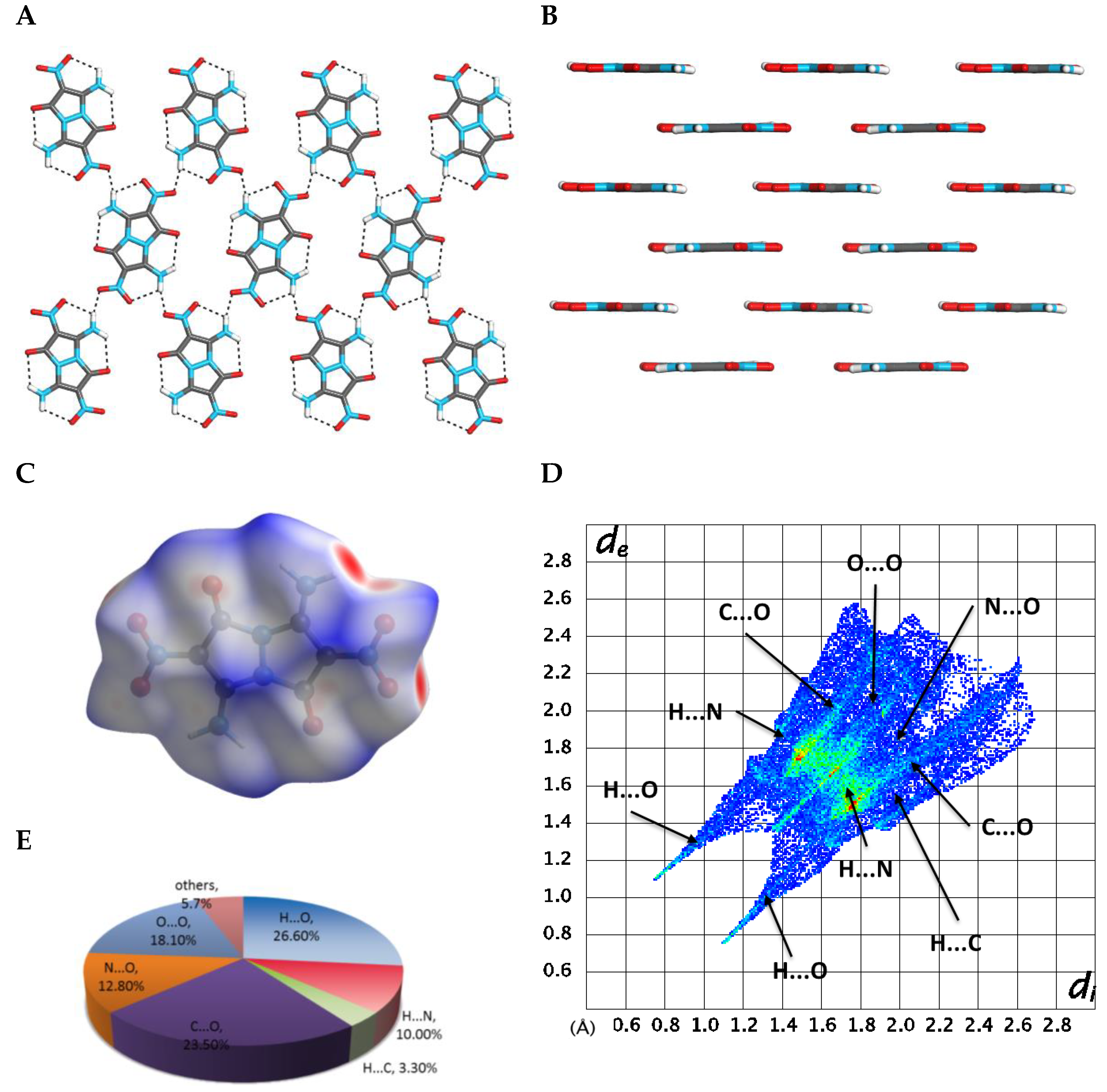
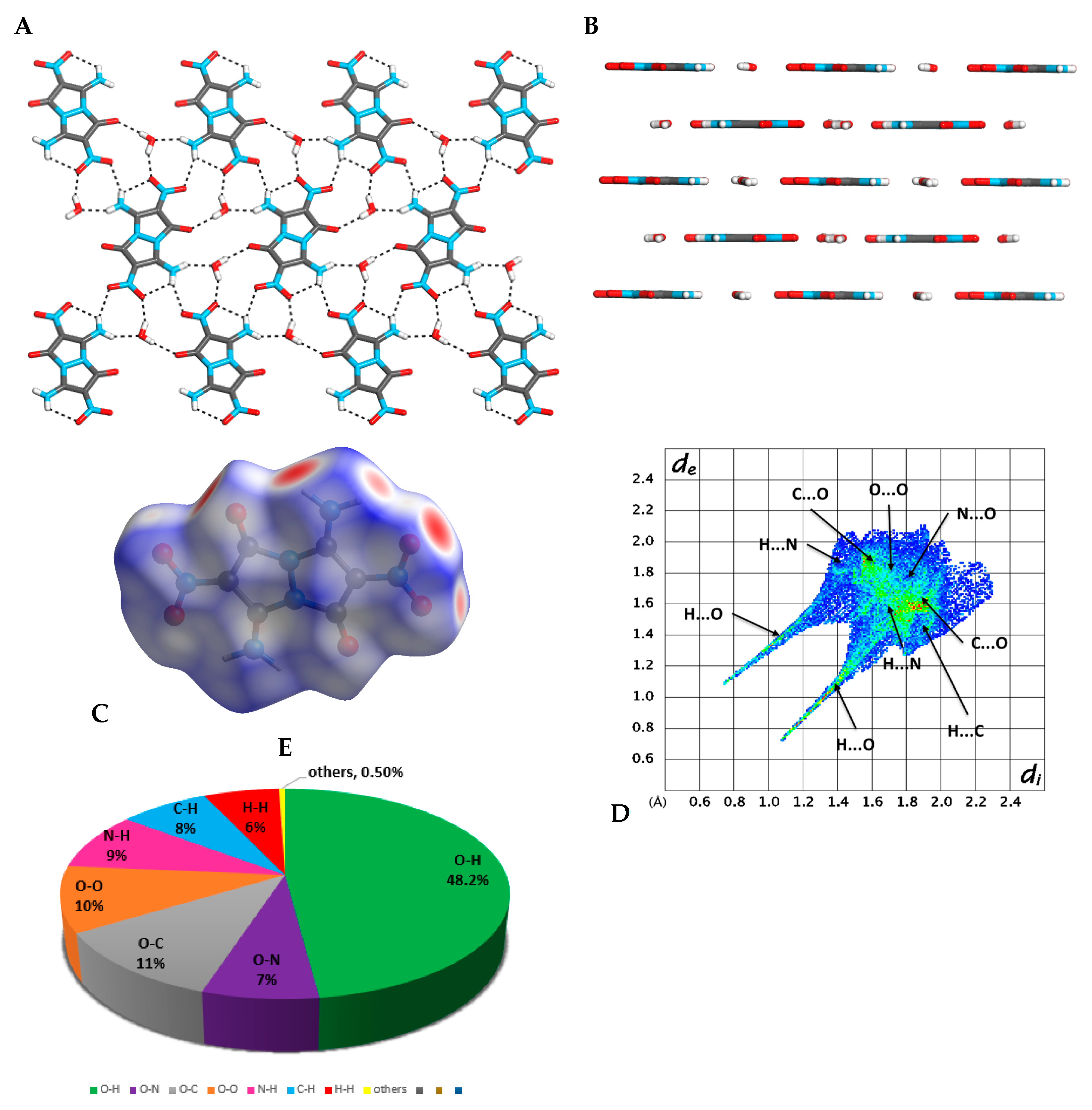
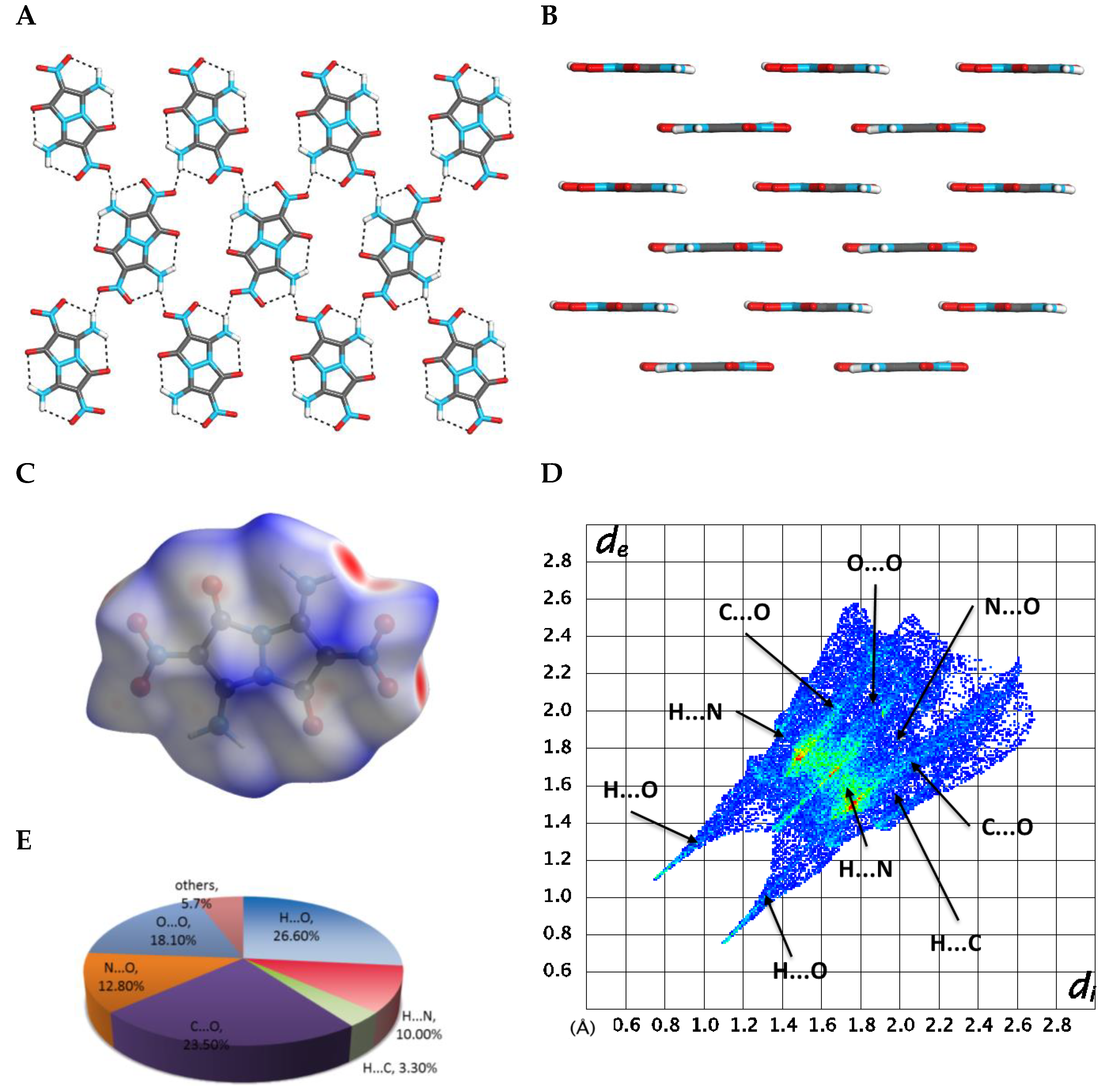
2.3. X-ray Crystallography
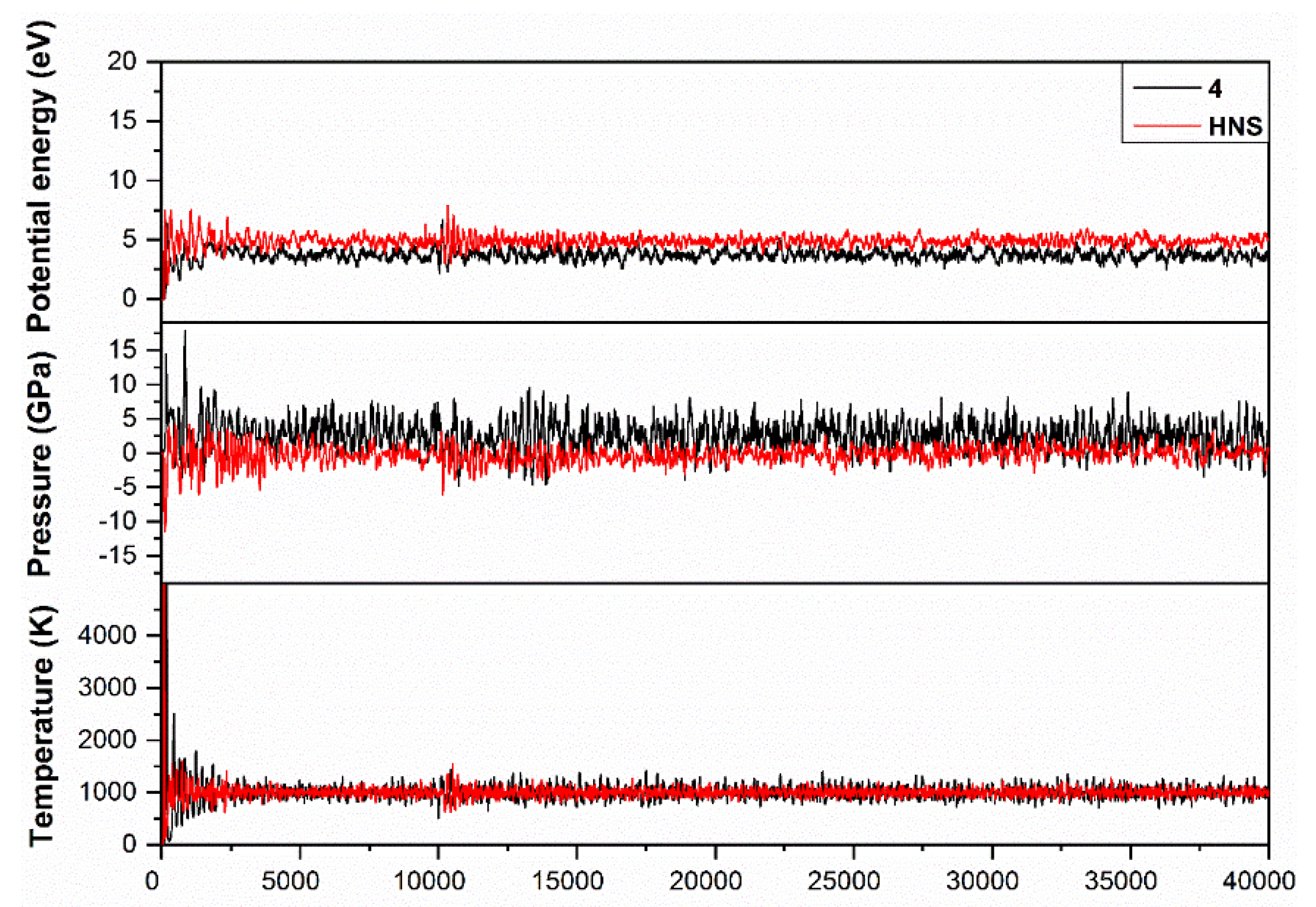
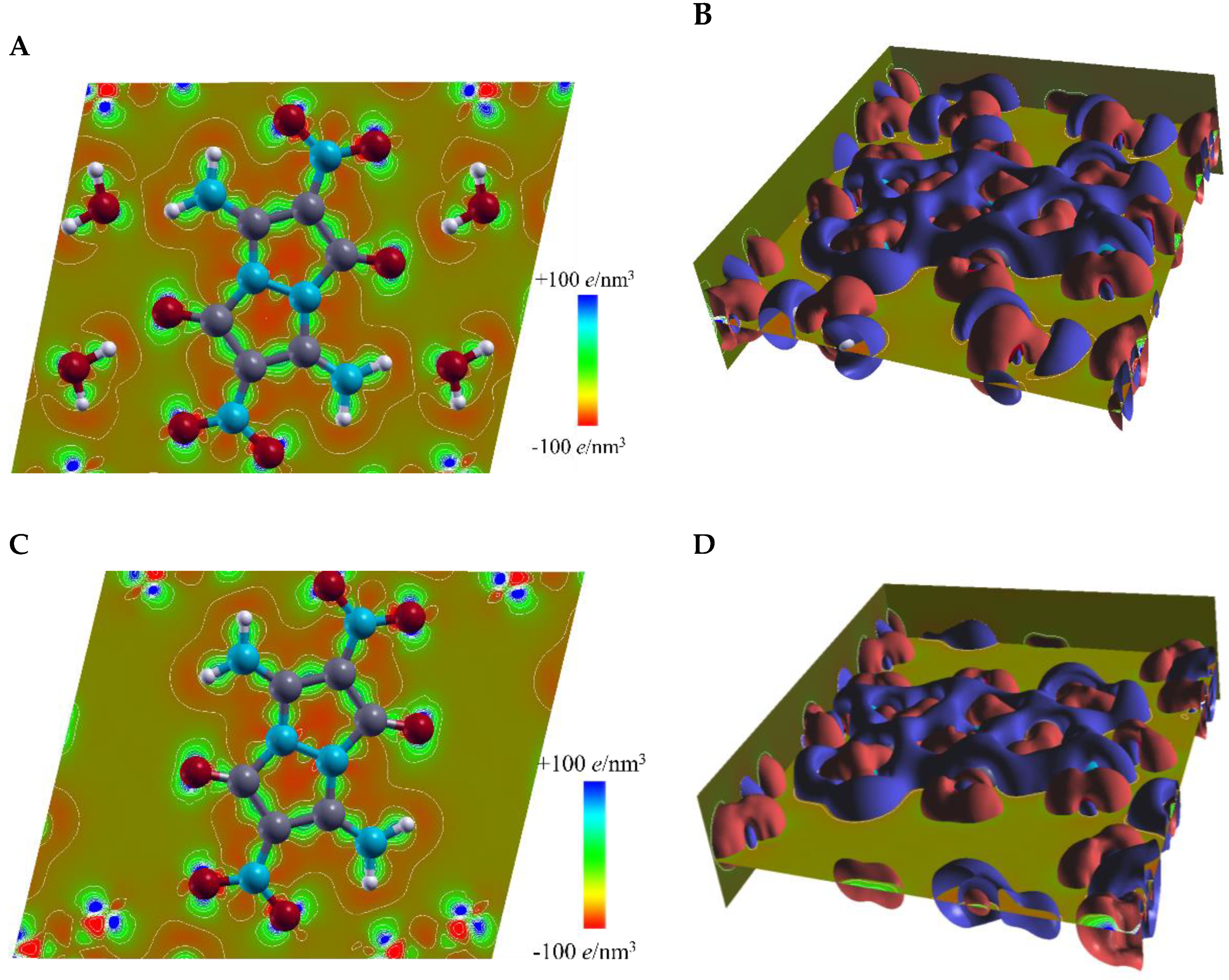
2.4. DFT Structure Calculations
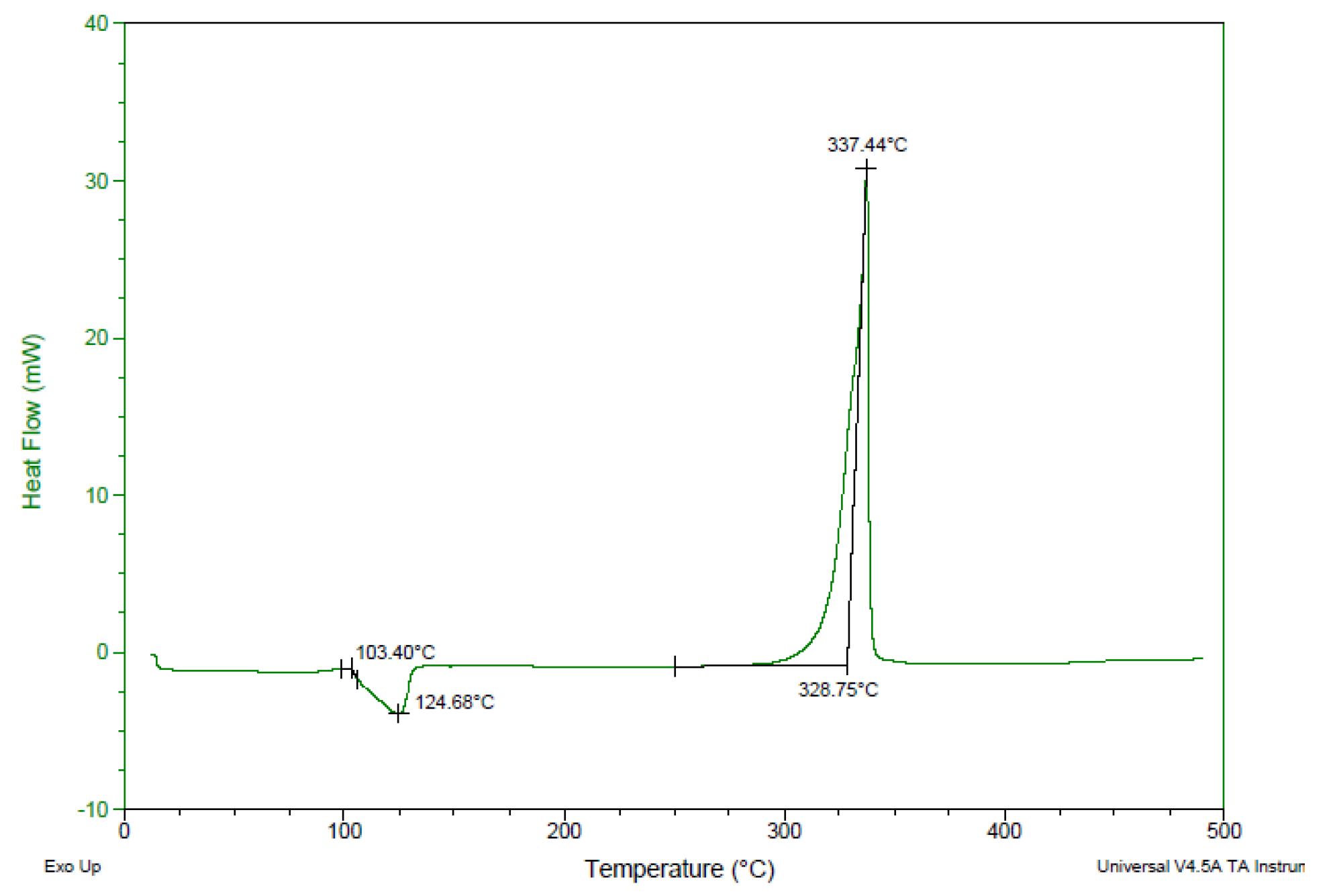
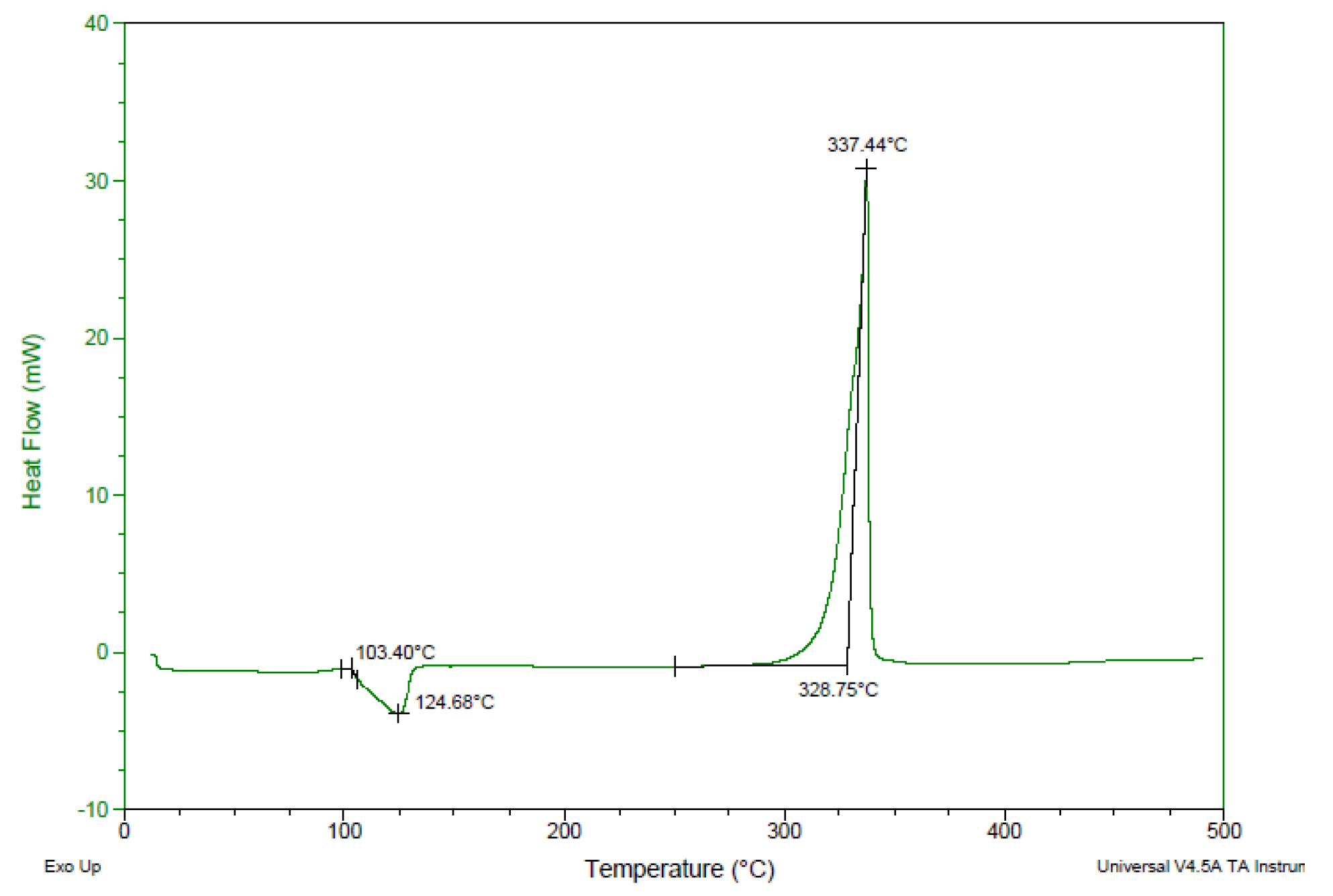
2.5. Physiochemical and Energetic Properties
3. Materials and Methods
General Information
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kilmer, E.E. Heat-resistant explosives for space applications. J. Spacecr. Rocket. 1968, 5, 1216–1219. [Google Scholar] [CrossRef]
- Tang, Y.; He, C.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. Aminonitro Groups Surrounding a Fused Pyrazolotriazine Ring: A Superior Thermally Stable and Insensitive Energetic Material. ACS Appl. Energy Mater. 2019, 2, 32263–32267. [Google Scholar] [CrossRef]
- Brauer, K. Present and Future Applications of Pyrotechnic Devices and Pyrotechnic Systems for Spacecraft. In Proceedings of the 19th Congress of the International Astronautical Federation, New York, NY, USA, 13–18 October 1968. [Google Scholar]
- Abdel-Aal, H.K.; Aggour, M.A.; Fahim, M.A. Petroleum and Gas Field Processing, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- James, H.; Barker, M. Thermally Stable Explosive System for Ultra-High-Temperature Perforating. In Proceedings of the SPE Annual Technical Conference and Exhibition, New Orleans, LA, USA, 30 September–2 October 2013. [Google Scholar]
- Zhang, L.; Wu, J.-Z.; Jiang, S.-L.; Yu, Y.; Chen, J. From intermolecular interactions to structures and properties of a novel cocrystal explosive: A first-principles study. Phys. Chem. Chem. Phys. 2016, 18, 26960–26969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jiao, F.; Li, H. Crystal Engineering for Creating Low Sensitivity and Highly Energetic Materials. Cryst. Growth Des. 2018, 18, 5713–5726. [Google Scholar] [CrossRef]
- Pagoria, P.; Zhang, M.; Zuckerman, N.; Lee, G.; Mitchell, A.; DeHope, A.; Gash, A.; Coon, C.; Gallagher, P. Synthetic Studies of 2,6-Diamino-3,5-Dinitropyrazine- 1-Oxide (LLM-105) from Discovery to Multi-Kilogram Scale. Propellants Explos. Pyrotech. 2017, 42, 1–14. [Google Scholar] [CrossRef]
- Subramanian, G.; Boyer, J.H.; Buzatu, D.; Stevens, E.D.; Trudell, M.L. Reactions of Benzotriazolo[2,l-α] benzotriazole Derivatives. 1.Synthesis of New Insensitive High-Density Energetic Compounds. J. Org. Chem. 1995, 60, 6110–6113. [Google Scholar] [CrossRef]
- Mitchell, A.R.; Pagoria, P.F.; Schmidt, R.D.; Coburn, M.D.; Lee, G.S.; Hsu, P.C. A Versatile Synthesis of 1,3,5-Triamino-2,4,6-Trinitrobenzene (TATB). In Proceedings of the 37th International Institute of Chemical Technology (ICT) Conference, Karlsruhe, Germany, 27–30 June 2006. [Google Scholar]
- Shevelev, S.A.; Dalinger, I.L.; Shkineva, T.K.; Ugrak, B.I.; Gulevskaya, V.I.; Kanishev, M.I. Nitropyrazoles. 1. Synthesis, conversion, and physicochemical properties of nitro derivatives of 1H,4H-pyrazolo[4,3-c]pyrazole. Izv. Akad. Nauk Seriya Khimicheskaya 1993, 6, 1108–1113. [Google Scholar]
- Pagoria, P.F.; Lee, G.S.; Mitchell, A.R.; Schmidt, R.D. A review of energetic materials synthesis. Thermochim. Acta 2002, 384, 187–204. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Schmid, P.C.; Schnell, S.; Stierstorfer, J. 3,6,7-Triamino-[1,2,4]triazolo[4,3-b][1,2,4]triazole: A Non-toxic, High-Performance Energetic Building Block with Excellent Stability. Chem. Eur. J. 2015, 21, 9219–9228. [Google Scholar] [CrossRef]
- Dheeraj, K.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. A Highly Stable and Insensitive Fused Triazolo-Triazine Explosive (TTX). Chem. Eur. J. 2017, 23, 1743–1747. [Google Scholar]
- Liu, Y.; Zhao, G.; Tang, Y.; Zhang, J.; Hu, L.; Imler, G.H.; Parrishc, D.A.; Shreeve, J.M. Multipurpose [1,2,4]triazolo[4,3-b][1,2,4,5] tetrazine-based energetic materials. J. Mater. Chem. A 2019, 7, 7875–7884. [Google Scholar] [CrossRef]
- Lau, C.M.; Thangaraj, K.; Kumar, G.; Ramakrishnan, V.T.; Stevens, E.D.; Boyer, J.H.; Politzer, I.R.; Pavlopoulos, T.G. Bimanes (1,5-Diazabicyclo[3.3.0]octadienediones): Laser Activity in syn-Bimanes. Heteroat. Chem. 1990, 1, 195–209. [Google Scholar] [CrossRef]
- Iraji, A.; Firuzi, O.; Khoshneviszadeh, M.; Tavakkoli, M.; Mahdavi, M.; Nadri, H.; Edraki, N.; Miri, R. Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur. J. Med. Chem. 2017, 141, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Schoenafinger, K. 1,5-Diaminopyrazolo[1,2-a]pyrazole-3,7-dione. Ger. Offen. Patent DE Patent No.: 2855193, 1980. [Google Scholar]
- Li, J.; Wang, S.; Liao, L.; Ma, Q.; Zhang, Z.; Fan, G. Stabilization of an intramolecular hydrogen-bond block in an s-triazine insensitive high-energy material. New J. Chem. 2019, 43, 10675–10679. [Google Scholar] [CrossRef]
- Hestenes, M.R.; Stiefel, E. Methods of Conjugate Gradients for Solving Linear Systems. J. Res. Natl. Bur. Stand. 1952, 49, 409–436. [Google Scholar] [CrossRef]
- Akst, I.B. Heat of detonation, the cylinder test, and performance munitions. In Proceedings of the 9th International Symposium on Detonation, Portland, OR, USA, 28 August–1 September 1989. [Google Scholar]
- Department of the Army Technical Manual. TM 9-1300-214 MILITARY EXPLOSIVES; Department of the Army Headquarters: Washington, DC, USA, 1984; p. 145.
- Politzer, P. Murray, J.S. Some Perspectives on Estimating Detonation Properties of C, H, N, O Compounds. Cent. Eur. J. Energ. Mater. 2011, 8, 209–220. [Google Scholar]
- Hobbs, M.L.; Baer, M.R. Calibration of the BKW-EOS with a Large Product Species Data Base and Measured CJ Properties. In Proceedings of the 10th Symposium (International) on Detonation, Boston, MA, USA, 12–16 July 1993. [Google Scholar]
- Gospodinov, I.; Klapötke, T.M.; Stierstörfer, J. Energetic Functionalization of the Pyridazine Scaffold: Synthesis and Characterization of 3,5-Diamino-4,6-dinitropyridazine-1-Oxide. Eur. J. Org. Chem. 2018, 8, 1004–1010. [Google Scholar] [CrossRef]
- Domasevitch, K.V.; Gospodinov, I.; Krautscheid, H.; Klapötke, T.M.; Stierstörfer, J. Facile and selective polynitrations at the 4-pyrazolyl dual backbone: straightforward access to a series of high-density energetic materials. New J. Chem. 2019, 43, 1305–1312. [Google Scholar] [CrossRef]
- Klapötke, T.M. Chemistry of High-Energy Materials, 4th ed.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2009; p. 350. [Google Scholar]
- Zhang, L.; Jiang, S.L.; Yu, Y.; Long, Y.; Zhao, H.Y.; Peng, L.J.; Chen, J. Phase Transition in Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) under Static Compression: An Application of the First Principles Method Specialized for C, H, N, O Solid Explosive. J. Phys. Chem. B 2016, 120, 11510–11522. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, L.; Sun, C.; Zhang, C.; Yang, C.; Chen, J.; Hu, B. Response to Comment on “Synthesis and characterization of the pentazolate anion cyclo-N5 – in (N5)6(H3O)3(NH4)4Cl”. Science 2018, 359, aas8953. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, S.-L.; Yu, Y.; Chen, J. Revealing Solid Properties of High-energy-density Molecular Cocrystals from the Cooperation of Hydrogen Bonding and Molecular Polarizability. Sci. Rep. 2019, 9, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yao, C.; Yu, Y.; Jiang, S.-L.; Sun, C.Q.; Chen, J. Stabilization of the Dual-Aromatic cyclo-N5 – Anion by Acidic Entrapment. J. Phys. Chem. Lett. 2019, 10, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Volume (Å3) | |
|---|---|---|---|---|---|---|---|
| Calculated | 8.29 | 9.65 | 6.41 | 90.00 | 93.21 | 90.00 | 512.44 |
| Experimental | 8.22 | 9.61 | 6.55 | 90.00 | 92.30 | 90.00 | 516.65 |
| Computational error | 0.85% | 0.42% | −2.14% | 0.00% | 0.99% | 0.00% | −0.81% |
| Solvent-removed | 7.81 | 9.38 | 6.32 | 90.00 | 94.08 | 90.00 | 461.32 |
| Compound | ΔfH°gas (kJ·mol−1) | E0 (Hartree) | ZPE (Hartree/Particle) | ΔHT (Hartree/Particle) | |
|---|---|---|---|---|---|
| 1. | 4 | −1010.4391593 | 0.1334850 | 0.0151990 | |
| 2. | CH4 | −74.8700000 | −40.4826803 | 0.0441530 | 0.0038100 |
| 3. | CH3NO2 | −81.0000000 | −244.9469106 | 0.0491150 | 0.0052990 |
| 4. | CH3NH2 | −23.5000000 | −95.8039027 | 0.0631100 | 0.0043620 |
| 5. | CH2O | −115.9000000 | −114.4669424 | 0.0263370 | 0.0041480 |
| 6. | C6H8N2 | +423.4445285 | −342.6714900 | 0.1295460 | 0.0074120 |
| Compounds and Computation Methods | △Hf c (kJ·mol−1) | VOD (km·s−1) | Pd (GPa) | Heat of Detonation (kJ·kg−1) | Detonation Temp. (K) | |
|---|---|---|---|---|---|---|
| 4 | HASEM | −707.18 | 6.88 | 22.04 | 1471.81 | 2136 |
| EXPLO 5 | 7.72 | 24.33 | −3639.187 | 2810.308 | ||
| 4-Water Removed | HASEM | 7.02 | 22.59 | 2295.46 | 2787 | |
| TATB | HASEM | −596.55 | 7.68 | 28.17 | −3721.24 | 2155 |
| EXPLO 5 | 8.17 | 28.86 | −3876.04 | 2768 | ||
| Experiment | 7.76 | 26.8 | −3912.04 [21] | -- | ||
| HNS | HASEM | +173.93 | 7.34 | 24.12 | −5687.27 | 4053 |
| EXPLO 5 | 7.22 | 21.98 | −4633.30 | 3444 | ||
| Experiment | 7.0–7.13 [22] | 26.20 [22] | −5690.24 [22] | 4150 (other cal.) [23] | ||
| Compound | Td/onset a (°C) | ρb (g·cm−3) | IS c (J) | FS d (N) | ESD e (J) | |
|---|---|---|---|---|---|---|
| 1. | 4 | 329 | 1.88 | 8.23 | >360 | 1.22 |
| 2. | TATB | 330 | 1.94 | 50 [24] | >353 [24] | 2.5–4.24 [24] |
| 3. | LLM-105 | 298 | 1.913 [7] | 20[25] | 360[25] | 0.6 [25] |
| 4. | HNS | 320 | 1.74 | 5 [26,27] | 240 [26,27] | 0.8 [26,27] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Kumar, D.; Zhang, L.; Shem-Tov, D.; Petrutik, N.; Chinnam, A.K.; Yao, C.; Pang, S.; Gozin, M. Energetic Butterfly: Heat-Resistant Diaminodinitro trans-Bimane. Molecules 2019, 24, 4324. https://doi.org/10.3390/molecules24234324
Zhang P, Kumar D, Zhang L, Shem-Tov D, Petrutik N, Chinnam AK, Yao C, Pang S, Gozin M. Energetic Butterfly: Heat-Resistant Diaminodinitro trans-Bimane. Molecules. 2019; 24(23):4324. https://doi.org/10.3390/molecules24234324
Chicago/Turabian StyleZhang, Pengcheng, Dheeraj Kumar, Lei Zhang, Daniel Shem-Tov, Natan Petrutik, Ajay Kumar Chinnam, Chuang Yao, Siping Pang, and Michael Gozin. 2019. "Energetic Butterfly: Heat-Resistant Diaminodinitro trans-Bimane" Molecules 24, no. 23: 4324. https://doi.org/10.3390/molecules24234324
APA StyleZhang, P., Kumar, D., Zhang, L., Shem-Tov, D., Petrutik, N., Chinnam, A. K., Yao, C., Pang, S., & Gozin, M. (2019). Energetic Butterfly: Heat-Resistant Diaminodinitro trans-Bimane. Molecules, 24(23), 4324. https://doi.org/10.3390/molecules24234324