3.3.1. Step A
Triethyl orthoformate or trimethyl orthoformate were used in the synthesis of 2a, 2b, 2c, 2d, 2e, 2f and 2g and triethyl orthoacetate or trimethyl orthoacetate for 2h, 2i, 2j, 2k, 2l, 2m, 2n and 2o. The orthoester (triethyl orthoformate, triethyl orthoacetate, trimethyl orthoformate or trimethyl orthoacetate, 330.0 mmol) and Meldrum’s acid (1.7 g, 12.0 mmol) was brought to a gentle reflux for 15 min. The resulting greenish solution was cooled to 80 °C and the appropriate benzene-1,2-diamine 1 (5.5 mmol) was added portionwise (caution: exothermic reaction). The resulting mixture was stirring up to reflux for 2 h, and left under r.t. for 16 h. Subsequently, diethyl ether or hexane was added and the solution was cooled to −35 °C where a precipitate formed. The precipitate was filtered off, washed with diethyl ether (4 × 100 mL) and dried to afford a solid:
1,2-Bis-[(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidenemethyl)amino]benzene (
2a) [
23,
41]; white solid 2.0 g (4.7 mmol, 86%); m.p
dec. = 207.6 °C.
5,5′-(((4-Fluoro-1,2-phenylene)bis(azanediyl))bis(methaneylylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (2b); white; 2.0 g (4.6 mmol, 83%); m.pdec. = 216 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.73 (s, 6H, 2CH3), 1.74 (s, 6H, 2CH3), 7.06–7.12 (dt, 3JH,H = 8.1 Hz, 4JF,H = 2.6 Hz, 1H, aromatic), 7.20 (dd, 3JF,H = 8.7 Hz, 4JH,H = 2.5 Hz, 1H, aromatic), 7.38 (dd, 3JF,H = 8.9 Hz, 3JH,H = 5.1 Hz, aromatic), 8.38 (d, 3JH,H = 13.5 Hz, 1H, vinyl), 8.52 (d, 3JH,H = 13.4 Hz, 1H, vinyl), 11.15 (d, 3JH,H = 13.5 Hz, 1H, NH), 11.42 (d, 3JH,H = 13.3 Hz, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 27.2, 27.3, 89.6, 90.3, 105.8 (d, 3JC,F = 14.3 Hz), 107.4 (d, 2JC,F = 26.8 Hz), 115.0 (d, 2JC,F = 22.9 Hz), 124.9 (d, 4JC,F = 9.5 Hz), 126.9, 133.3 (d, 3JC,F = 10.1 Hz), 161.1, 155.1 (d, 1JC,F = 342.9 Hz), 162.9, 163.2, 165.4, 165.5; 19F{1H}-NMR (CDCl3; 470.5 MHz) δ = −108.94; 19F- NMR (CDCl3; 470.5 MHz) δ = −108.94 (dd, 3JF,H = 12.9 Hz, 3JF,H = 7.8 Hz); UV-Vis (methanol; λ [nm] (logε)): 329 (4.48), 292 (4.56), 212 (4.43).
5,5′-(((4-Chloro-1,2-phenylene)bis(azanediyl))bis(methaneylylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (
2c) [
23]; white; 2.1 g (4.6 mmol, 84%); m.p
dec. = 202.7 °C; UV-Vis (methanol; λ [nm] (logε)): 332 (4.59), 296 (4.75), 223 (4.47), 206 (4.46).
5,5′-(((4-Methyl-1,2-phenylene)bis(azanediyl))bis(methaneylylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (
2d); white; 2.2 g (5.2 mmol, 94%) [
41]; m.p
dec. = 212 °C; UV-Vis (methanol; λ [nm] (logε)): 330 (4.85), 296 (4.93), 221 (4.83).
3,4-Bis(((2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)methyl)amino)benzonitrile (2e); beige, 1.7 g (3.8 mmol, 69%); m.pdec. = 250 °C; 1H-NMR (DMSO-d6; 400.2 MHz) δ = 1.68 (s, 6H, 2CH3), 1.68 (s, 6H, 2CH3), 7.81-7.84 (m, 2H, aromatic), 8.16 (dd, 3JH,H = 4.3 Hz, 4JH,H = 0.8 Hz, 1H, aromatic), 8.33 (d, 3JH,H = 14.1 Hz, 1H, vinyl), 8.41 (d, 3JH,H = 13.9 Hz, 1H, vinyl), 11.33 (d, 3JH,H = 14.1 Hz, 1H, NH), 11.43 (d, 3JH,H = 14.0 Hz, 1H, NH); 13C{1H}-NMR (DMSO-d6; 125.78 MHz) δ = 26.5, 26.6, 88.3, 88.8, 104.1, 104.3, 110.0, 117.4, 122.7, 128.0, 132.0, 133.6, 137.5, 155.6, 157.4, 162.3, 162.5, 163.5, 163.6; UV-Vis (methanol; λ [nm] (logε)): 342 (4.11), 296 (4.19), 219 (4.00).
3,4-Bis(((2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)methyl)amino)benzoic acid (2f); beige, 0.8 g (1.8 mmol, 32%); m.pdec. = 225 °C; 1H NMR (CDCl3; 400.2 MHz) δ = 1.77 (s, 6H, 2CH3), 1.79 (s, 6H, 2CH3), 7.50 (d, 3JH,H = 8.5 Hz, 1H, aromatic), 8.14 (d, 3JH,H = 8.9 Hz, 1H, aromatic), 8.50 (s, 1H, aromatic), 8.59 (d, 3JH,H = 13.1 Hz, 1H, vinyl), 8.99 (d, 3JH,H = 12.7 Hz, 1H, vinyl), 11.51 (d, 3JH,H = 12.6 Hz, 1H, NH), 11.66 (d, 3JH,H = 12.9 Hz, 1H, NH); 1H-NMR (DMSO-d6; 500.2 MHz) δ = 1.68 (s, 6H, 2CH3), 1.68 (s, 6H, 2CH3), 7.74 (d, 3JH,H = 8.4 Hz, 1H, aromatic), 7.96 (dd, 3JH,H = 8.3 Hz, 4JH,H = 1.7 Hz, 1H, aromatic), 8.03 (d, 4JH,H = 1.7 Hz, 1H, aromatic), 8.24 (d, 3JH,H = 14.2 Hz, 1H, vinyl), 8.40 (d, 3JH,H = 14.1 Hz, 1H, vinyl), 11.35 (d, 3JH,H = 14.3 Hz, 1H, NH), 11.44 (d, 3JH,H = 14.1 Hz, 1H, NH); 13C{1H}-NMR (DMSO-d6; 125.78 MHz) δ = 26.50, 26.52, 87.8, 88.2, 104.0, 104.1, 121.9, 125.1, 129.3, 130.0, 133.1, 137.2, 155.7, 157.5, 162.4, 162.7, 163.4, 163.6, 165.9, 168.2; UV-Vis (methanol; λ [nm] (logε)): 340 (3.96),303 (3.87) 274 (3.82), 220 (4.00).
Methyl 3,4-bis(((2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)methyl)amino)benzoate (2g); beige, 1.6 g (3.5 mmol, 63%); m.pdec. = 225 °C; 1H-NMR (DMSO-d6; 500.2 MHz) δ = 1.68 (s, 6H, 2CH3), 1.69 (s, 6H, 2CH3), 4.00 (s, 3H, OCH3), 7.39 (d, 3JH,H = 8.3 Hz, 1H, aromatic), 7.76 (dd, 3JH,H = 8.2 Hz, 4JH,H = 1.7 Hz, 1H, aromatic), 8.18 (d, 4JH,H = 1.6 Hz, 1H, aromatic), 8.24 (d, 3JH,H = 14.5 Hz, 1H, vinyl), 8.81 (d, 3JH,H = 14.7 Hz, 1H, vinyl), 11.40 (d, 3JH,H = 14.1 Hz, 2H, NH), 11.84 (d, 3JH,H = 14.8 Hz, 2H, NH); 13C{1H}-NMR (DMSO-d6; 125.78 MHz) δ = 26.47, 26.52, 54.2, 87.2, 87.7, 104.39, 104.38, 116.0, 119.9, 127.2, 128.0, 131.3, 140.1, 151.3, 159.3, 162.5, 164.7, 166.6, 172.6; UV-Vis (methanol; λ [nm] (logε)): 340 (3.96),300 (3.87) 275 (3.82), 220 (4.00).
5,5′-((1,2-Phenylenebis(azanediyl))bis(ethan-1-yl-1-ylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (
2h) [
22,
23]; white; 1.2 g (2.8 mmol, 51%); m.p
dec. = 189.6 °C.
5,5′-(((4-Fluoro-1,2-phenylene)bis(azanediyl))bis(ethan-1-yl-1-ylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (2i); white, 0.9 g (2.0 mmol, 36%); m.pdec.. = 218.0 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.70 (s, 6H, 2CH3), 1.71 (s, 6H, 2CH3), 2.50 (s, 3H, CH3), 2.58 (s, 3H, CH3), 7.12 (dd, 3JH,F = 8.2 Hz, 4JH,H = 2.3 Hz, 1H, aromatic), 7.18-7.20 (m, 1H, aromatic), 7.35 (dd, 3JH,F = 8.7 Hz, 3JH,H = 5.4 Hz, 1H, aromatic), 12.68 (s, 1H, NH), 12.85 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 19.6, 19.7, 26.7, 26.7, 87.6, 88.0, 103.4, 103.5, 115.4 (d, 2JC,F = 24.8 Hz), 116.4 (d, 2JC,F = 22.4 Hz), 128.5 (d, 4JC,F = 3.7 Hz), 129.7 (d, 3JC,F = 9.5 Hz), 134.1 (d, 3JC,F = 10.3 Hz), 161.8 (d, 1JC,F = 252.9 Hz), 162.3, 162.5, 167.7, 167.6, 172.8, 173.5; 19F{1H}-NMR (CDCl3; 470.5 MHz) δ = −108.53; 19F-NMR (CDCl3; 470.5 MHz) δ = −108.53 (dd, 3JF,H = 13.3 Hz, 4JF,H = 7.7 Hz); UV-Vis (methanol; λ [nm] (logε)): 311 (4.30), 283 (4.66), 226 (4.38), 201 (4.27).
5,5′-(((4-Chloro-1,2-phenylene)bis(azanediyl))bis(ethan-1-yl-1-ylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (2j); white, 1.4 g (2.9 mmol, 52%); mpdec. = 203.8 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.71 (s, 12H, 4CH3), 2.53 (s, 3H, CH3), 2.56 (s, 3H, CH3), 7.30 (d, 3JH,H = 8.5 Hz, 1H, aromatic), 7.38 (d, 4JH,H = 1.6 Hz, 1H, aromatic), 7.48 (dd, 3JH,H = 8.5 Hz, 4JH,H = 1.8 Hz, 1H, aromatic), 12.76 (s, 1H, NH), 12.81 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 100.5 MHz) δ = 19.7, 19.7, 26.7, 87.9, 88.0, 103.5, 103.5, 128.1, 129.0, 129.5, 131.1, 133.6, 135.0, 162.3, 162.4, 167.7, 167.7, 173.0, 173.1; UV-Vis (methanol; λ [nm] (logε)): 315 (4.36), 285 (4.74), 226 (4.46), 202 (4.38).
5,5′-(((4-Bromo-1,2-phenylene)bis(azanediyl))bis(ethan-1-yl-1-ylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (2k); white, 0.9 g (1.8 mmol, 32%); m.pdec. = 204.6 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.71 (s, 12H, 4CH3), 2.53 (s, 3H, CH3), 2.56 (s, 3H, CH3), 7.23 (d, 3JH,H = 8.5 Hz, 1H, aromatic), 7.53 (d, 4JH,H = 1.5 Hz, 1H, aromatic), 7.63 (dd, 3JH,H = 8.5 Hz, 4JH,H = 1.7 Hz, 1H, aromatic), 12.76 (s, 1H, NH), 12.81 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 19.6, 19.7, 26.6, 87.7, 87.8, 103.4, 103.4, 122.3, 129.1, 131.0, 131.6, 132.4, 133.7, 162.2, 162.3, 167.5, 172.9; UV-Vis (methanol; λ [nm] (logε)): 313 (4.29), 284 (4.65), 226 (4.38), 201 (4.37).
5,5′-(((4-Methyl-1,2-phenylene)bis(azanediyl))bis(ethan-1-yl-1-ylidene))bis(2,2-dimethyl-1,3-dioxane-4,6-dione) (2l); white, 1.3 g (2.9 mmol, 52%); m.pdec. = 204.2 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.70 (s, 12H, 4CH3), 2.45 (s, 3H, CH3), 2.51 (s, 3H, CH3), 2.52 (s, 3H, CH3), 7.17 (s, 1H, aromatic), 7.23 (d, 3JH,H = 8.1 Hz, 1H, aromatic), 7.30 (d, 3JH,H = 8.3 Hz, 1H, aromatic), 12.68 (s, 1H, NH), 12.71 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 19.6, 19.6, 21.2, 26.6, 87.1, 87.2, 103.2, 127.7, 128.5, 129.8, 130.1, 132.2, 140.1, 162.5, 162.5, 167.6, 173.2, 173.4; UV-Vis (methanol; λ [nm] (logε)): 319 (4.12), 283 (4.60), 227 (4.38), 203 (4.23).
3,4-Bis((1-(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)ethyl)amino)benzonitrile (2m); white, 1.0 g (2.2 mmol, 40%); m.pdec. = 205.6 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.72 (bs, 12H, 4CH3), 2.55 (s, 3H, CH3), 2.61 (s, 3H, CH3), 7.49 (d, 3JH,H = 8.2 Hz, 1H, aromatic), 7.68 (s, 1H, aromatic), 7.78 (d, 3JH,H = 8.2 Hz, 1H, aromatic), 12.87 (s, 1H, NH), 13.04 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 19.7, 19.9, 26.8, 26.8, 88.6, 89.0, 103.7, 103.7, 112.7, 116.7, 128.2, 131.6, 132.6, 133.1, 136.9, 162.1, 162.2, 167.7, 167.7, 172.2, 172.9; UV-Vis (methanol; λ [nm] (logε)): 324 (4.27), 289 (4.58), 224 (4.40), 204 (4.40).
Methyl 3,4-bis((1-(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)ethyl)amino)benzoate (2n); white, 1.2 g (2.3 mmol, 42%); m.pdec. = 184.8 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.72 (s, 6H, 2CH3), 1.73 (s, 6H, 2CH3), 2.55 (s, 3H, CH3), 2.60 (s, 3H, CH3), 3.98 (s, 3H, OCH3), 7.44 (d, 3JH,H = 8.3 Hz, 1H, aromatic), 8.03 (s, 1H, aromatic), 8.15 (d, 3JH,H = 8.2 Hz, 1H, aromatic), 12.84 (s, 1H, NH), 12.97 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 19.7, 19.9, 26.7, 26.8, 53.0, 88.0, 88.4, 103.5, 103.6, 127.5, 129.2, 130.2, 130.8, 132.3, 136.4, 162.3, 162.4, 165.0, 167.7, 167.7, 172.6; UV-Vis (methanol; λ [nm] (logε)): 323 (4.61), 289 (4.96), 226 (4.75), 203 (4.65).
Ethyl 3,4-bis((1-(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)ethyl)amino)benzoate (2o); white, 0.9 g (1.7 mmol, 31%); m.pdec. = 171.4 °C; 1H-NMR (CDCl3; 400.2 MHz) δ = 1.42 (t, 3JH,H = 7.0 Hz, 3H, CH3), 1.71 (s, 6H, 2CH3), 1.72 (s, 6H, 2CH3), 2.54 (s, 3H, CH3), 2.59 (s, 3H, CH3), 4.43 (q, 3JH,H = 7.1 Hz, 2H, OCH2), 7.43 (d, 3JH,H = 8.2 Hz, 1H, aromatic), 8.02 (s, 1H, aromatic), 8.15 (d, 3JH,H = 8.1 Hz, 1H, aromatic), 12.82 (s, 1H, NH), 12.95 (s, 1H, NH); 13C{1H}-NMR (CDCl3; 125.78 MHz) δ = 14.4, 19.7, 19.9, 26.7, 26.7, 62.1, 87.9, 88.3, 103.5, 103.5, 127.5, 129.2, 130.2, 131.2, 132.2, 136.3, 162.3, 162.4, 164.4, 167.7, 167.7, 172.6, 173.3; UV-Vis (methanol; λ [nm] (logε)): 321 (4.25), 287 (4.63), 225 (4.44), 204 (4.35).
1H-benzo[d]imidazole-6-carboxylic acid; m.pdec. = 278.0-280.0 °C; 1H-NMR (DMSO-d6/KOD; 500.2 MHz) δ = 7.74 (d, 3JH,H = 8.7 Hz, 1H, aromatic), 7.96 (d, 3JH,H = 8.7 Hz, 4JH,H = 1.4 Hz, 1H, aromatic), 8.23 (s, 1H, aromatic), 9.08 (s, 1H, aromatic); 1H-NMR (D2O/KOD; 500.18 MHz) δ = 7.66 (d, 3JH,H = 8.5 Hz, 1H, aromatic), 7.81 (dd, 3JH,H = 8.5 Hz, 4JH,H = 1.5 Hz, 1H, aromatic), 8.21 (s, 1H, aromatic), 8.36 (s, 1H, aromatic); 13C{1H}-NMR (DMSO-d6/KOD; 125.78 MHz) δ = 116.0, 117.9, 128.4, 130.4, 132.1, 135.0, 143.1, 170.8; 13C{1H}-NMR (D2O/KOD; 125.78 MHz) δ = 115.3 (bs), 118.3 (bs), 123.8, 124.8, 144.7, 168.4; HRMS (IT TOF): m/z Calcd for C8H7N2O2 [M + H]+= 163.0508, Found 163.0509.
3.3.2. Step B
Modifications to existing procedures described in the literature [
22,
23] rely on purifications. The crude products were extracted with hexane or toluene at Soxhlet apparatus. Into freshly distillated diphenyl ether (50 mL) at 240 °C was added compound
2 (10.0 mmol) in small portions, resulting in vigorous gas evolution. The resulting orange solution was brought to reflux for 30 min, and was then allowed to cool to 70 °C. The acetone or hexane (25 mL) was added and a dark-brown solid precipitated was filtered, washed with acetone (2 × 10 mL). The crude product was extracted with hexane or toluene at Soxhlet apparatus to yield solid as follows:
1,10-Dihydro-1,10-phenanthroline-4,7-dione (
3a); [
41] brown; 1.4 g (6.8 mmol, 68%); m.p
dec. > 350 °C;
1H- NMR (CD
3OD/KOD/D
2O; 400.2 MHz) δ = 6.62 (d,
3JH,H = 5.6 Hz, 2H, aromatic), 8.08 (s, 2H, aromatic), 8.36 (d,
3JH,H = 5.6 Hz, 2H, aromatic);
13C{
1H}-NMR (CD
3OD/KOD/D
2O; 100.5 MHz) δ = 111.6, 118.8, 127.0, 148.8, 150.5, 174.7.
5-Fluoro-1,10-dihydro-1,10-phenanthroline-4,7-dione (3b); brown; 1.8 g (7.7 mmol, 77%); m.pdec. >350.0 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 6.45 (d, 3JH,H = 6.5 Hz, 1H, aromatic), 6.50 (d, 3JH,H = 6.2 Hz, 1H, aromatic), 7.31 (d, 3JH,F = 14.1 Hz, 1H, aromatic), 8.01 (d, 3JH,H = 6.4 Hz, 1H, aromatic), 8.10 (d, 3JH,H = 6.1 Hz, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 101.1 (d, 2JC,F = 24.1 Hz), 113.0 (d, 1JC,F = 307.7 Hz), 117.6 (d, 2JC,F = 11.3 Hz), 125.0 (d, 3JC,F = 9.6 Hz), 130.7, 136.4, 142.9, 143.4, 147.9, 156.4, 158.9, 175.7, 176.9; GC-MS: tr = 5.1 min, (EI) M+ = 230 (< 1%); 19F{1H}-NMR (CD3OD/KOD/D2O; 470.5 MHz) δ = −119.68; 19F-NMR (CD3OD/KOD/D2O; 470.5 MHz) δ = −119.68 (d, 3JF,H = 13.8 Hz); UV-Vis (methanol; λ [nm] (logε)): 352 (3.40), 329 (3.61), 308 (3.55), 273 (3.56), 251 (3.63), 224 (3.91), 208 (3.96).
5-Chloro-1,10-dihydro-1,10-phenanthroline-4,7-dione (
3c); brown; [
23]; 1.5 g (6.1 mmol, 61%); m.p
dec. = 272.8-273.6 °C;
1H-NMR (CD
3OD/KOD/D
2O; 400.2 MHz) δ = 6.62 (d,
3JH,H = 6.9 Hz, 1H, aromatic), 6.64 (d,
3JH,H = 6.4 Hz, 1H, aromatic), 8.27 (d,
3JH,H = 5.9 Hz, 1H, aromatic), 8.31 (d,
3JH,H = 5.8 Hz, 1H, aromatic);
13C{
1H}-NMR (CD
3OD/KOD/D
2O; 100.5 MHz) δ = 112.0, 114.5, 120.3, 122.5, 125.4, 126.1, 144.0, 147.1, 148.6, 149.4, 169.3, 173.7, 173.8, 176.0, 176.1; GC-MS: t
r = 11.5 min, (EI) M
+ = 246 (100%), 248 (32%), [M + H]
+ = 247 (8%), [M-CO]
+ = 218 (90%); UV-Vis (methanol; λ [nm] (logε)): 356 (3.83), 338 (4.06), 324 (4.06), 285 (3.97), 273 (4.03), 249 (4.42), 231 (4.28), 215 (4.42).
5-Methyl-1,10-dihydro-1,10-phenanthroline-4,7-dione (3d); brown; 1.9 g (8.6 mmol, 86%); m.pdec. > 350.0 °C; 1H-NMR (CD3OD/KOD/D2O; 500.2 MHz) δ = 2.73 (d, 4JH,H = 0.8 Hz, 3H, CH3), 6.36 (d, 3JH,H = 6.6 Hz, 1H, aromatic), 6.48 (d, 3JH,H = 6.0 Hz, 1H, aromatic), 7.47 (d, 4JH,H = 1.0 Hz, 1H, aromatic), 7.88 (d, 3JH,H = 6.6 Hz, 1H, aromatic), 8.08 (d, 3JH,H = 6.0 Hz, 1H, aromatic); 13C{1H}-NMR(CD3OD/KOD/D2O; 125.78 MHz) δ = 24.5, 112.0, 114.0, 119.9, 124.9, 125.9, 132.6, 139.8, 141.0, 142.5, 148.0, 174.6, 180.9; GC-MS: tr = 7.0 min, (EI) [M-H]+ = 225 (1%); UV-Vis (methanol; λ [nm] (logε)): 355 (3.66), 327 (3.89), 306 (3.83), 275 (3.93), 247 (4.14), 224 (4.23), 209 (4.28).
4,7-Dioxo-1,4,7,10-tetrahydro-1,10-phenanthroline-5-carbonitrile (3e); beige, 1.5 g (6.4 mmol, 64%); m.pdec. > 350 °C; 1H-NMR (DMSO–d6/KOD/D2O; 400.2 MHz) δ = 6.37 (d, 3JH,H = 6.8 Hz, 1H, aromatic), 6.41 (d, 3JH,H = 6.3 Hz, 1H, aromatic), 8.02 (d, 3JH,H = 6.7 Hz, 1H, aromatic), 8.16 (d, 3JH,H = 6.3 Hz, 1H, aromatic), 8.26 (s, 1H, aromatic); 13C{1H}-NMR (DMSO–d6/KOD/D2O; 125.78 MHz) δ = 101.1, 113.7, 114.2, 122.2, 123.3, 124.8, 131.2, 139.6, 142.2, 144.6, 149.7, 176.1, 176.8; UV-Vis (methanol; λ [nm] (logε)): 382 (3.27), 350 (3.50), 335 (3.44), 277 (3.39), 242 (3.78), 214 (3.81).
5-Fluoro-2,9-dimethyl-1,5,10,10a-tetrahydro-1,10-phenanthroline-4,7-dione (3f); brown; 2.2 g (8.7 mmol, 87%); m.pdec. = 370.2–372.8 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.40 (bs, 6H, CH3), 6.26 (bs, 1H, aromatic), 6.35 (bs, 1H, aromatic), 7.19 (bd, 3JH,F = 11.1 Hz, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 21.4, 22.9, 100.4 (d, 2JC,F = 24.2 Hz), 112.2 (d, 1JC,F = 274.4 Hz), 115.5 (d, 2JC,F = 11.1 Hz), 123.1 (d, 3JC,F = 9.7 Hz), 135.0, 141.5 (d, 4JC,F = 3.7 Hz), 154.0, 155.8, 157.8, 158.3, 175.0, 176.2, 176.2; GC-MS: tr = 10.5 min, (EI) M+ = 258 (100%), [M + H-CO]+ = 229 (98%).
2,9-Dimethyl-1,10-phenanthroline-4,7(1H,10H)-dione (
3g); brown; [
22]; 1.6 g (6.8 mmol, 68%); m.p
dec. = 291.8–292.5 °C;
1H-NMR (CD
3OD/KOD/D
2O; 400.2 MHz) δ = 2.29 (s, 6H, CH
3), 6.24 (s, 2H, aromatic), 7.64 (s, 2H, aromatic);
13C{
1H}-NMR (CD
3OD/KOD/D
2O; 100.5 MHz) δ = 21.8, 100.0, 110.1, 117.0, 122.9, 138.9, 155.9, 174.6.
5-Fluoro-2,9-dimethyl-1,5,10,10a-tetrahydro-1,10-phenanthroline-4,7-dione (3h); brown; 2.2 g (8.7 mmol, 87%); m.pdec. = 370.2-372.8 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.40 (bs, 6H, CH3), 6.26 (bs, 1H, aromatic), 6.35 (bs, 1H, aromatic), 7.19 (bd, 3JH,F = 11.1 Hz, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 21.4, 22.9, 100.4 (d, 2JC,F = 24.2 Hz), 112.2 (d, 1JC,F = 274.4 Hz), 115.5 (d, 2JC,F = 11.1 Hz), 123.1 (d, 3JC,F = 9.7 Hz), 135.0, 141.5 (d, 4JC,F = 3.7 Hz), 154.0, 155.8, 157.8, 158.3, 175.0, 176.2, 176.2; GC-MS: tr = 10.5 min, (EI) M+ = 258 (100%), [M + H-CO]+ = 229 (98%); 19F{1H}-NMR (CD3OD/KOD/D2O; 470.5 MHz) δ = −119.99; 19F-NMR (CD3OD/KOD/D2O; 470.5 MHz) δ = −119.99 (d, 3JF,H = 13.9 Hz); UV-Vis (methanol; λ [nm] (logε)): 353 (3.76), 337 (3.85), 319 (4.00), 311 (3.97), 270 (4.21), 254 (4.46), 225 (4.31), 214 (4.44).
5-Chloro-2,9-dimethyl-1,5,10,10a-tetrahydro-1,10-phenanthroline-4,7-dione (3i); 2.3 g (8.3 mmol, 83%); brown; m.pdec. = 332.7–334.8 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.46 (s, 3H, CH3), 2.49 (s, 3H, CH3), 6.40 (s, 1H, aromatic), 6.41 (s, 1H, aromatic), 7.78 (s, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 21.8, 22.6, 112.0, 114.3, 120.1, 121.1, 124.2, 126.2, 138.4, 141.6, 155.2, 156.8, 175.9, 178.1; GC-MS: tr = 11.3 min, (EI) M+ = 273.9 (100%), [M-CO]+ = 245 (80%); UV-Vis (methanol; λ [nm] (logε)): 357 (3.65), 340 (3.84), 327 (3.96), 316 (3.94), 280 (4.00), 254 (4.35), 230 (4.25), 216 (4.35).
5-Bromo-2,9-dimethyl-1,5,10,10a-tetrahydro-1,10-phenanthroline-4,7-dione (3j); brown; 2.7 g (8.5 mmol, 85%); m.pdec. = 325.9–326.6 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.48 (s, 3H, CH3), 2.50 (s, 3H, CH3), 6.40 (s, 1H, aromatic), 6.43 (s, 1H, aromatic), 8.09 (s, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 21.6, 22.7, 112.2, 112.7, 113.9, 121.4, 124.4, 124.9, 138.9, 141.1, 154.4, 157.1, 175.8, 175.8, 178.2; GC-MS: tr = 13.5 min, (EI) M+ = 318 (100%), 320 (96%), [M-CO]+ = 290 (63%); UV-Vis (methanol; λ [nm] (logε)): 356 (3.87), 339 (4.08), 328 (4.17), 315 (4.13), 281 (4.19), 254 (4.55), 231 (4.46), 217 (4.53).
2,5,9-Trimethyl-1,10-dihydro-1,10-phenanthroline-4,7-dione (3k); brown; 2.3 g (9.2 mmol, 92%); m.pdec. = 337.4–338.9 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.31 (s, 3H, CH3), 2.36 (s, 3H, CH3), 2.66 (s, 3H, OCH3), 6.16 (s, 1H, aromatic), 6.31 (s, 1H, aromatic), 7.28 (s, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 21.1, 23.5, 24.4, 111.4, 113.2, 119.3, 123.0, 123.9, 131.6, 138.2, 139.6, 152.8, 157.5, 174.2, 180.3; GC-MS: tr = 12.1 min, (EI) M+ = 254 (100 %), [M + H-CO]+ = 225 (56%); UV-Vis (methanol; λ [nm] (logε)): 354 (3.72), 338 (3.95), 320 (4.14), 282 (4.30), 257 (4.45), 226 (4.40), 217 (4.42), 204 (4.40).
2,9-Dimethyl-4,7-dioxo-1,4,7,10-tetrahydro-1,10-phenanthroline-5-carbonitrile (3l); brown; 1.6 g (6.2 mmol, 62%); m.pdec. = 390.7–392.5 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.45 (s, 3H, CH3), 2.46 (s, 3H, CH3), 6.45 (s, 1H, aromatic), 6.47 (s, 1H, aromatic), 8.21 (s, 1H, aromatic); 13C{1H}-NMR (CD3OD/KOD/D2O; 100.5 MHz) δ = 23.8, 100.0, 112.1, 112.2, 122.0, 122.9, 123.3, 130.5, 144.5, 146.7, 159.1, 159.2, 162.1, 169.4, 173.8, 174.1; GC-MS: tr = 9.4 min, (EI) M+ = 265 (5%); UV-Vis (methanol; λ [nm] (logε)): 350 (3.90), 343 (3.93), 328 (3.87), 286 (3.82), 276 (3.90), 252 (4.31), 228 (4.15), 217 (4.28).
Methyl 2,9-dimethyl-4,7-dioxo-1,4,7,10-tetrahydro-1,10-phenanthroline-5-carboxylate (3m); brown; 2.3 g (7.8 mmol, 78%); m.pdec. = 293.8–294.3 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 2.44 (s, 6H, 2CH3), 3.85 (s, 3H, OCH3), 6.32 (s, 1H, aromatic), 6.44 (s, 1H, aromatic), 7.78 (s, 1H, aromatic); GC-MS: tr = 9.3 min, (EI) [M-MeOH]+ = 266 (100%); UV-Vis (methanol; λ [nm] (logε)): 345 (3.83), 330 (4.04), 314 (3.98), 280 (4.03), 252 (4.43), 215 (4.36).
Ethyl 2,9-dimethyl-4,7-dioxo-1,4,7,10-tetrahydro-1,10-phenanthroline-5-carboxylate (3n); brown; 2.0 g (6.5 mmol, 65%); m.pdec. = 301.4–301.8 °C; 1H-NMR (CD3OD/KOD/D2O; 400.2 MHz) δ = 1.41 (t, 3JH,H = 7.1 Hz, 3H, OCH2CH3), 2.36 (s, 3H, CH3), 2.38 (s, 3H, CH3), 4.46 (q, 3JH,H = 7.0 Hz, 2H, OCH2), 6.29 (s, 1H, aromatic), 6.37 (s, 1H, aromatic), 7.79 (s, 1H, aromatic); GC-MS: tr = 17.9 min, (EI) M+ = 312 (50%); UV-Vis (methanol; λ [nm] (logε)): 354 (3.58), 345 (3.81), 330 (4.04), 316 (3.99), 279 (4.03), 252 (4.44), 227 (4.26), 214 (4.36).
3.3.3. Step C
Modifications to existing procedure described in the literature [
22,
23] rely on the evaporation of excess phosphoryl chloride under reduced pressure, and the addition of CH
2Cl
2 or CHCl
3 into reaction mixture after alkalified by NaOH solution, because of the very exothermic hydrolysis of residual phosphoryl chloride. Next the crude products were purified by chromatography and crystallization from CH
2Cl
2.
Freshly distillated phosphoryl chloride (82.0 g, 50 mL, 534.8 mmol) was mixed under argon with compounds 3 (5.0 mmol) and the resulting solutions were stirred at 90 °C for 4 h. The excess of phosphoryl chloride was slowly evaporated, under reduced pressure. The reaction mixture was slowly added to a well stirred mixture of ice (50 g) in water (100 mL). After stirring for 15 min. the resulting reaction mixture was carefully brought to pH 13–14 (pH 7 in the case of molecule 4g) by adding NaOH solution (40%). The aqueous layer was extracted with CH2Cl2 (4 × 10 mL). The combined organic layers was separated and dried over MgSO4. Evaporation of the brown colored solvent afforded products 4 as light tan crystals. Next, the crude products were purified by chromatography on silica gel using methanol/dichloromethane as eluent and finally crystallization from CH2Cl2 to yield precipitates as follows:
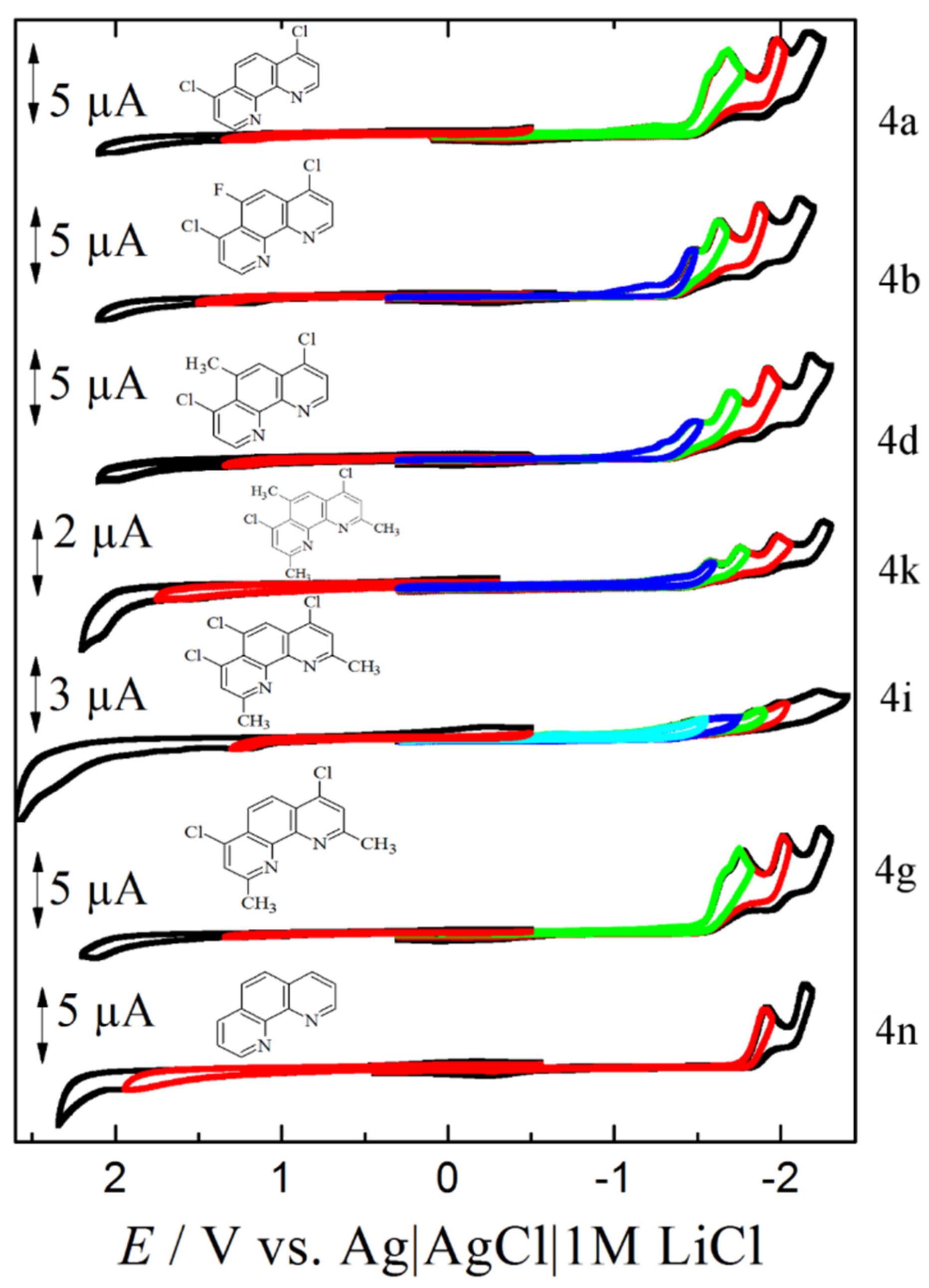
4,7-Dichloro-1,10-phenanthroline (
4a) [
42,
43]; beige; 1.0 g (4.2 mmol, 84%); m.p
dec. = 254.8–255.0 °C; UV-Vis (methanol; λ [nm] (logε)): 300 (3.83), 264 (4.40), 239 (4.33), 225 (4.29), 202 (4.36); IR (KBr):
= 3422, 3034, 1563, 1487, 1418, 835, 720 cm
−1 (
Supporting Information Figure S1).
4,7-Dichloro-5-fluoro-1,10-phenanthroline (
4b); beige; 1.1 g (4.2 mmol, 85%); m.p
dec. = 217.8–218.0 °C;
1H- NMR (CDCl
3; 600.2 MHz) δ = 7.75 (d,
3JH,H = 4.7 Hz, 1H, aromatic), 7.77 (d,
3JH,H = 4.8 Hz, 1H, aromatic), 7.94 (d,
3JH,F = 13.3 Hz, 1H, aromatic), 9.02 (dd,
3JH,H = 4.8 Hz,
5JH,F = 0.5 Hz, 1H, aromatic), 9.08 (d,
3JH,H = 4.8 Hz, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 150.0 MHz) δ = 106.7 (d,
2JC,F = 25.9 Hz), 119.9 (d,
2JC,F = 12.6 Hz), 124.4, 126.3, 126.8 (d,
3JC,F = 10.8 Hz), 140.3, 142.0 (d,
3JC,F = 6.2 Hz), 144.7, 148.9 (d,
4JC,F = 2.3 Hz), 149.7 (d,
4JC,F = 2.6 Hz), 151.0, 156.3 (d,
1JC,F = 263.6 Hz);
19F{
1H}-NMR (CDCl
3; 470.5 MHz) δ = −108.98;
19F-NMR (CDCl
3; 470.5 MHz) δ = −108.98 (d,
3JF,H = 13.4 Hz); GC-MS: t
r = 9.0 min, (EI) M
+ = 266.0 (100%); Anal. Calcd for C
12H
5N
2Cl
2F: C, 53.96; H, 1.89; N, 10.49; Found: C, 53.90; H, 1.93; N, 10.45; UV-Vis (methanol; λ [nm] (logε)): 305 (4.01), 268 (4.46), 240 (4.47), 225 (4.40), 203 (4.45); IR (KBr):
= 3422, 3035, 1626, 1571,1546, 1410, 842, 752 cm
−1 (
Supporting Information Figure S2).
4,5,7-Trichloro-1,10-phenanthroline (
4c) [
42]; beige; 1.0 g (3.8 mmol, 75%); m.p
dec. = 194.6–195.0 °C; GC-MS: t
r = 9.4 min, (EI) M
+ = 281.9 (100%), 283.9 (95%), [M-Cl]
+ = 247 (46%); UV-Vis (methanol; λ [nm] (logε)): 314 (3.82), 302 (3.87), 270 (4.32), 247 (4.31), 209 (4.30); IR (KBr):
= 3356, 3021, 2920, 2191, 1567, 1403, 833, 716 cm
−1 (
Supporting Information Figure S3).
4,7-Dichloro-5-methyl-1,10-phenanthroline (
4d) [
23]; beige; 1.2 g (4.6 mmol, 91%); m.p
dec. = 166.8–167.0 °C; GC-MS: t
r = 9.6 min, (EI) M
+ = 262.1 (100%); UV-Vis (methanol; λ [nm] (logε)): 304 (4.09), 271 (4.50), 241 (4.49), 225 (4.42), 205 (4.47); IR (KBr):
= 3454, 3089, 2933, 1685, 1550,1432, 1400, 1085, 844, 744 cm
−1 (
Supporting Information Figure S4).
4,7-Dichloro-1,10-phenanthroline-5-carbonitrile (
4e); beige; 0.6 g (2.4 mmol, 48%); m.p. = 249.7–250.0 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 7.84 (m, 2H, aromatic), 8.88 (s, 1H, aromatic), 9.13 (d,
3JH,H = 4.7 Hz, 1H, aromatic), 9.19 (d,
3JH,H = 4.7 Hz, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 125.78 MHz) δ = 108.1, 118.1, 124.2, 125.2, 125.3, 126.5, 135.4, 142.3, 143.6, 147.5, 148.1, 151.5, 153.4; GC-MS: t
r = 10.1 min, (EI) M
+ = 273.0 (100%); Anal. Calcd for C
13H
5N
3Cl
2: C, 56.96; H, 1.84; N, 15.33; Found: C, 56.90; H, 1.94; N, 15.29; UV-Vis (methanol; λ [nm] (logε)): 315 (3.67), 302 (3.76), 292 (3.68), 268 (4.15), 249 (4.11), 231 (4.02), 210 (4.11); IR (KBr):
= 3406, 3029, 2224, 1569, 1498, 1406, 1084, 846, 798 cm
−1 (
Supporting Information Figure S5).
7-Chloropyrrolo [2,3,4-de][1,10]phenanthrolin-5(4H)-one (
4f) beige; m.p
dec. = 310–330 °C;
1H-NMR (D
2O/KOD; 500.18 MHz) δ = 6.47 (d,
3JH,H = 6.9 Hz, 1H, aromatic), 6.61 (d,
3JH,H = 5.9 Hz, 1H, aromatic), 7.69 (s, 1H, aromatic), 8.05 (d,
3JH,H = 6.9 Hz, 1H, aromatic), 8.28 (d,
3JH,H = 5.9 Hz, 1H, aromatic);
1H- NMR (D
2O/D
2SO
4; 400.2 MHz) δ = 7.29 (d,
3JH,H = 6.8 Hz, 1H, aromatic), 7.42 (d,
3JH,H = 6.8 Hz, 1H, aromatic), 8.28 (s, 1H, aromatic), 8.78 (d,
3JH,H = 6.8 Hz, 1H, aromatic), 8.87 (d,
3JH,H = 6.8 Hz, 1H, aromatic);
13C{
1H}-NMR (D
2O/KOD; 125.78 MHz) δ = 111.5, 111.6, 114.8, 120.7, 125.1, 132.5, 138.1, 139.2, 140.7, 149.3, 173.8, 178.0, 179.7; MS-IT-TOF [M-H]
- = 254 (80%), [M + H]
+ = 256 (30%), HRMS (IT TOF): m/z Calcd for C
13H
7N
3ClO [M + H]
+ = 256.0278, Found 256.0279; UV-Vis (methanol; λ [nm] (logε)): 348 (3.28), 334 (3.27), 278 (3.31), 245 (3.64), 221 (3.36); IR (KBr):
= 3086, 1560, 1522, 1402, 1385, 1363, 1026 cm
−1 (
Supporting Information Figure S6).
4,7-Dichloro-2,9-dimethyl-1,10-phenanthroline (
4g) [
22,
42,
44,
45]; beige; 1.3 g (4.6 mmol, 92%); m.p
dec. = 201.0–201.5 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 2.93 (s, 6H, 2CH
3), 7.63 (s, 2H, aromatic), 8.24 (s, 2H, aromatic);
13C{
1H}-NMR (CDCl
3; 100.5 MHz) δ = 26.0, 122.3, 124.4, 125.1, 143.0, 146.2, 160.1; CP/MAS
13C-NMR δ = 25.1, 121.7, 141.3, 143.8, 159.3; CP/MAS
15N-NMR δ = −76.15; IR (KBr):
= 2963, 1637, 1568, 1522, 1402, 1385, 1363, 1026 cm
−1 (
Supporting Information Figure S7).
4,7-Dichloro-5-fluoro-2,9-dimethyl-1,10-phenanthroline (
4h) [
44]; beige; 1.4 g (4.6 mmol, 93%); m.p
dec. = 213.0–213.5 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 2.97 (s, 3H, CH
3), 3.01 (s, 3H, CH
3), 7.67 (s, 1H, aromatic), 7.68 (s, 1H, aromatic), 7.86 (d,
3JH,F = 13.1 Hz, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 100.5 MHz) δ = 25.3, 25.5, 105.6 (d,
2JC,F = 26.0 Hz), 118.0 (d,
3JC,F = 12.5 Hz), 125.1 (d,
3JC,F = 11.1 Hz), 126.1 (d,
1JC,F = 184.1 Hz), 140.3, 142.8 (d,
2JC,F = 68.6 Hz), 147.0, 154.9, 157.5, 159.1, 161.3;
19F{
1H}-NMR (CDCl
3; 470.5 MHz) δ = −110.97;
19F-NMR (CDCl
3; 470.5 MHz) δ = −110.97 (d,
3JF,H = 11.5 Hz); GC-MS: t
r = 8.2 min, (EI) M
+ = 294 (100%), 296. (64%), [M-Cl]
+ = 259 (12%); Anal. Calcd for C
14H
9N
2Cl
2F: C, 56.97; H, 3.07; N, 9.49; Found: C, 56.90; H, 3.19; N, 9.32; UV-Vis (methanol; λ [nm] (logε)): 309 (3.88), 296 (3.95), 273 (4.46), 241 (4.47), 211 (4.40); IR (KBr):
= 3471, 3075, 1628, 1579, 1375, 1145, 858 cm
−1 (
Supporting Information Figure S8).
4,5,7-Trichloro-2,9-dimethyl-1,10-phenanthroline (
4i); beige; 1.2 g (3.9 mmol, 78%); m.p
dec. = 162.9–163.7 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 2.92 (s, 3H, CH
3), 2.94 (s, 3H, CH
3), 7.63 (s, 1H, aromatic), 7.66 (s, 1H, aromatic), 8.27 (s, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 100.5 MHz) δ = 25.3, 25.7, 122.7, 124.5, 124.6, 125.0, 128.0, 129.0, 142.1, 142.3, 144.8, 147.6, 160.2, 160.3; GC-MS: t
r = 9.9 min, (EI) M
+ = 310 (100%), 312 (95%), [M-Cl]
+ = 275 (11%), [M-2Cl]
+ = 240 (40%); Anal. Calcd for C
14H
9N
2Cl
3: C, 53.97; H, 2.91; N, 8.99; Found: C, 53.90; H, 3.01; N, 8.87; UV-Vis (methanol; λ [nm] (logε)): 316 (4.16), 304 (4.21), 274 (4.64), 247 (4.67), 210 (4.55); IR (KBr):
= 3456, 3065, 2922, 1594, 1576, 1528, 1332, 905, 874 cm
−1 (
Supporting Information Figure S9).
5-Bromo-4,7-dichloro-2,9-dimethyl-1,10-phenanthroline (
4j); beige; 1.2 g (3.4 mmol, 68%); m.p
dec. = 180.1–181.7 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 2.93 (s, 3H, CH
3), 2.95 (s, 3H, CH
3), 7.63 (s, 1H, aromatic), 7.69 (s, 1H, aromatic), 8.58 (s, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 100.5 MHz) δ = 25.2, 25.8, 116.2, 123.2, 125.0, 125.1, 127.8, 129.2, 142.1, 143.0, 145.0, 147.2, 160.0, 160.5; GC-MS: t
r = 12.5 min, (EI) M
+ = 354 (63.7%), 356 (100%), 358 (44%), [M-Br]
+ = 275 (28%); Anal. Calcd for C
14H
9N
2Cl
2Br: C, 47.23; H, 2.55; N, 7.87; Found: C, 47.90; H, 2.67; N, 7.86; UV-Vis (methanol; λ [nm] (logε)): 318 (3.97), 306 (4.02), 275 (4.43), 249 (4.43), 213 (4.34); IR (KBr):
= 3452, 2923, 1588, 1438, 1117, 900, 876, 718 cm
−1 (
Supporting Information Figure S10).
4,7-Dichloro-2,5,9-trimethyl-1,10-phenanthroline (
4k) [
42,
44]; beige; 1.2 g (4.3 mmol, 86%); m.p. = 111.9–112.6 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 2.92 (s, 3H, CH
3), 2.96 (s, 3H, CH
3), 3.11 (s, 3H, CH
3), 7.62 (bs, 2H, aromatic), 7.95 (s, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 100.5 MHz) δ = 25.2, 25.6, 26.3, 123.9, 124.5, 124.6, 125.4, 126.9, 133.8, 142.3, 143.1, 144.9, 147.2, 159.0, 159.1; CP/MAS
13C-NMR δ = 25.0, 27.5, 122.5, 123.5, 131.0, 139.0, 143.3, 145.3, 158.3; CP MAS
15N-NMR δ = −75.52; GC-MS: t
r = 9.7 min, (EI) M
+ = 290 (100 %), [M-Cl]
+ = 255 (12%); Anal. Calcd for C
15H
12N
2Cl
2: C, 61.88; H, 4.15; N, 9.62; Found: C, 61.89; H, 4.20; N, 9.60; UV-Vis (methanol; λ [nm] (logε)): 343 (3.57), 311 (3.73), 275 (4.30), 254 (4.34), 213 (4.24); IR (KBr):
= 3452, 2924, 1609, 1579, 1530, 1439, 1380, 1236, 902, 871, 716 cm
−1 (
Supporting Information Figure S11).
4,7-Dichloro-2,9-dimethyl-1,10-phenanthroline-5-carbonitrile (
4l); beige; 0.5 g (1.6 mmol, 32%); m.p
dec. = 188.6–189.9 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 2.96 (s, 3H, CH
3), 2.99 (s, 3H, CH
3), 7.72 (s, 1H, aromatic), 7.73 (s, 1H, aromatic), 8.79 (s, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 125.78 MHz) δ = 25.6, 26.2, 106.9, 118.4, 122.6, 123.7, 125.6, 127.0, 134.3, 142.2, 143.7, 146.1, 146.8, 161.6, 163.8; GC-MS: t
r = 10.5 min, (EI) M
+ = 301 (100%), [M-Cl]
+ = 266 (14%); Anal. Calcd for C
15H
9N
3Cl
2: C, 59.63; H, 3.00; N, 13.91; Found: C, 59.89; H, 3.27; N, 13.87; UV-Vis (methanol; λ [nm] (logε)): 353 (3.07), 337 (3.68), 319 (4.24), 307 (4.31), 275 (4.67), 249 (4.60), 235 (4.54), 211 (4.59); IR (KBr):
= 3471, 3406, 2223, 1587, 1443, 1357, 1338, 908, 847 cm
−1 (
Supporting Information Figure S12).
Ethyl 4,7-dichloro-2,9-dimethyl-1,10-phenanthroline-5-carboxylate (
4m); beige; 0.7 g (1.9 mmol, 38%); m.p. = 115.9–116.2 °C;
1H-NMR (CDCl
3; 400.2 MHz) δ = 1.43 (t,
3JH,H = 7.1 Hz, 3H, OCH
2C
H3), 2.93 (s, 3H, CH
3), 2.94 (s, 3H, CH
3), 4.49 (q,
3JH,H = 7.1 Hz, 2H, OCH
2), 7.65 (2s, 2H, aromatic), 8.31 (s, 1H, aromatic);
13C{
1H}-NMR (CDCl
3; 100.5 MHz) δ = 14.1, 25.5, 25.9, 62.6, 122.1, 123.5, 123.8, 124.9, 126.0, 129.3, 141.7, 143.6, 146.3, 146.6, 160.5, 161.5, 169.0; GC-MS: t
r = 10.9 min, (EI) M
+ = 348 (45%), [M-Cl]
+ = 313 (21%); Anal. Calcd for C
17H
14N
2Cl
2O
2: C, 58.47; H, 4.04; N, 8.02; Found: C, 58.90; H, 4.25; N, 7.98; UV-Vis (methanol; λ [nm] (logε)): 308 (3.88), 295 (3.95), 272 (4.43), 245 (4.44), 212 (4.33); IR (KBr):
= 3432, 2980, 1727, 1577, 1275, 1248, 1214,1190, 1122, 1031 cm
−1 (
Supporting Information Figure S13).


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









