
The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
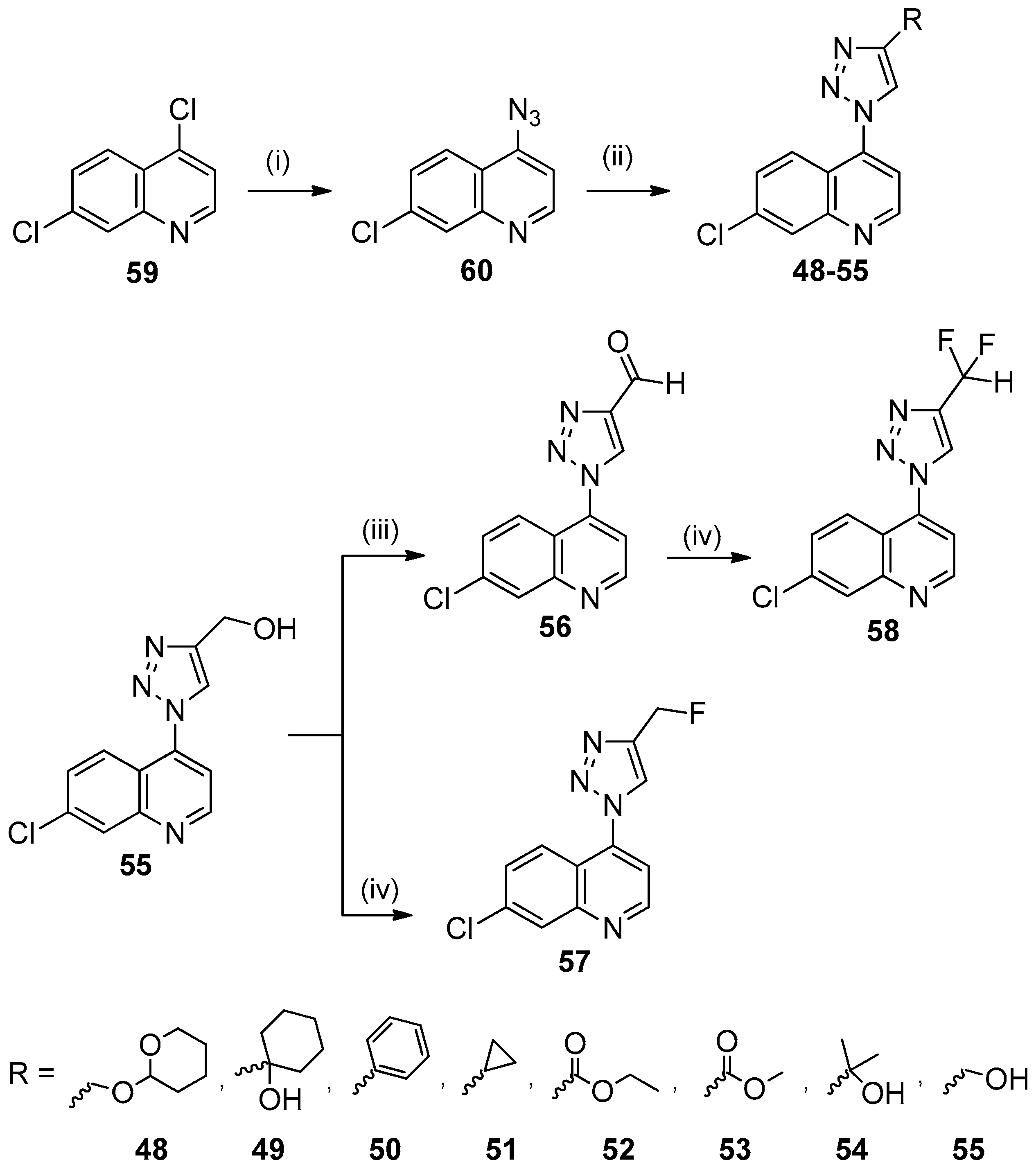{kind=link}
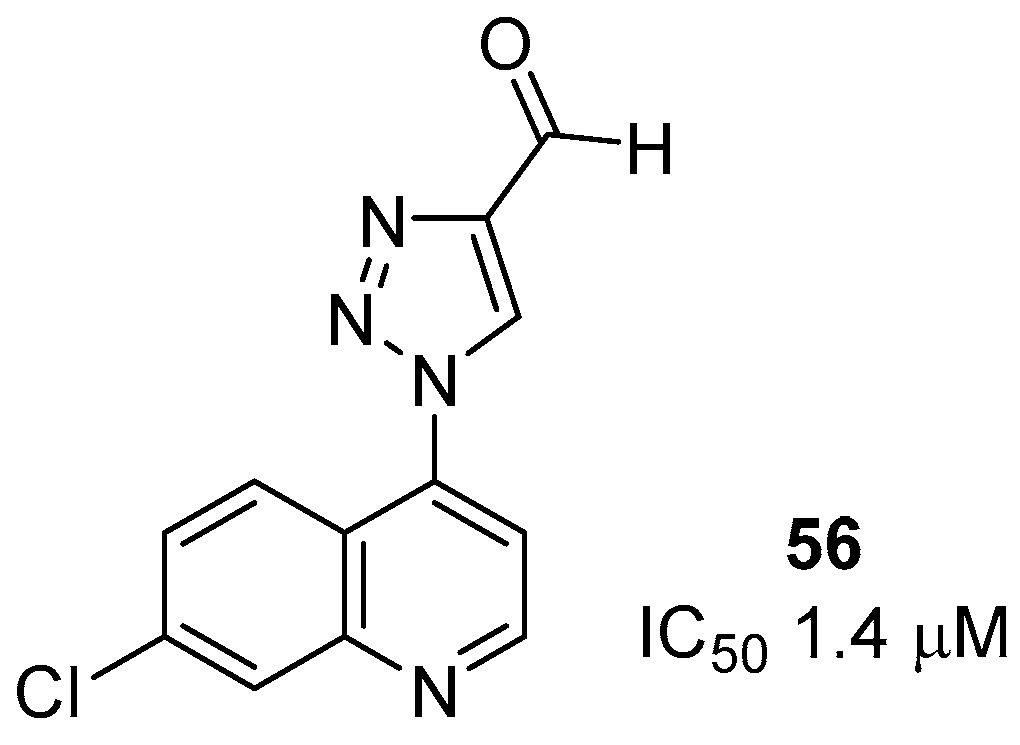{kind=link}
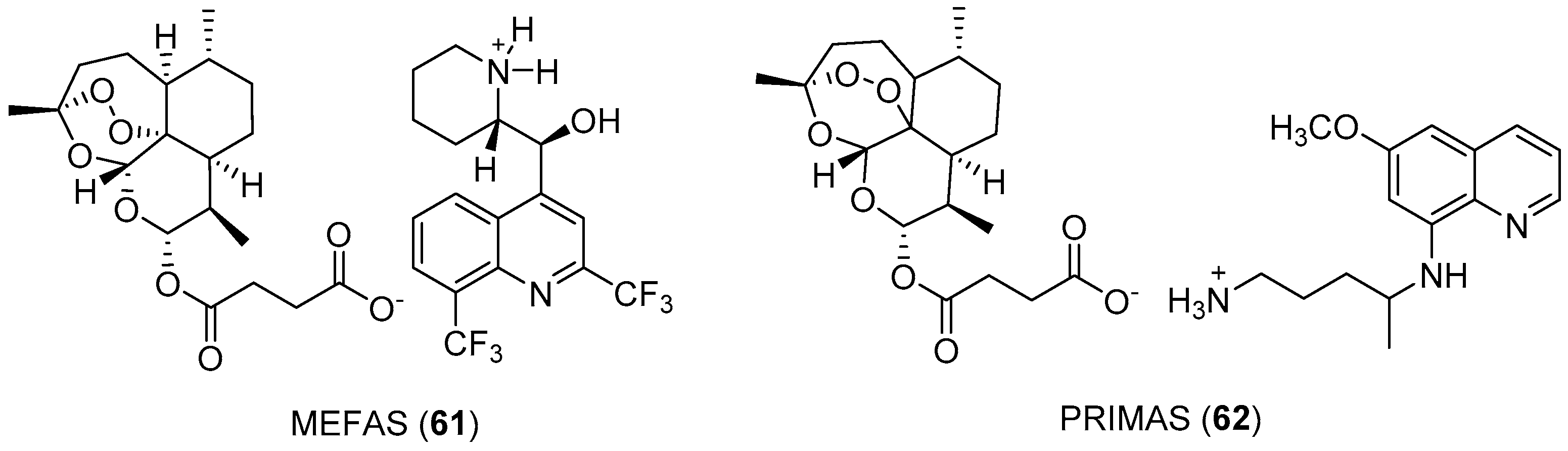{kind=link}
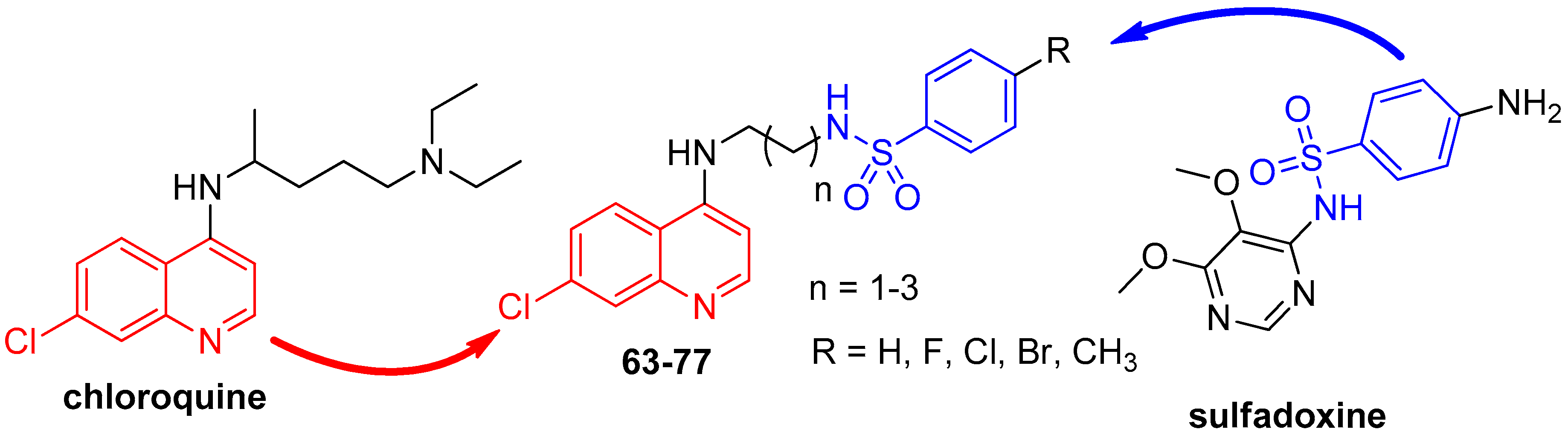{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). World Malaria Report. Available online: https://apps.who.int/iris/bitstream/handle/10665/275867/9789241565653-eng.pdf?ua=1 (accessed on 6 July 2019).
- World Health Organization (WHO). Guidelines for the Treatment of Malaria. 3rd ed.; Available online: http://www.who.int/malaria/publications/atoz/9789241549127/en/ (accessed on 6 July 2019).
- Hott, A.; Tucker, M.S.; Casandra, D.; Sparks, K.; Kyle, D.E. Fitness of artemisinin-resistant Plasmodium falciparum in vitro. J. Antimicrob. Chemother. 2015, 70, 2787–2796. [Google Scholar] [CrossRef] [PubMed]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.V.; Rizk, S.S.; Njimoh, D.L.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.C.; Ubben, D.; Wells, T.N.C. A framework for assessing the risk of resistance for anti-malarials in development. Malaria J. 2012, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Naß, J.; Efferth, T. Development of artemisinin resistance in malaria therapy. Pharmacol. Res. 2019, 146, 104275. [Google Scholar] [CrossRef]
- Anthony, M.P.; Burrows, J.N.; Duparc, S.; Moehrle, J.J.; Wells, T.N.C. The global pipeline of new medicines for the control and elimination of malaria. Malaria J. 2012, 11, 316. [Google Scholar] [CrossRef]
- Alven, S.; Aderibigbe, B. Combination Therapy Strategies for the Treatment of Malaria. Molecules 2019, 24, 3601. [Google Scholar] [CrossRef]
- Xhamla Nqoro, X.; Naki Tobeka, N.; Aderibigbe, B.A. Quinoline-Based Hybrid Compounds with Antimalarial Activity. Molecules 2017, 22, 2268. [Google Scholar] [CrossRef]
- Medicines for Malaria Venture (MMV). Global Portfolio of Antimalarial Medicines. Available online: https://www.mmv.org/research-development/mmv-supported-projects (accessed on 6 July 2019).
- Narula, A.K.; Azad, C.S.; Nainwal, L.M. New dimensions in the field of antimalarial research against malaria resurgence. Eur. J. Med. Chem. 2019, 181, 111353. [Google Scholar] [CrossRef]
- Marella, A.; Verma, G.; Shaquiquzzaman, M.D.; Khan, M.D.F.; Akhtar, W.; Alam, M.D.M. Malaria Hybrids: A Chronological Evolution. Mini-Rev. Med. Chem. 2019, 19, 1144–1177. [Google Scholar] [CrossRef]
- Wamae, K.; Okanda, D.; Ndwiga, L.; Osoti, V.; Kimenyi, K.M.; Abdi, A.I.; Bejon, P.; Sutherland, C.; Ochola-Oyier, L.I. No evidence of P. falciparum K13 artemisinin conferring mutations over a 24-year analysis in Coastal Kenya, but a near complete reversion to chloroquine wild type parasites. Antimicrob. Agents Chemother. 2010, 45, 3245–3264. [Google Scholar] [CrossRef]
- Wells, T.N.; Hooft van Huijsduijnen, R.; Van Voorhis, W.C. Malaria medicines: A glass half full? Nat. Rev. Drug Discov. 2015, 14, 424–442. [Google Scholar] [CrossRef]
- Boechat, N.; Pinheiro, L.C.S.; Silva, T.S.; Aguiar, A.C.C.; Carvalho, A.S.; Bastos, M.M.; Costa, C.C.P.; Pinheiro, S.; Pinto, A.C.; Mendonça, J.S.; et al. New Trifluoromethyl Triazolopyrimidines as Anti-Plasmodium falciparum Agents. Molecules 2012, 17, 8285–8302. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorination in medicinal chemistry: Methods, strategies, and recent developments. Org. Process. Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Boechat, N.; Bastos, M.M. Trifluoromethylation of carbonyl compounds. Curr. Org. Synth. 2010, 7, 403–413. [Google Scholar] [CrossRef]
- Zohdi, H.F. Reactions with 3-amino-5-(trifluoromethyl)-1,2,4-triazole: A simple route to fluorinated polysubstituted triazolo[1,5-a]pyrimidine and triazolo[5,1-c]triazine derivatives. J. Chem. Res. (S) 1997, 392–393. [Google Scholar] [CrossRef]
- Leal, B.; Afonso, I.F.; Rodrigues, C.R.; Abreu, P.A.; Garrett, R.; Pinheiro, L.C.S.; Azevedo, A.R.; Borges, J.C.; Vegi, P.F.; Santos, C.C.C.; et al. Antibacterial profile against drug-resistant Staphylococcus epidermidis clinical strain and structure–activity relationship studies of 1H-pyrazolo[3,4-b]pyridine and thieno[2,3-b]pyridine derivatives. Bioorg. Med. Chem. 2008, 16, 8196–8204. [Google Scholar] [CrossRef]
- Pinheiro, M.P.; Iulek, J.; Nonato, M.C. Crystal structure of Trypanosoma cruzi dihydroorotate dehydrogenase from Y strain. Biochem. Biophys. Res. Commun. 2008, 369, 812–817. [Google Scholar] [CrossRef]
- Hoelz, L.V.B.; Calil, F.A.; Nonato, M.C.; Pinheiro, L.C.S.; Boechat, N. Plasmodium falciparum dihydroorotate dehydrogenase: A drug target against malaria. Future Med. Chem. 2018, 10, 1853–1874. [Google Scholar] [CrossRef]
- Phillips, M.A.; Gujjar, R.; Malmquist, N.A.; White, J.; El Mazouni, F.; Baldwin, J.; Rathod, P.K. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 2008, 51, 3649–3653. [Google Scholar] [CrossRef]
- Gujjar, R.; El Mazouni, F.; White, K.L.; White, J.; Creason, S.; Shackleford, D.M.; Deng, X.; Charman, W.N.; Bathurst, I.; Burrows, J.; et al. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J. Med. Chem. 2011, 54, 3935–3949. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; White, K.L.; Kokkonda, S.; Deng, X.; White, J.; El Mazouni, F.; Marsh, K.; Tomchick, D.R.; Manjalanagara, K.; Rudra, K.R.; et al. A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria. ACS Infect. Dis. 2016, 2, 945–957. [Google Scholar] [CrossRef]
- Pavadai, E.; El Mazouni, F.; Wittlin, S.; de Kock, C.; Phillips, M.A.; Chibale, K. Identification of new human malaria parasite Plasmodium falciparum dihydroorotate dehydrogenase inhibitors by pharmacophore and structure-based virtual screening. J. Chem. Inf. Model. 2016, 56, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, L.F.S.P.; Coutinho, J.P.; Jabor, V.A.P.; Feliciano, P.R.; Nonato, M.C.; Kaiser, C.R.; Menezes, C.M.S.; Hammes, A.S.O.; Caffarena, E.R.; Hoelz, L.V.B.; et al. Evaluation of 7-arylaminopyrazolo[1,5-a]pyrimidines as anti-Plasmodium falciparum, antimalarial, and Pf-dihydroorotate dehydrogenase inhibitors. Eur. J. Med. Chem. 2017, 126, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Nagle, A.S.; Chatterjee, A.K. Road towards new antimalarials—Overview of the strategies and their chemical progress. Curr. Med. Chem. 2011, 18, 853–871. [Google Scholar] [CrossRef]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef]
- Mushtaque, M.D. Shahjahan Reemergence of chloroquine (CQ) analogs as multi-targeting antimalarial agents: A review. Eur. J. Med. Chem. 2015, 90, 280–295. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, H.; Smith, P.; de Kock, C.; Chibale, K.; Balzarini, J. Quinoline-pyrimidine hybrids: Synthesis, antiplasmodial activity, SAR, and mode of action studies. J. Med. Chem. 2014, 57, 435–448. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, H.; Chibale, K.; Balzarini, J. Synthesis of 4-aminoquinoline-pyrimidine hybrids as potent antimalarials and their mode of action studies. Eur. J. Med. Chem. 2013, 66, 314–323. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, H.; Chibale, K.; Balzarini, J.; Little, S.; Bharatam, P.V. 2-Aminopyrimidine based 4-aminoquinoline anti-plasmodial agents. Synthesis, biological activity, structure-activity relationship and mode of action studies. Eur. J. Med. Chem. 2012, 52, 82–97. [Google Scholar] [CrossRef]
- Vandekerckhove, S.; D’hooghe, M. Quinoline-based antimalarial hybrid compounds. Bioorg. Med. Chem. 2015, 23, 5098–5119. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, P.N.; Karad, S.C.; Raval, D.K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936. [Google Scholar] [CrossRef] [PubMed]
- Boechat, N.; Ferreira, M.L.G.; Pinheiro, L.C.S.; Jesus, A.M.L.; Leite, M.M.M.; Aguiar, A.C.C.; Andrade, I.M.; Krettli, A.U. New compounds hybrids 1H-1,2,3-triazole-quinoline against Plasmodium falciparum. Chem. Biol. Drug. Des. 2014, 84, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bakunov, S.A.; Bakunova, S.M.; Wenzler, T.; Ghebru, M.; Werbovetz, K.A.; Brun, R.; Tidwell, R.R. Synthesis and antiprotozoal activity of cationic 1,4-Diphenyl-1H-1,2,3-triazoles. J. Med. Chem. 2010, 53, 254–272. [Google Scholar] [CrossRef]
- Boechat, N.; Ferreira, V.F.; Ferreira, S.B.; Ferreira, M.L.G.; Silva, F.C.; Bastos, M.M.; Costa, M.S.; Lourenço, M.C.; Pinto, A.C.; Krettli, A.U.; et al. Novel 1,2,3-triazole derivatives for use against Mycobacterium tuberculosis H37Rv (ATCC 27294) strain. J. Med. Chem. 2011, 54, 5988–5999. [Google Scholar] [CrossRef]
- Ferreira, S.B.; Costa, M.S.; Boechat, N.; Bezerra, S.R.J.; Genestra, M.S.; Canto-Cavalheiro, M.M.; Kover, W.B.; Ferreira, V.F. Synthesis and evaluation of new difluoromethyl azoles as antileishmanial agents. Eur. J. Med. Chem. 2007, 42, 1388–1395. [Google Scholar] [CrossRef]
- Muregi, F.W.; Ishih, A. Next-Generation antimalarial drugs: Hybrid molecules as a new strategy in drug design. Drug. Dev. Res. 2010, 71, 20–32. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- de Pilla Varotti, F.; Botelho, A.C.C.; Andrade, A.A.; de Paula, R.C.; Fagundes, E.M.; Valverde, A.; Mayer, L.M.; Mendonça, J.S.; de Souza, M.V.; Boechat, N.; et al. Synthesis, antimalarial activity, and intracellular targets of Mefas, a new hybrid compound derived from mefloquine and artesunate. Antimicrob. Agents Chemother. 2008, 52, 3868–3874. [Google Scholar] [CrossRef]
- Penna-Coutinho, J.; Almela, M.J.; Miguel-Blanco, C.; Herreros, E.; Sá, P.M.; Boechat, N.; Krettli, A.U. Transmission-blocking potential of MEFAS, a hybrid compound derived fromartesunate and mefloquine. Antimicrob. Agents Chemother. 2016, 60, 3145–3147. [Google Scholar] [CrossRef]
- Boechat, N.; Souza, M.V.N.; Valverde, A.L.; Krettli, A.U. Compounds derived from artesunate, preparation process, pharmaceutical composition and use of the respective medicine. U.S. Patent 8,802,701 B2, 12 August 2014. [Google Scholar]
- Teixeira, C.; Vale, N.; Pérez, B.; Gomes, A.; Gomes, J.R.B.; Gomes, P. “Recycling” classical drugs for malaria. Chem. Rev. 2014, 114, 11164–11220. [Google Scholar] [CrossRef] [PubMed]
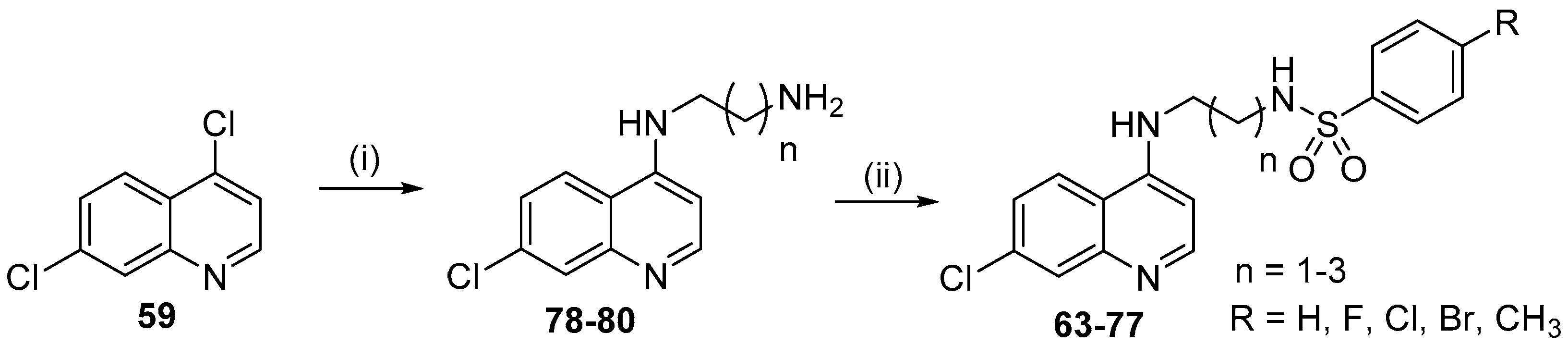
- Pinheiro, L.C.S.; Boechat, N.; Ferreira, M.L.G.; Junior, C.C.S.; Jesus, A.M.L.; Leite, M.M.M.; Souza, N.; Krettli, A.U. Anti-Plasmodium falciparum activity of quinoline-sulfonamide hybrids. Bioorg. Med. Chem. 2015, 23, 5979–5984. [Google Scholar] [CrossRef] [PubMed]
- Nava-Zuazo, C.; Estrada-Soto, S.; Guerrero-Álvarez, J.; León-Rivera, I.; Molina-Salinas, G.M.; Said-Fernández, S.; Chan-Bacab, M.J.; Cedillo-Rivera, R.; Moo-Puc, R.; Mirón-López, G.; et al. Design, synthesis, and in vitro antiprotozoal, antimycobacterial activities of N-{2-[(7-chloroquinolin-4-yl)amino]ethyl}ureas. Bioorg. Med. Chem. 2010, 18, 6398–6403. [Google Scholar] [CrossRef] [PubMed]
- Boechat, N.; Pinheiro, L.C.S.; Santos-Filho, O.A.; Silva, I.C. Design and synthesis of new N-(5-trifluoromethyl)-1H-1,2,4-triazol-3-yl benzenesulfonamides as possible antimalarial prototypes. Molecules 2011, 16, 8083–8097. [Google Scholar] [CrossRef]
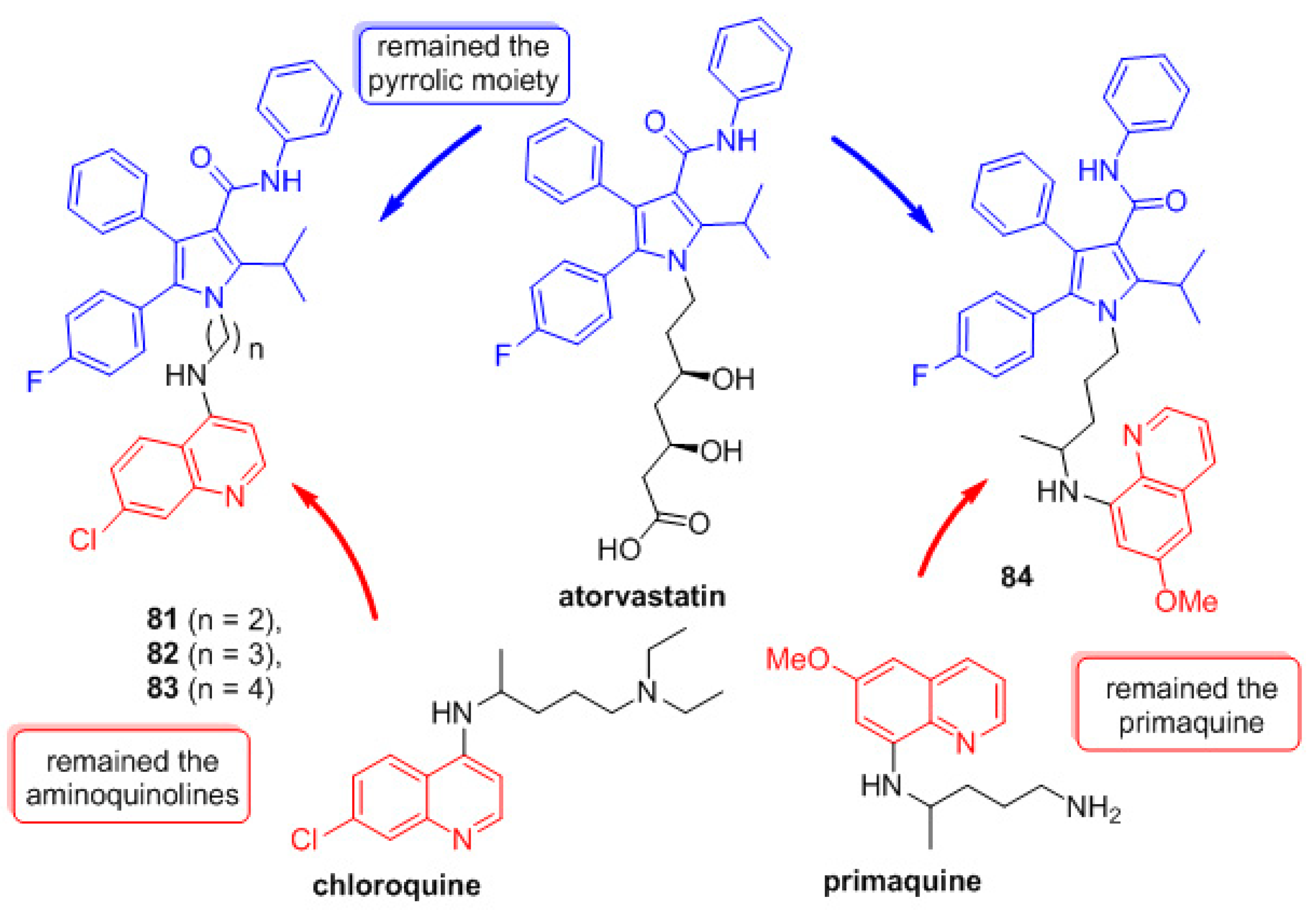
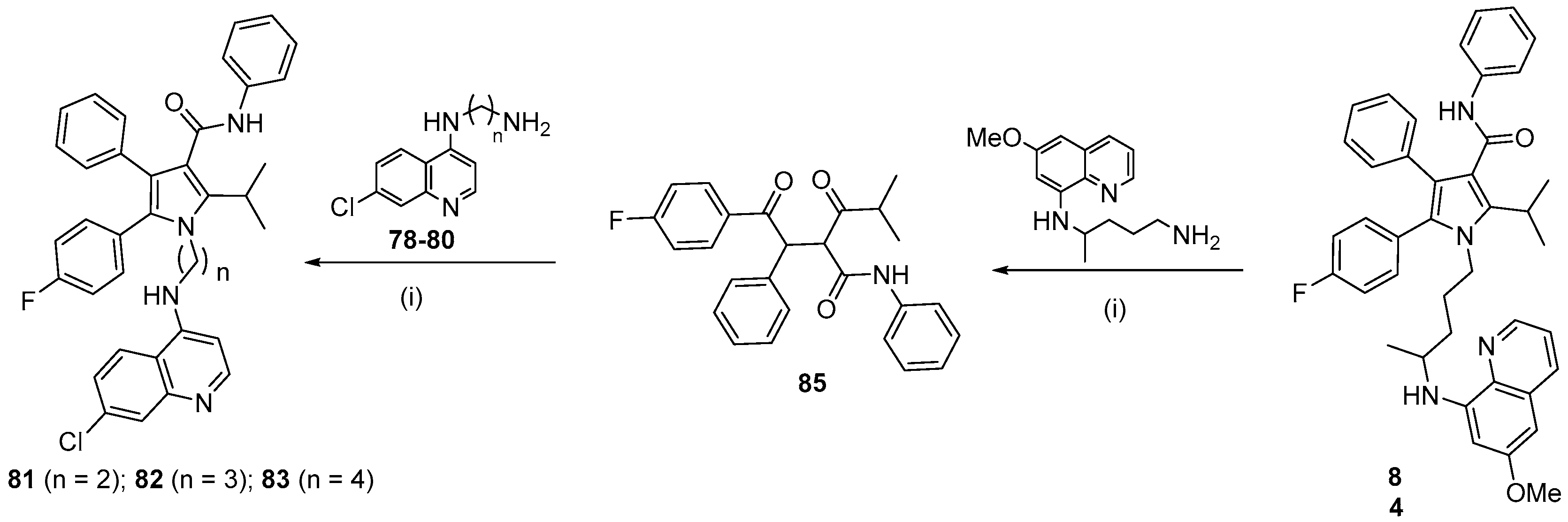
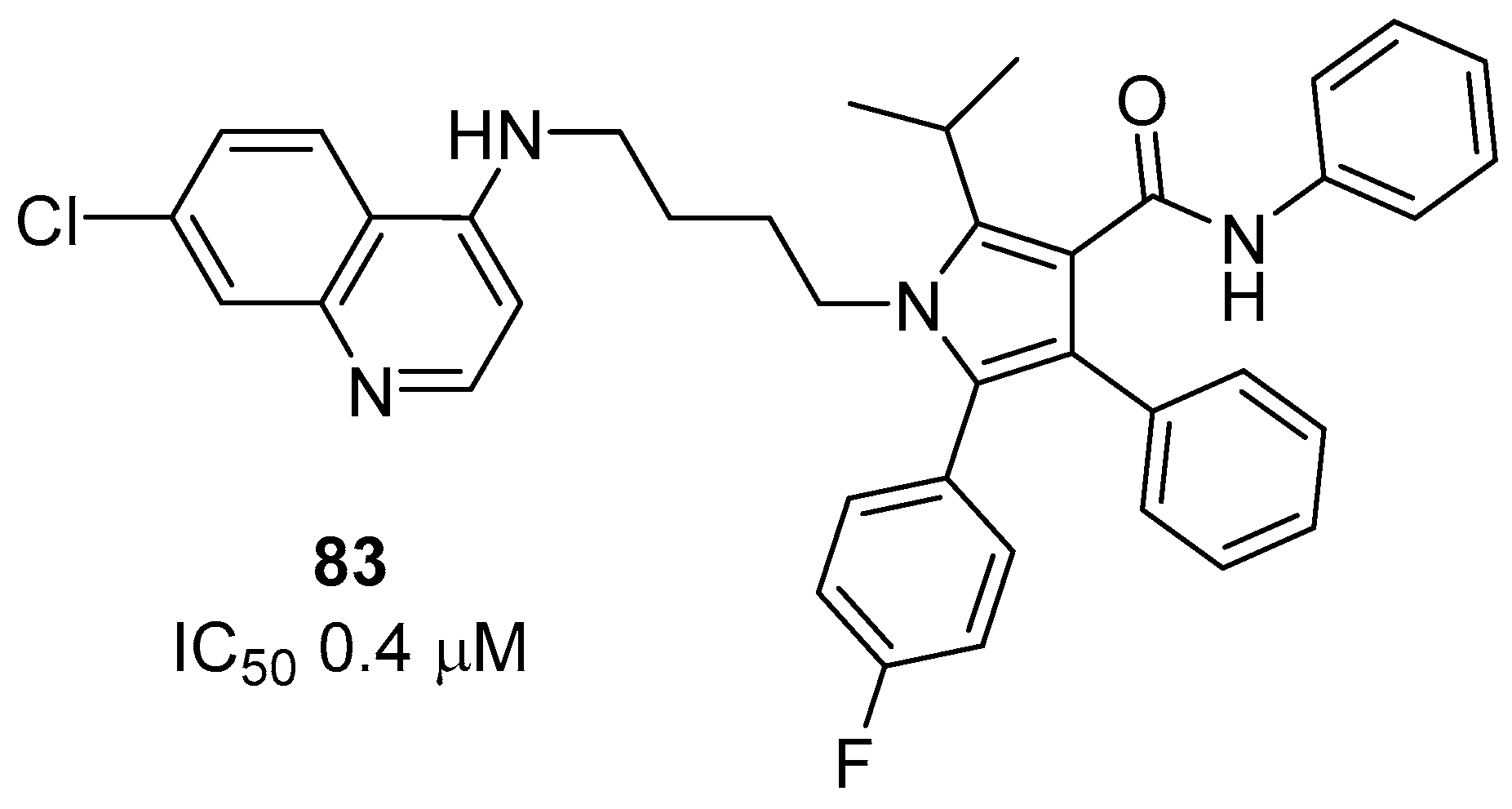
- Carvalho, R.C.C.; Martins, W.A.; Silva, T.P.; Kaiser, C.R.; Bastos, M.M.; Pinheiro, L.C.S.; Krettli, A.U.; Boechat, N. New pentasubstituted pyrrole hybrid atorvastatin–quinoline derivatives with antiplasmodial activity. Bioorg. Med. Chem. Lett. 2016, 26, 1881–1884. [Google Scholar] [CrossRef]
- Gudipati, S.; Katram, S.; Komati, S.; Kudavalli, S.J. Amorphous atorvastatin calcium. WO Patent 2,006,039,441 A2, 13 April 2006. [Google Scholar]
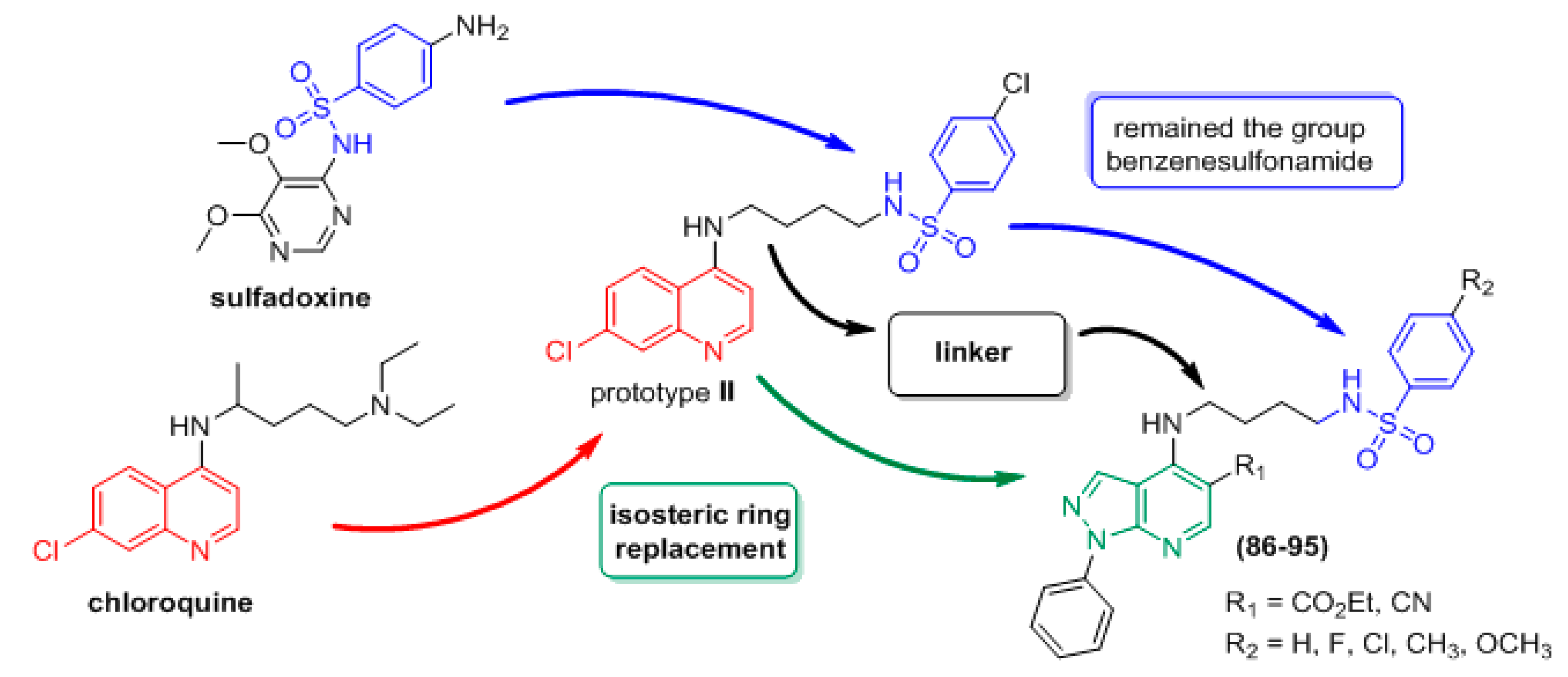
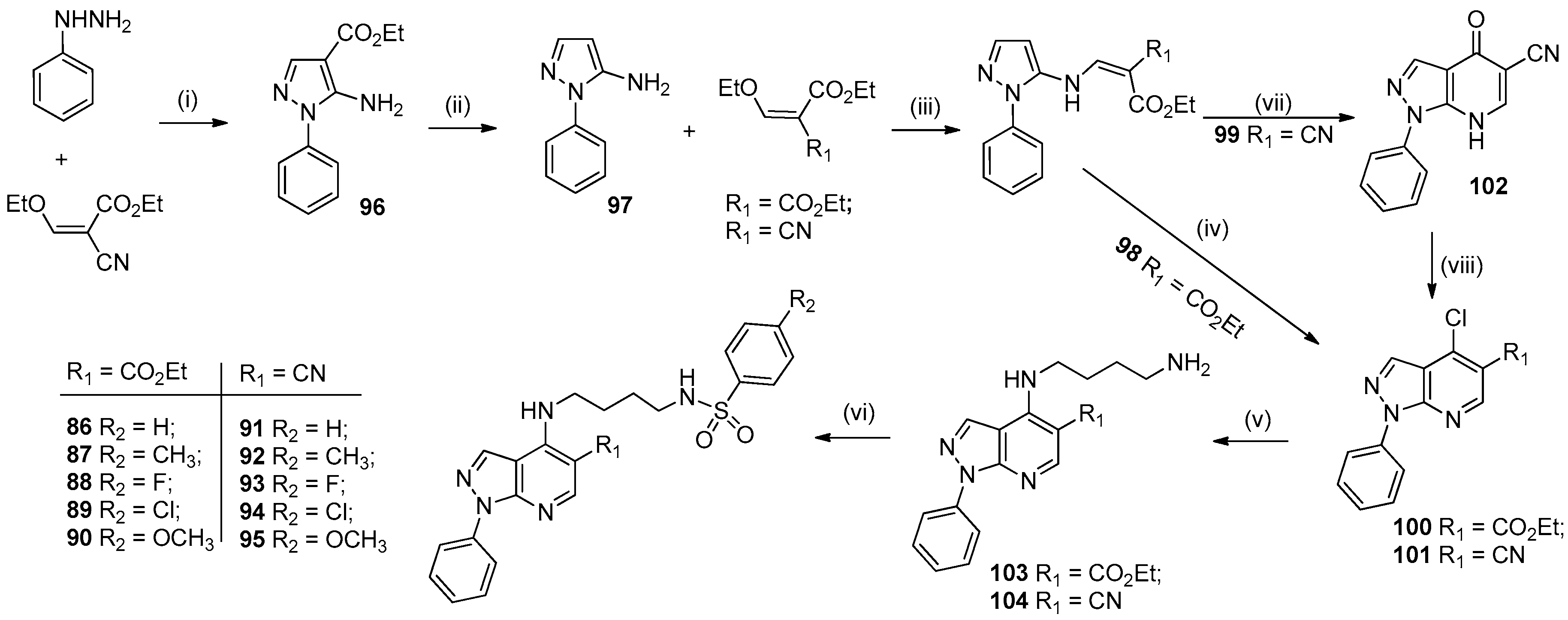
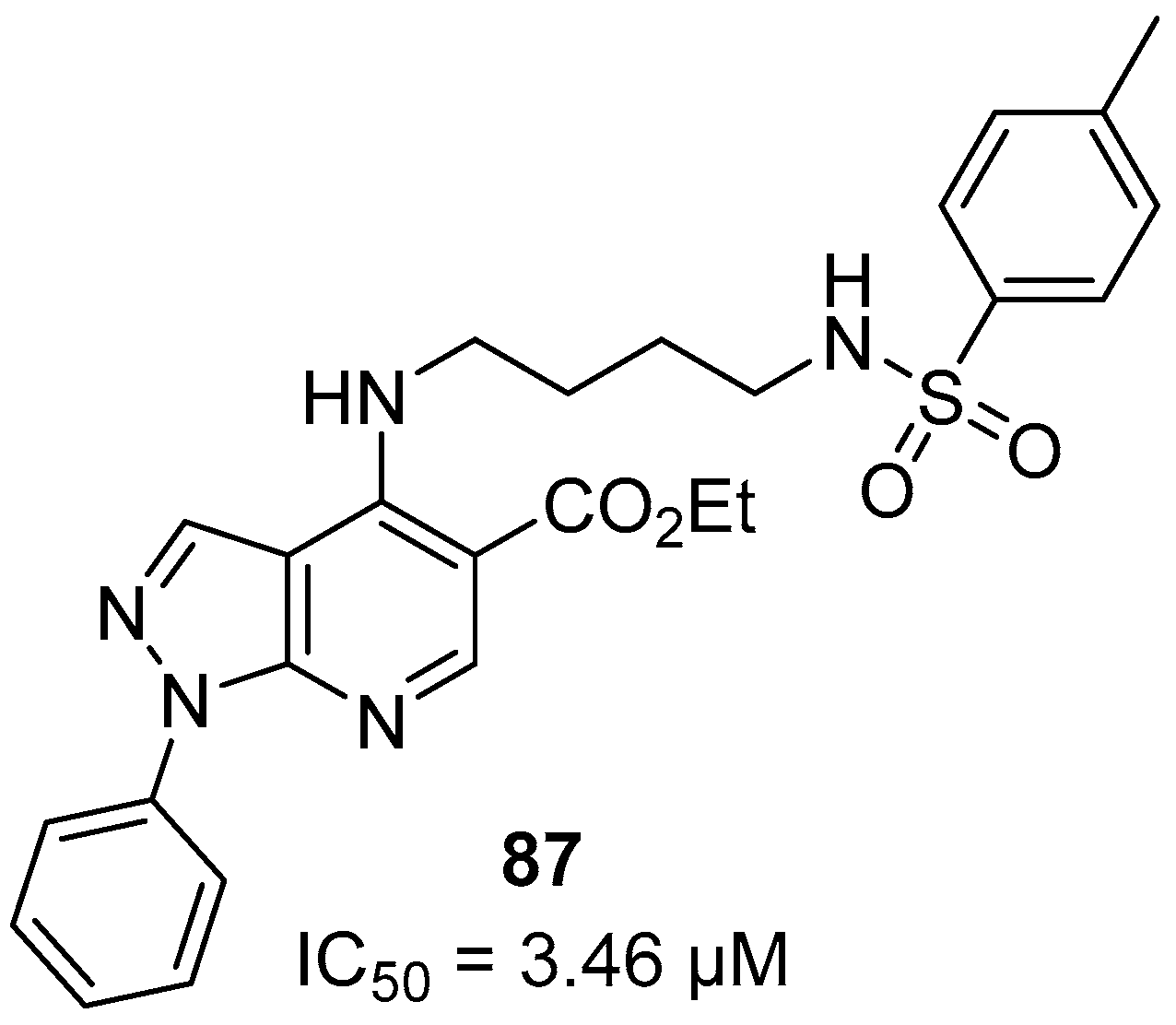
- Silva, T.B.; Bernardino, A.M.R.; Ferreira, M.L.G.; Rogerio, K.R.; Carvalho, L.J.M.; Boechat, N.; Pinheiro, L.C.S. Design, synthesis and anti-P. falciparum activity of pyrazolopyridine-sulfonamide derivatives. Bioorg. Med. Chem. 2016, 24, 4492–4498. [Google Scholar] [CrossRef]
- Manach, C.L.; Paquet, T.; Brunschwig, C.; Njoroge, M.; Han, Z.; Cabrera, D.G.; Bashyam, S.; Dhinakaran, R.; Taylor, D.; Reader, J.; et al. A novel pyrazolopyridine with in vivo activity in Plasmodium berghei- and Plasmodium falciparum-infected mouse models from structure–activity relationship studies around the core of recently identified antimalarial imidazopyridazines. J. Med. Chem. 2015, 58, 8713–8722. [Google Scholar] [CrossRef]
- Bernardino, A.M.R.; Azevedo, A.R.; Pinheiro, L.C.S.; Borges, J.C.; Carvalho, V.L.; Miranda, M.D.; Meneses, M.D.F.; Nascimento, M.; Ferreira, D.; Rebello, M.A.; et al. Synthesis and antiviral activity of new 4-(phenylamino)/4-[(methylpyridin-2-yl)amino]-1-phenyl-1Hpyrazolo[3,4-b]pyridine-4-carboxylic acids derivatives. Med. Chem. Res. 2007, 16, 352–369. [Google Scholar] [CrossRef]
- Bernardino, A.M.R.; Pinheiro, L.C.S.; Rodrigues, C.R.; Loureiro, N.I.; Castro, H.C.; Lanfredi-Rangel, A.; Sabatini-Lopes, J.; Borges, J.C.; Carvalho, J.M.; Romeiro, G.A.; et al. Design, synthesis, SAR, and biological evaluation of new 4-(phenylamino)thieno[2,3-b]pyridine derivatives. Bioorg. Med. Chem. 2006, 14, 5765–5770. [Google Scholar] [CrossRef]
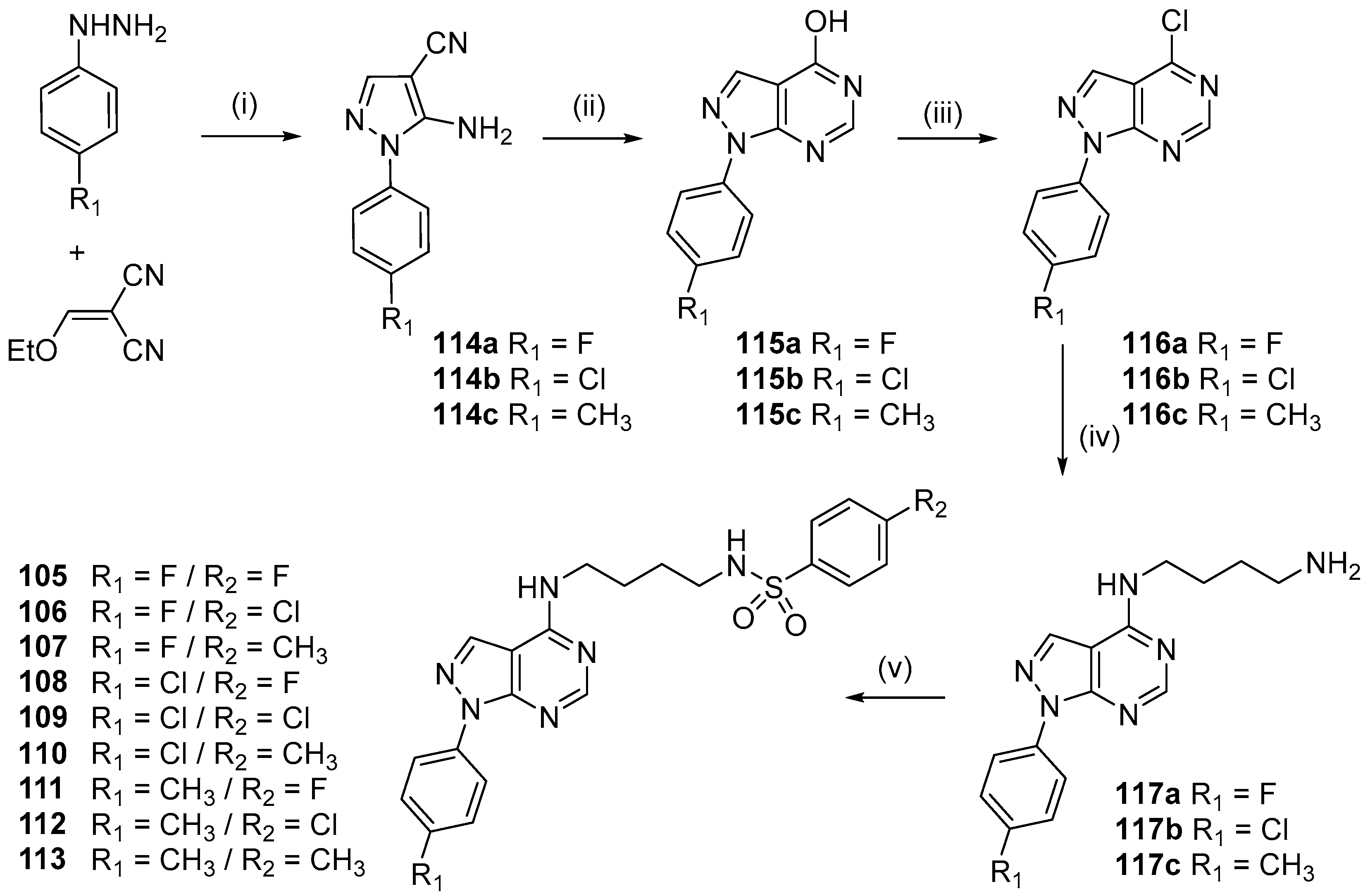
- Silveira, F.F.; Feitosa, L.M.; Mafra, J.C.M.; Ferreira, M.L.G.; Rogerio, K.R.; Carvalho, L.J.M.; Boechat, N.; Pinheiro, L.C.S. Synthesis and anti-Plasmodium falciparum evaluation of novel pyrazolopyrimidine derivatives. Med. Chem. Res. 2018, 27, 1876–1884. [Google Scholar] [CrossRef]
- Santos, M.S.; Bernardino, A.M.R.; Pinheiro, L.C.S.; Canto-Cavalheiro, M.M.; Leon, L.L. An efficient synthesis of new 5-(1-aryl-1H-pyrazole-4-yl)-1H-tetrazoles from 1-aryl-1H-pyrazole-4-carbonitriles via [3 + 2]cycloaddition reaction. J. Heterocyclic Chem. 2012, 49, 1425–1428. [Google Scholar] [CrossRef]
- Soliman, A.M.; Sultan, A.A.; El Remaily, M.A.A.; Abdel-Ghany, H. Synthesis of some novel fused azole derivatives. Synth. Commun. 2012, 42, 2748–2762. [Google Scholar] [CrossRef]
- da Silva, R.M.R.J.; Gandi, M.O.; Mendonça, J.S.; Carvalho, A.S.; Coutinho, J.P.; Aguiar, A.C.C.; Krettli, A.U.; Boechat, N. New hybrid trifluoromethylquinolines as antiplasmodium agents. Bioorg. Med. Chem. 2019, 27, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Ohnmacht, C.J.; Patel, A.R.; Lutz, R.E. Antimalarials. 7. Bis(trifluoromethyl)-α-(2-piperidyl)-4-quinolinemethanols. J. Med. Chem. 1971, 14, 926–928. [Google Scholar] [PubMed]
- Meshram, H.M.; Reddy, B.C.; Kumar, D.A.; Kalyan, M.; Ramesh, P.; Kavitha, P.; Rao, J.V. Synthesis and cytotoxicity of new quinoline derivatives. Ind. J. Chem. 2012, 51B, 1411–1416. [Google Scholar]
- Witkowski, B.; Khim, N.; Chim, P.; Kim, S.; Ke, S.; Kloeung, N.; Chy, S.; Duong, S.; Leang, R.; Ringwald, P.; et al. Reduced Artemisinin Susceptibility of Plasmodium falciparum Ring Stages in Western Cambodia. Antimicrob. Agents Chemother. 2013, 57, 914–923. [Google Scholar] [CrossRef] [PubMed]


























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
C. S. Pinheiro, L.; M. Feitosa, L.; O. Gandi, M.; F. Silveira, F.; Boechat, N. The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines. Molecules 2019, 24, 4095. https://doi.org/10.3390/molecules24224095
C. S. Pinheiro L, M. Feitosa L, O. Gandi M, F. Silveira F, Boechat N. The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines. Molecules. 2019; 24(22):4095. https://doi.org/10.3390/molecules24224095
Chicago/Turabian StyleC. S. Pinheiro, Luiz, Lívia M. Feitosa, Marilia O. Gandi, Flávia F. Silveira, and Nubia Boechat. 2019. "The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines" Molecules 24, no. 22: 4095. https://doi.org/10.3390/molecules24224095
APA StyleC. S. Pinheiro, L., M. Feitosa, L., O. Gandi, M., F. Silveira, F., & Boechat, N. (2019). The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines. Molecules, 24(22), 4095. https://doi.org/10.3390/molecules24224095