
[b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
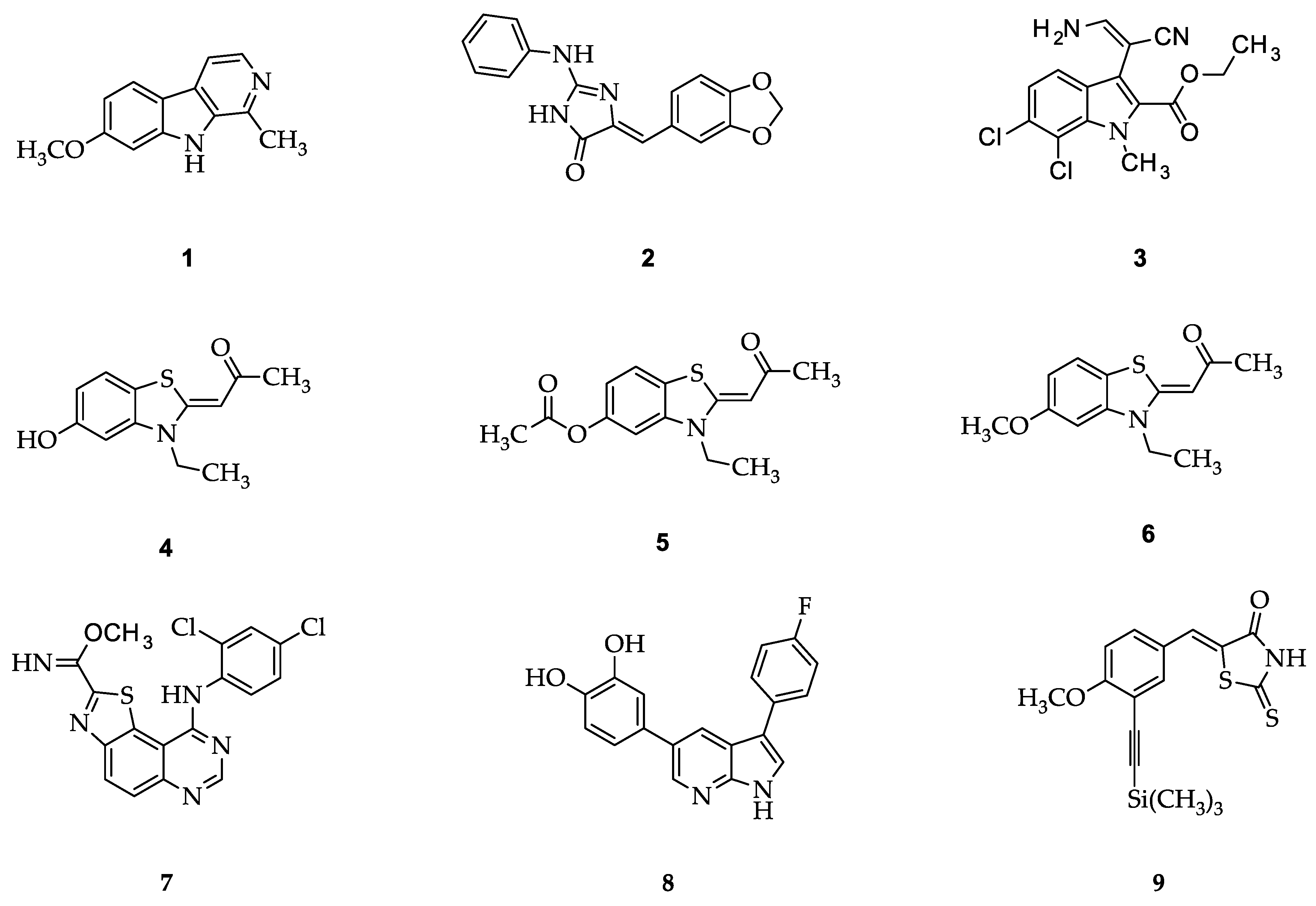
2.1. Structure Design Considerations and Docking Analyses
2.2. Syntheses
2.3. Kinase Inhibitory Activity
2.4. Solubility
2.5. Crystal Structure Analysis
3. Materials and Methods
3.1. General Information
3.2. Syntheses
3.2.1. General procedure for the synthesis of the Mannich base hydrochlorides (Procedure A)
3.2.2. Synthesis and Characterization of 11, 12, 13, 14, 15, 16, 20, 21, 22, 24, 25, 26, 27 and 28
3.3. Molecular Docking
3.4. Protein Kinase Assays
3.5. Determination of Thermodynamic Solubility
3.6. Determination of Kinetic Solubility
3.7. Crystallization of DYRK1A-11 Complex, Data Collection and Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Becker:, W.; Weber, Y.; Wetzel, K.; Eirmbter, K.; Tejedor, F.J.; Joost, H.G. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J. Biol. Chem. 1998, 273, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Hammerle, B.; Ulin, E.; Guimera, J.; Becker, W.; Guillemot, F.; Tejedor, F.J. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development 2011, 138, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Aranda, S.; Barallobre, M.J.; Barhoum, R.; Fernandez, E.; Fotaki, V.; Delabar, J.M.; de la Luna, S.; de la Villa, P.; Arbones, M.L. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 2008, 15, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle 2012, 11, 3389–3394. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jeong, B.C.; Lee, J.H.; Kee, H.J.; Kook, H.; Kim, N.S.; Kim, Y.H.; Kim, J.K.; Ahn, K.Y.; Kim, K.K. A repressor complex, AP4 transcription factor and geminin, negatively regulates expression of target genes in nonneuronal cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13074–13079. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.O.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef]
- Pozo, N.; Zahonero, C.; Fernandez, P.; Linares, J.M.; Ayuso, A.; Hagiwara, M.; Perez, A.; Ricoy, J.R.; Hernandez-Lain, A.; Sepulveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef]
- Grau, C.; Arato, K.; Fernandez-Fernandez, J.M.; Valderrama, A.; Sindreu, C.; Fillat, C.; Ferrer, I.; de la Luna, S.; Altafaj, X. DYRK1A-mediated phosphorylation of GluN2A at Ser(1048) regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front. Cell. Neurosci. 2014, 8, 331. [Google Scholar] [CrossRef]
- Duchon, A.; Herault, Y. DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Soppa, U.; Tejedor, F.J. DYRK1A: A potential drug target for multiple Down syndrome neuropathologies. CNS Neurol. Disord. Drug Targets 2014, 13, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Fruit, C.; Herault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: A new avenue for a disease modifying treatment of Alzheimer’s? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef]
- Esvan, Y.J.; Zeinyeh, W.; Boibessot, T.; Nauton, L.; Thery, V.; Knapp, S.; Chaikuad, A.; Loaec, N.; Meijer, L.; Anizon, F.; et al. Discovery of pyrido[3,4-g]quinazoline derivatives as CMGC family protein kinase inhibitors: Design, synthesis, inhibitory potency and X-ray co-crystal structure. Eur. J. Med. Chem. 2016, 118, 170–177. [Google Scholar] [CrossRef]
- Kim, H.; Sablin, S.O.; Ramsay, R.R. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef]
- Souchet, B.; Audrain, M.; Billard, J.M.; Dairou, J.; Fol, R.; Orefice, N.S.; Tada, S.; Gu, Y.; Dufayet-Chaffaud, G.; Limanton, E.; et al. Inhibition of DYRK1A proteolysis modifies its kinase specificity and rescues Alzheimer phenotype in APP/PS1 mice. Acta Neuropathol. Commun. 2019, 7, 46. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Duchon, A.; Manousopoulou, A.; Loaec, N.; Villiers, B.; Pani, G.; Karatas, M.; Mechling, A.E.; Harsan, L.A.; Limanton, E.; et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Loaec, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roue, M.; Bourguet-Kondracki, M.L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef]
- Fant, X.; Durieu, E.; Chicanne, G.; Payrastre, B.; Sbrissa, D.; Shisheva, A.; Limanton, E.; Carreaux, F.; Bazureau, J.P.; Meijer, L. cdc-like/dual-specificity tyrosine phosphorylation-regulated kinases inhibitor leucettine L41 induces mTOR-dependent autophagy: Implication for Alzheimer’s disease. Mol. Pharmacol. 2014, 85, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Burgy, G.; Tahtouh, T.; Durieu, E.; Foll-Josselin, B.; Limanton, E.; Meijer, L.; Carreaux, F.; Bazureau, J.P. Chemical synthesis and biological validation of immobilized protein kinase inhibitory Leucettines. Eur. J. Med. Chem. 2013, 62, 728–737. [Google Scholar] [CrossRef]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef]
- Foucourt, A.; Hedou, D.; Dubouilh-Benard, C.; Girard, A.; Taverne, T.; Casagrande, A.S.; Desire, L.; Leblond, B.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef]
- Chaikuad, A.; Diharce, J.; Schroder, M.; Foucourt, A.; Leblond, B.; Casagrande, A.S.; Desire, L.; Bonnet, P.; Knapp, S.; Besson, T. An unusual binding model of the methyl 9-anilinothiazolo[5,4-f] quinazoline-2-carbimidates (EHT 1610 and EHT 5372) confers high selectivity for dual-specificity tyrosine phosphorylation-regulated kinases. J. Med. Chem. 2016, 59, 10315–10321. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.; Gourdain, S.; Albac, C.; Dekker, A.D.; Bui, L.C.; Dairou, J.; Schmitz-Afonso, I.; Hue, N.; Rodrigues-Lima, F.; Delabar, J.M.; et al. DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci. Rep. 2018, 8, 2859. [Google Scholar] [CrossRef] [PubMed]
- Kii, I.; Sumida, Y.; Goto, T.; Sonamoto, R.; Okuno, Y.; Yoshida, S.; Kato-Sumida, T.; Koike, Y.; Abe, M.; Nonaka, Y.; et al. Selective inhibition of the kinase DYRK1A by targeting its folding process. Nat. Commun. 2016, 7, 11391. [Google Scholar] [CrossRef] [PubMed]
- Falke, H.; Chaikuad, A.; Becker, A.; Loaec, N.; Lozach, O.; Abu Jhaisha, S.; Becker, W.; Jones, P.G.; Preu, L.; Baumann, K.; et al. 10-iodo-11H-indolo[3,2-c]quinoline-6-carboxylic acids are selective inhibitors of DYRK1A. J. Med. Chem. 2015, 58, 3131–3143. [Google Scholar] [CrossRef] [PubMed]
- Meine, R.; Becker, W.; Falke, H.; Preu, L.; Loaec, N.; Meijer, L.; Kunick, C. Indole-3-carbonitriles as DYRK1A inhibitors by fragment-based drug design. Molecules 2018, 23, 64. [Google Scholar] [CrossRef] [PubMed]
- Falke, H. Neue Selektive Hemmstoffe der Proteinkinase DYRK1A. Dissertation Technische Universität Braunschweig; Shaker Verlag: Aachen, Germany, 2014. [Google Scholar]
- Cuthbertson, T.J.; Ibanez, M.; Rijnbrand, C.A.; Jackson, A.J.; Mittapalli, G.K.; Zhao, F.; MacDonald, J.E.; Wong-Staal, F. Hepatitis c Virus Entry Inhibitors. WO 2008/021745, 21 February 2008. [Google Scholar]
- Yamane, K.; Fujimori, K. A convenient synthesis of indolotropones and 6-substituted 5-azabenz[b]azulenes. Bull. Soc. Chem. Jpn. 1976, 49, 1101–1104. [Google Scholar] [CrossRef]
- Gore, S.; Baskaran, S.; König, B. Fischer indole synthesis in low melting mixtures. Org. Lett. 2012, 14, 4568–4571. [Google Scholar] [CrossRef]
- Saal, C.; Petereit, A.C. Optimizing solubility: Kinetic versus thermodynamic solubility temptations and risks. Eur. J. Pharm. Sci. 2012, 47, 589–595. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- CCG. Molecular Operating Environment; 2015.1001; Chemical Computing Group Inc.: Montreal, QC, Canada, 2015. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Lobley, C.M.; Prince, S.M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J. Acknowledging Errors: Advanced Molecular Replacement with Phaser. Methods Mol. Biol. 2017, 1607, 421–453. [Google Scholar] [CrossRef]
- Emsley, P. Tools for ligand validation in Coot. Acta Crystallogr. D Struct. Biol. 2017, 73, 203–210. [Google Scholar] [CrossRef]
- Skubak, P.; Murshudov, G.N.; Pannu, N.S. Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2196–2201. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry. | CDK1 | CDK2 | CDK5 | CDK9 | CK1 | CLK1 | DYRK1A | GSK-3 |
|---|---|---|---|---|---|---|---|---|
| 10 | >10 | >10 | >10 | >10 | >10 | 0.50 | 0.006 | >10 |
| 11 | >10 | >10 | >10 | >10 | >10 | 0.17 | 0.20 | >10 |
| 12 | >10 | >10 | >10 | >10 | >10 | 0.89 | 0.71 | >10 |
| 13 | n.t. | n.t. | 8.2 | n.t. | >10 | 0.30 | 1.6 | 4.4 |
| 14 | 1.2 | 1.7 | 0.81 | 0.88 | 3.6 | 0.12 | 0.19 | 2.7 |
| 15 | 7.0 | 2.5 | 2.5 | 2.2 | 4.2 | 0.32 | 2.2 | >10 |
| 16 | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | >10 | n.t. |
| Compound | Sexp., therm., pH 7.4 [µM] a | Sexp., kin., pH 7.4 [µM] b |
|---|---|---|
| KuFal194 (10) | <5 | 5.28 (4.99–5.57) |
| 11 | <5 | 26.1 (25.6–26.5) |
| 16 | 13,650 (11,100–16,200) | 2310 * |
| PDB Accession Code | 6T6A |
|---|---|
| Data collection | |
| beamline | Diamond Light Source, i04-1 |
| wavelength (Å) | 0.9200 |
| Resolution a (Å) | 46.23–2.80 (2.95–2.80) |
| space group | C2 |
| cell dimensions | |
| a (Å) | 244.9 |
| b (Å) | 65.4 |
| c (Å) | 148.1 |
| α (deg) | 90 |
| β (deg) | 115.2 |
| γ (deg) | 90 |
| no. unique reflections a | 51,386 (7523) |
| Completeness a (%) | 97.6 (98.5) |
| I/σI a | 7.5 (2.1) |
| Rmerge a | 0.122 (0.535) |
| CC(1/2) | 0.991 (0.757) |
| Redundancy a | 3.6 (3.7) |
| Refinement | |
| no. atoms in refinement (P/L/O) b | 11,336/48/436 |
| Rfact (%) | 20.5 |
| Rfree (%) | 24.2 |
| Bf (P/L/O) b (Å2) | 49/55/39 |
| rmsd bond c (Å) | 0.010 |
| rmsd deviation angle c (deg) | 1.4 |
| Molprobity | |
| Ramachandran favored | 93.71 |
| Ramachandran outlier | 0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lechner, C.; Flaßhoff, M.; Falke, H.; Preu, L.; Loaëc, N.; Meijer, L.; Knapp, S.; Chaikuad, A.; Kunick, C. [b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors. Molecules 2019, 24, 4090. https://doi.org/10.3390/molecules24224090
Lechner C, Flaßhoff M, Falke H, Preu L, Loaëc N, Meijer L, Knapp S, Chaikuad A, Kunick C. [b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors. Molecules. 2019; 24(22):4090. https://doi.org/10.3390/molecules24224090
Chicago/Turabian StyleLechner, Christian, Maren Flaßhoff, Hannes Falke, Lutz Preu, Nadége Loaëc, Laurent Meijer, Stefan Knapp, Apirat Chaikuad, and Conrad Kunick. 2019. "[b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors" Molecules 24, no. 22: 4090. https://doi.org/10.3390/molecules24224090
APA StyleLechner, C., Flaßhoff, M., Falke, H., Preu, L., Loaëc, N., Meijer, L., Knapp, S., Chaikuad, A., & Kunick, C. (2019). [b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors. Molecules, 24(22), 4090. https://doi.org/10.3390/molecules24224090