Structural Analysis, Molecular Modelling and Preliminary Competition Binding Studies of AM-DAN as a NMDA Receptor PCP-Site Fluorescent Ligand


Abstract

1. Introduction
2. Materials and Methods
2.1. Synthesis of AM-DAN
2.2. Energy Minima Studies
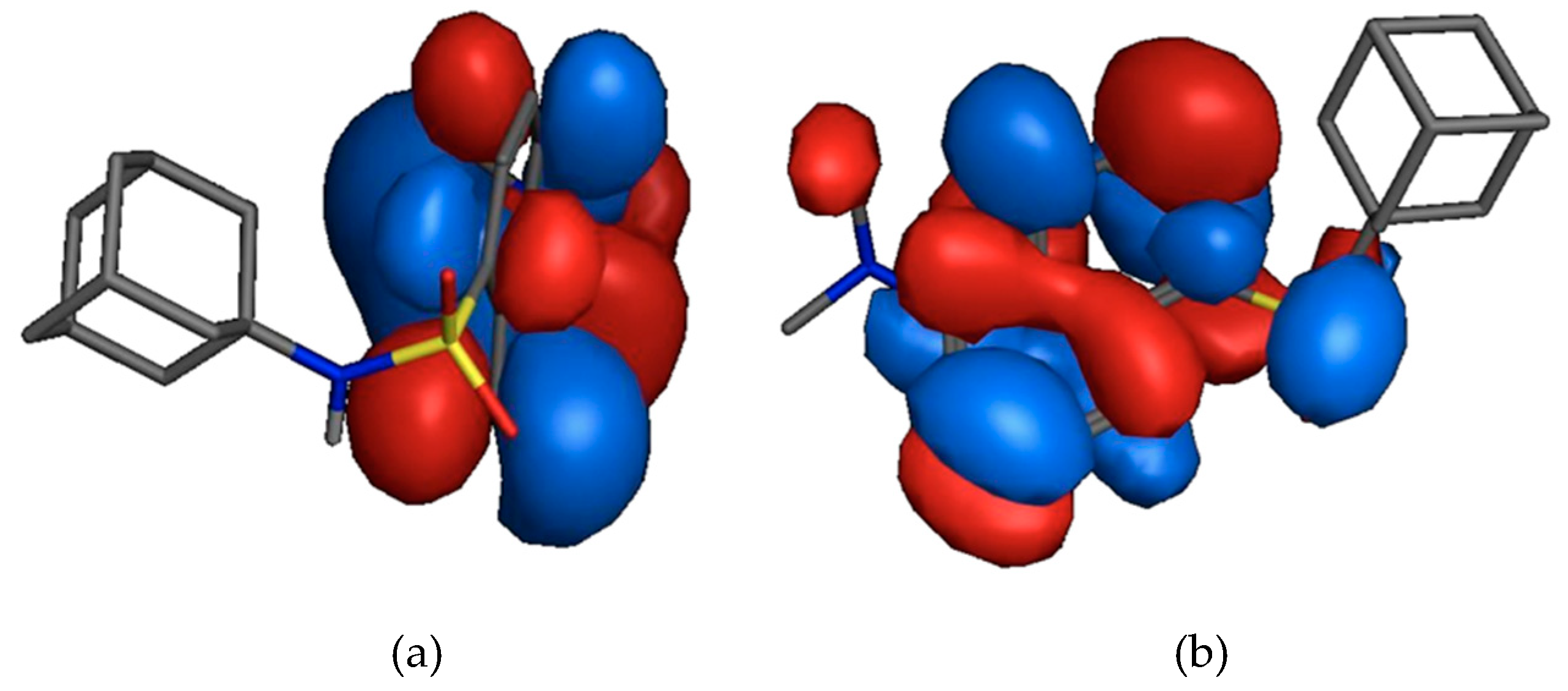
2.3. HOMO and LUMO Calculations
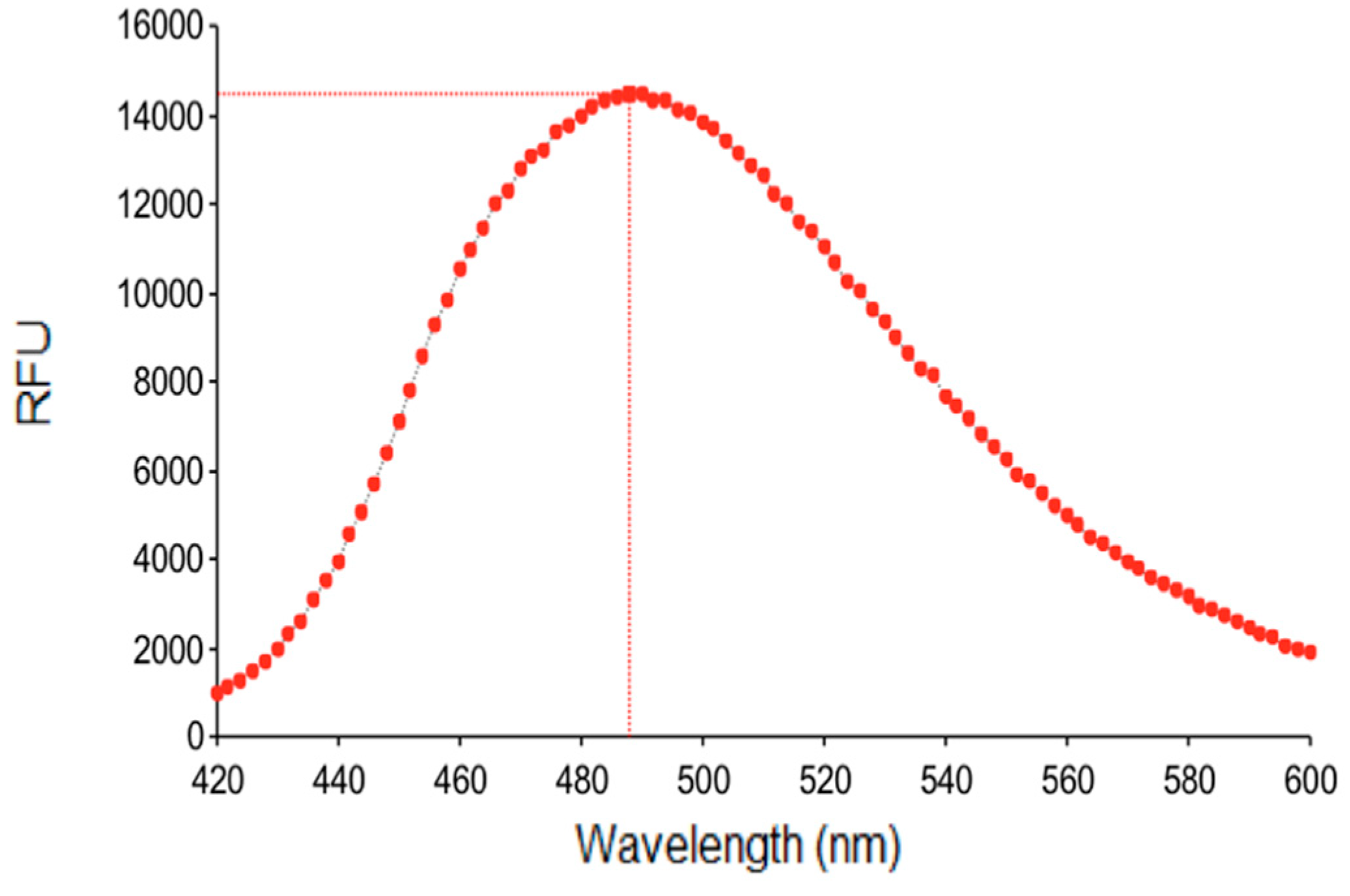
2.4. Fluorescence Studies
2.5. Molecular Modelling
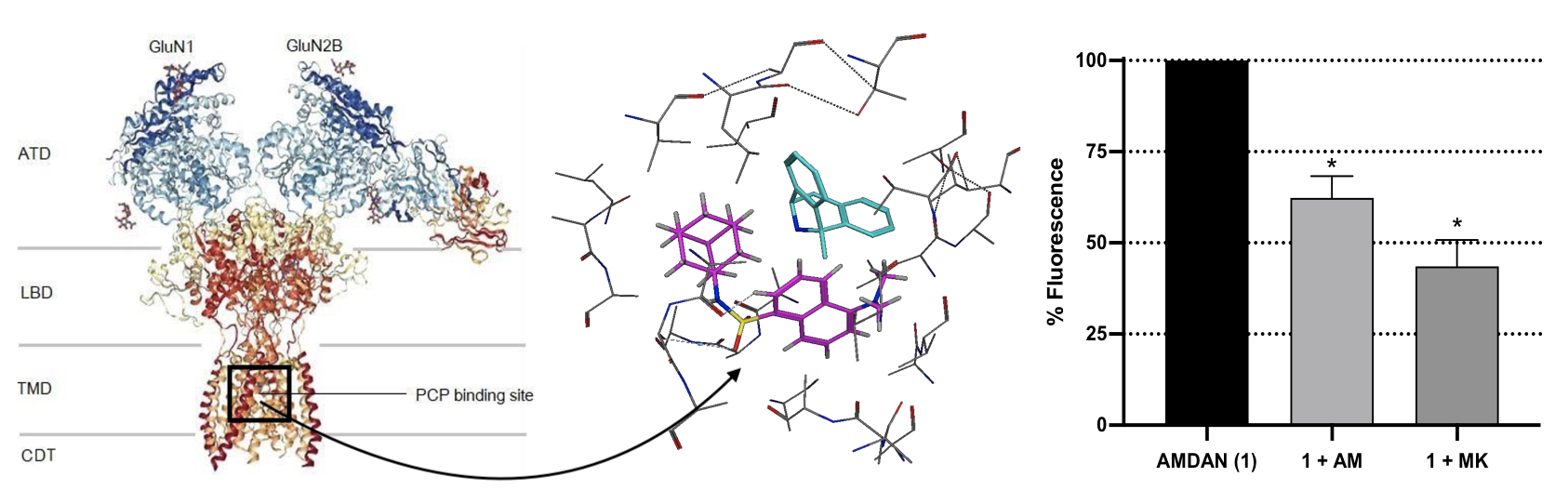
2.5.1. Blind Docking
2.5.2. In Silico Structure-based Docking Studies
2.6. NMDAR Fluorescent Competition-Binding Study Using AM-DAN
3. Results and Discussion
3.1. Energy Minima Studies
3.2. HOMO and LUMO Calculations
3.3. Fluorescence Studies
3.4. Molecular Modelling
3.4.1. Blind Docking
3.4.2. In Silico Structure-Based Docking Studies
3.5. NMDAR Fluorescent Competition Binding Study Using AM-DAN
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, S.; Stein, R.A.; Yoshioka, C.; Lee, C.-H.; Goehring, A.; Mchaourab, H.S.; Gouaux, E. Mechanism of NMDA Receptor Inhibition and Activation. Cell 2016, 165, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; van Dyk, S.; Green, I.R.; Malan, S.F. Synthesis, evaluation and application of polycyclic fluorescent analogues as N-methyl-D-aspartate receptor and voltage gated calcium channel ligands. Eur. J. Med. Chem. 2011, 46, 5010–5020. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Olney, J.W. Excitotoxity and the NMDA receptor. Trends Neurosci. 1987, 10, 299–302. [Google Scholar] [CrossRef]
- Vanle, B.; Olcott, W.; Jimenez, J.; Bashmi, L.; Danovitch, I.; IsHak, W.W. NMDA antagonists for treating the non-motor symptoms in Parkinson’s disease. Transl. Psychiatry 2018, 8, 117. [Google Scholar] [CrossRef]
- Adamsky, A.; Goshen, I. Astrocytes in Memory Function: Pioneering Findings and Future Directions. Neuroscience 2018, 370, 14–26. [Google Scholar] [CrossRef]
- Kroemer, R.T.; Koutsilieri, E.; Hecht, P.; Liedl, K.R.; Riederer, P.; Kornhuber, J. Quantitative Analysis of the Structural Requirements for Blockade of theN-Methyl-d-aspartate Receptor at the Phencyclidine Binding Site. J. Med. Chem. 1998, 41, 393–400. [Google Scholar] [CrossRef]
- Bristow, L.J.; Hutson, P.H.; Thorn, L.; Tricklebank, M.D. The glycine/NMDA receptor antagonist, R-(+)-HA-966, blocks activation of the mesolimbic dopaminergic system induced by phencyclidine and dizocilpine (MK-801) in rodents. Br. J. Pharmacol. 1993, 108, 1156–1163. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lü, W.; Michel, J.C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 2014, 511, 191–197. [Google Scholar] [CrossRef]
- Lü, W.; Du, J.; Goehring, A.; Gouaux, E. Cryo-EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science 2017, 355. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Terre’Blanche, G.; Van der Schyf, C.J.; Malan, S.F. Screening of novel pentacyclo-undecylamines for neuroprotective activity. Eur. J. Pharmacol. 2003, 458, 73–79. [Google Scholar] [CrossRef]
- Jirgensons, A.; Kauss, V.; Kalvinsh, I.; Gold, M.R.; Danysz, W.; Parsons, C.G.; Quack, G. Synthesis and structure–affinity relationships of 1,3,5-alkylsubstituted cyclohexylamines binding at NMDA receptor PCP site. Eur. J. Med. Chem. 2000, 35, 555–565. [Google Scholar] [CrossRef]
- Reuman, M.; Mallamo, J.P.; DeHaven-Hudkins, D.L. Synthesis and binding affinity of 2,3,3a,4,9,9a-hexahydro-9,4-(iminomethano)-1H-benz[f]indenes. Ligands for the PCP site of the NMDA receptor. Bioorg. Med. Chem. Lett. 1995, 5, 371–376. [Google Scholar] [CrossRef]
- Salabert, A.-S.; Fonta, C.; Fontan, C.; Adel, D.; Alonso, M.; Pestourie, C.; Belhadj-Tahar, H.; Tafani, M.; Payoux, P. Radiolabeling of [18F]-fluoroethylnormemantine and initial in vivo evaluation of this innovative PET tracer for imaging the PCP sites of NMDA receptors. Nucl. Med. Biol. 2015, 42, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Auer, M.; Moore, K.J.; Meyer-Almes, F.J.; Guenther, R.; Pope, A.J.; Stoeckli, K.A. Fluorescence correlation spectroscopy: Lead discovery by miniaturized HTS. Drug Disc. Today 1998, 3, 457–465. [Google Scholar] [CrossRef]
- Leopoldo, M.; Lacivita, E.; Berardi, F.; Perrone, R. Developments in fluorescent probes for receptor research. Drug Disc. Today 2009, 14, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Dyk, S.V.; Malan, S.F. Small molecule fluorescent ligands as central nervous system imaging probes. Mini Rev. Med. Chem. 2013, 13, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.J.; Kellam, B. Fluorophore-tagged GPCR ligands. Curr. Opin. Chem. Biol. 2005, 9, 517–525. [Google Scholar] [CrossRef]
- McCallum, M.M.; Pawlak, A.J.; Shadrick, W.R.; Simeonov, A.; Jadhav, A.; Yasgar, A.; Maloney, D.J.; Arnold, L.A. A fluorescence-based high throughput assay for the determination of small molecule- human serum albumin protein binding. Anal. Bioanal. Chem. 2014, 406, 1867–1875. [Google Scholar] [CrossRef]
- Joubert, J. Crystal structure of N-(adamantan-1-yl)-5-(dimethylamino)naphthalene-1-sulfonamide. Z. Kristallogr.-New Cryst. Struct. 2019. [Google Scholar] [CrossRef]
- Joubert, J.; van Dyk, S.; Green, I.R.; Malan, S.F. Synthesis and evaluation of fluorescent heterocyclic aminoadamantanes as multifunctional neuroprotective agents. Bioorg. Med. Chem. 2011, 19, 3935–3944. [Google Scholar] [CrossRef]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. Version 1.2.0. Available online: http://avogadro.cc/ (accessed on 8 November 2019).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), Version 2018.01. Available online: http://www. chemcomp.com (accessed on 8 November 2019).
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Greenwood, J.R.; Gottfries, J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J. Mol. Graph. Model. 2003, 21, 449–462. [Google Scholar] [CrossRef]
- Joubert, J. Synthesis, Crystal Structure, DFT Studies, Docking Studies, and Fluorescent Properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile. Crystals 2019, 9, 24. [Google Scholar] [CrossRef]
- Joubert, J.; Foxen, E.B.; Malan, S.F. Microwave Optimized Synthesis of N-(adamantan-1-yl)-4-[(adamantan-1-yl)-sulfamoyl]benzamide and Its Derivatives for Anti-Dengue Virus Activity. Molecules 2018, 23, 1678. [Google Scholar] [CrossRef]
- Zindo, F.T.; Malan, S.F.; Omoruyi, S.I.; Enogieru, A.B.; Ekpo, O.E.; Joubert, J. Design, synthesis and evaluation of pentacycloundecane and hexacycloundecane propargylamine derivatives as multifunctional neuroprotective agents. Eur. J. Med. Chem. 2019, 163, 83–94. [Google Scholar] [CrossRef]
- Denya, I.; Malan, S.F.; Enogieru, A.B.; Omoruyi, S.I.; Ekpo, O.E.; Kapp, E.; Zindo, F.T.; Joubert, J. Design, synthesis and evaluation of indole derivatives as multifunctional agents against Alzheimer’s disease. Medchemcomm 2018, 9, 357–370. [Google Scholar] [CrossRef]
- Kapp, E.; Visser, H.; Sampson, S.L.; Malan, S.F.; Streicher, E.M.; Foka, G.B.; Warner, D.F.; Omoruyi, S.I.; Enogieru, A.B.; Ekpo, O.E.; et al. Versatility of 7-Substituted Coumarin Molecules as Antimycobacterial Agents, Neuronal Enzyme Inhibitors and Neuroprotective Agents. Molecules 2017, 22, 1644. [Google Scholar] [CrossRef]
- Grirrane, A.; Pastor, A.; Galindo, A.; del Río, D.; Orlandini, A.; Mealli, C.; Ienco, A.; Caneschi, A.; Fernández Sanz, J. Supramolecular interactions as determining factors of the geometry of metallic building blocks: Tetracarboxylate dimanganese species. Angew. Chem. Int. Ed. Engl. 2005, 44, 3429–3432. [Google Scholar] [CrossRef]
- Grirrane, A.; Pastor, A.; Galindo, A. Thiodiacetate–Manganese Chemistry with N ligands: Unique Control of the Supramolecular Arrangement over the Metal Coordination Mode. Eur. J. 2011, 17, 10600–10617. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.; Domingo, L.R.; Andrés, J.; Pérez, P.; Tapia, O. Nonlocal (pair site) reactivity from second-order static density response function: Gas-and solution-phase reactivity of the acetaldehyde enolate as a test case. J. Phys. Chem. A 1999, 103, 1367–1375. [Google Scholar] [CrossRef]
- Clare, B.W. Frontier orbital energies in quantitative structure-activity relationships: A comparison of quantum chemical methods. Theor. Chim. Acta 1994, 87, 415–430. [Google Scholar] [CrossRef]
- Clare, B.W. The relationship of charge transfer complexes to frontier orbital energies in qsar. J. Mol. Struct. Theochem. 1995, 331, 63–78. [Google Scholar] [CrossRef]
- Clare, B.W. Charge transfer complexes and frontier orbital energies in qsar: A congeneric series of electron acceptors. J. Mol. Struct. Theochem. 1995, 337, 139–150. [Google Scholar] [CrossRef]
- Heaton, C.; Miller, A.; Powell, R. Predicting the reactivity of fluorinated compounds with copper using semi-empirical calculations. J. Fluor. Chem. 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef]
Sample Availability: The compounds are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pose | Binding Site | Binding Energy (kcal/mol) |
|---|---|---|
| 1 | PCP | −10.3 |
| 2 | PCP | −9.7 |
| 3 | PCP | −9.5 |
| 4 | PCP | −9.2 |
| 5 | PCP | −9.2 |
| 6 | PCP | −9.0 |
| 7 | PCP | −8.9 |
| 8 | Glutamate | −8.9 |
| 9 | PCP | −8.8 |
| 10 | Glutamate | −8.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndzibongwana, S.; Ngobese, S.; Sayed, A.; Shongwe, C.; White-Phillips, S.; Joubert, J. Structural Analysis, Molecular Modelling and Preliminary Competition Binding Studies of AM-DAN as a NMDA Receptor PCP-Site Fluorescent Ligand. Molecules 2019, 24, 4092. https://doi.org/10.3390/molecules24224092
Ndzibongwana S, Ngobese S, Sayed A, Shongwe C, White-Phillips S, Joubert J. Structural Analysis, Molecular Modelling and Preliminary Competition Binding Studies of AM-DAN as a NMDA Receptor PCP-Site Fluorescent Ligand. Molecules. 2019; 24(22):4092. https://doi.org/10.3390/molecules24224092
Chicago/Turabian StyleNdzibongwana, Sethu, Samukelo Ngobese, Ahmad Sayed, Ciniso Shongwe, Simon White-Phillips, and Jacques Joubert. 2019. "Structural Analysis, Molecular Modelling and Preliminary Competition Binding Studies of AM-DAN as a NMDA Receptor PCP-Site Fluorescent Ligand" Molecules 24, no. 22: 4092. https://doi.org/10.3390/molecules24224092
APA StyleNdzibongwana, S., Ngobese, S., Sayed, A., Shongwe, C., White-Phillips, S., & Joubert, J. (2019). Structural Analysis, Molecular Modelling and Preliminary Competition Binding Studies of AM-DAN as a NMDA Receptor PCP-Site Fluorescent Ligand. Molecules, 24(22), 4092. https://doi.org/10.3390/molecules24224092