Catalytic Hydrolysis Mechanism of Cocaine by Human Carboxylesterase 1: An Orthoester Intermediate Slows Down the Reaction

Abstract
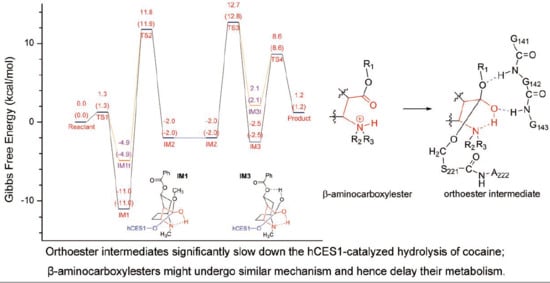
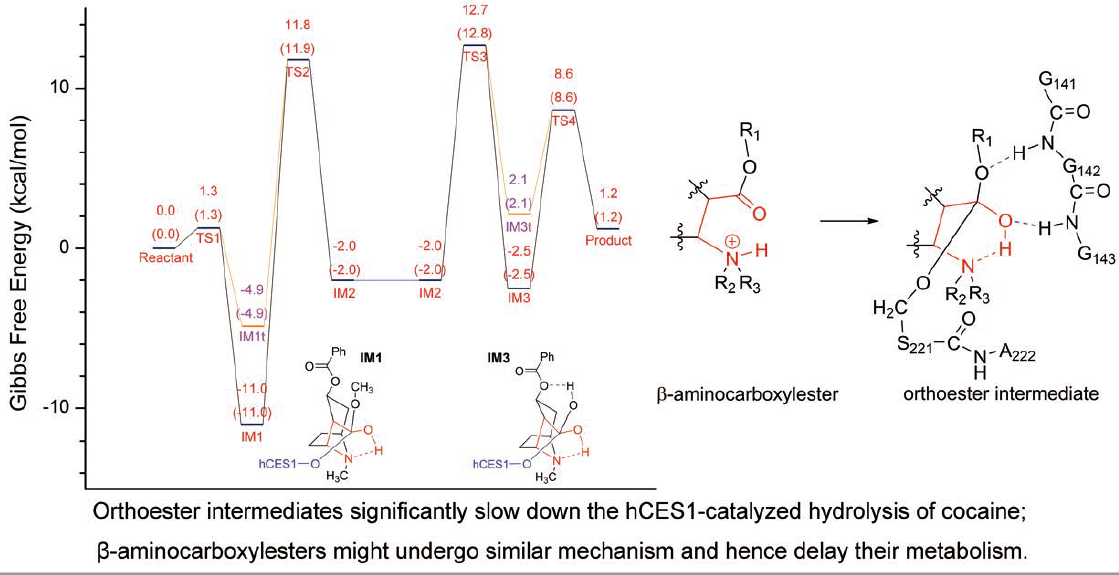{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
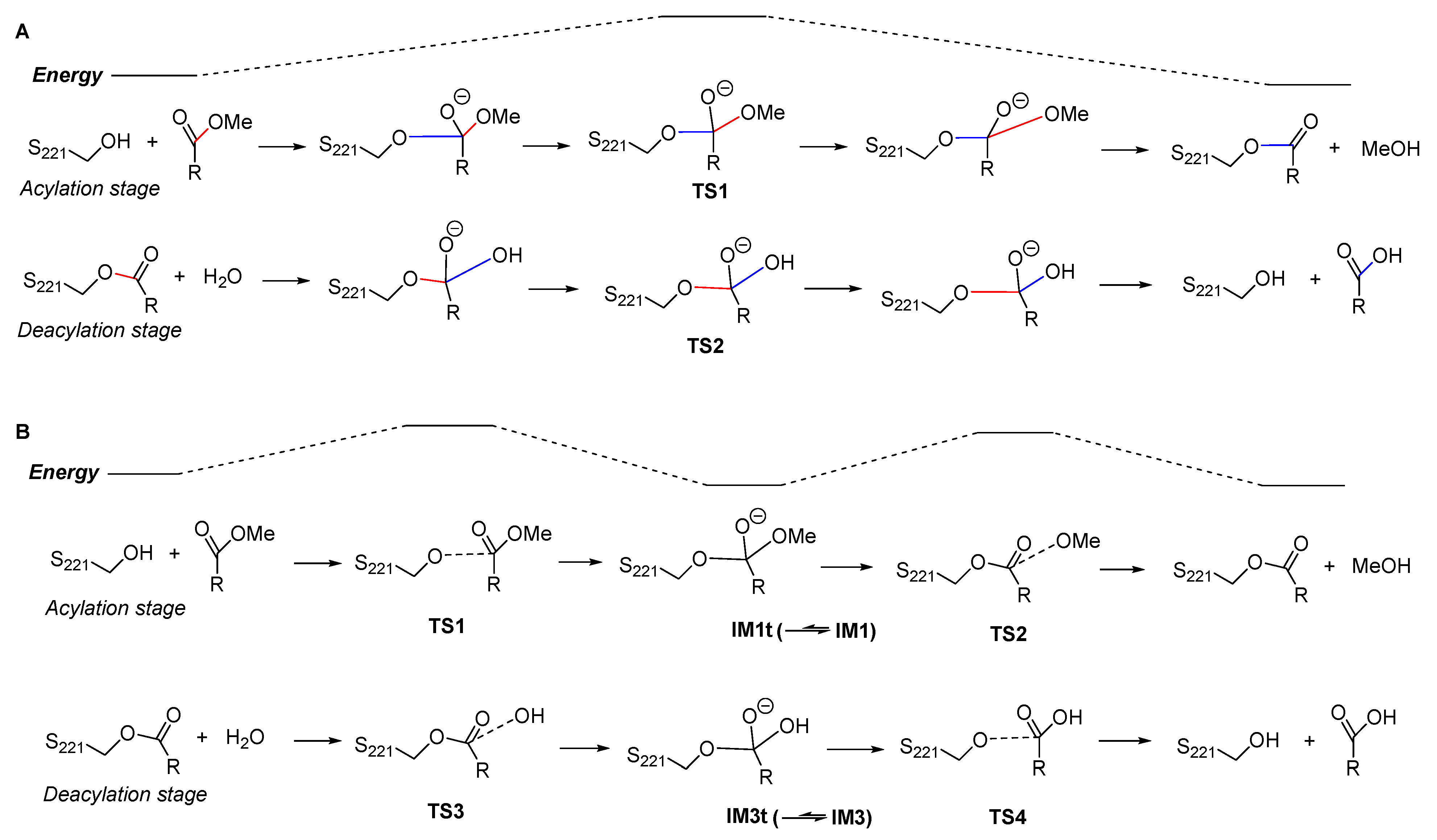
2.1. Gibbs Free Energy Profile of the Whole Reaction Pathway
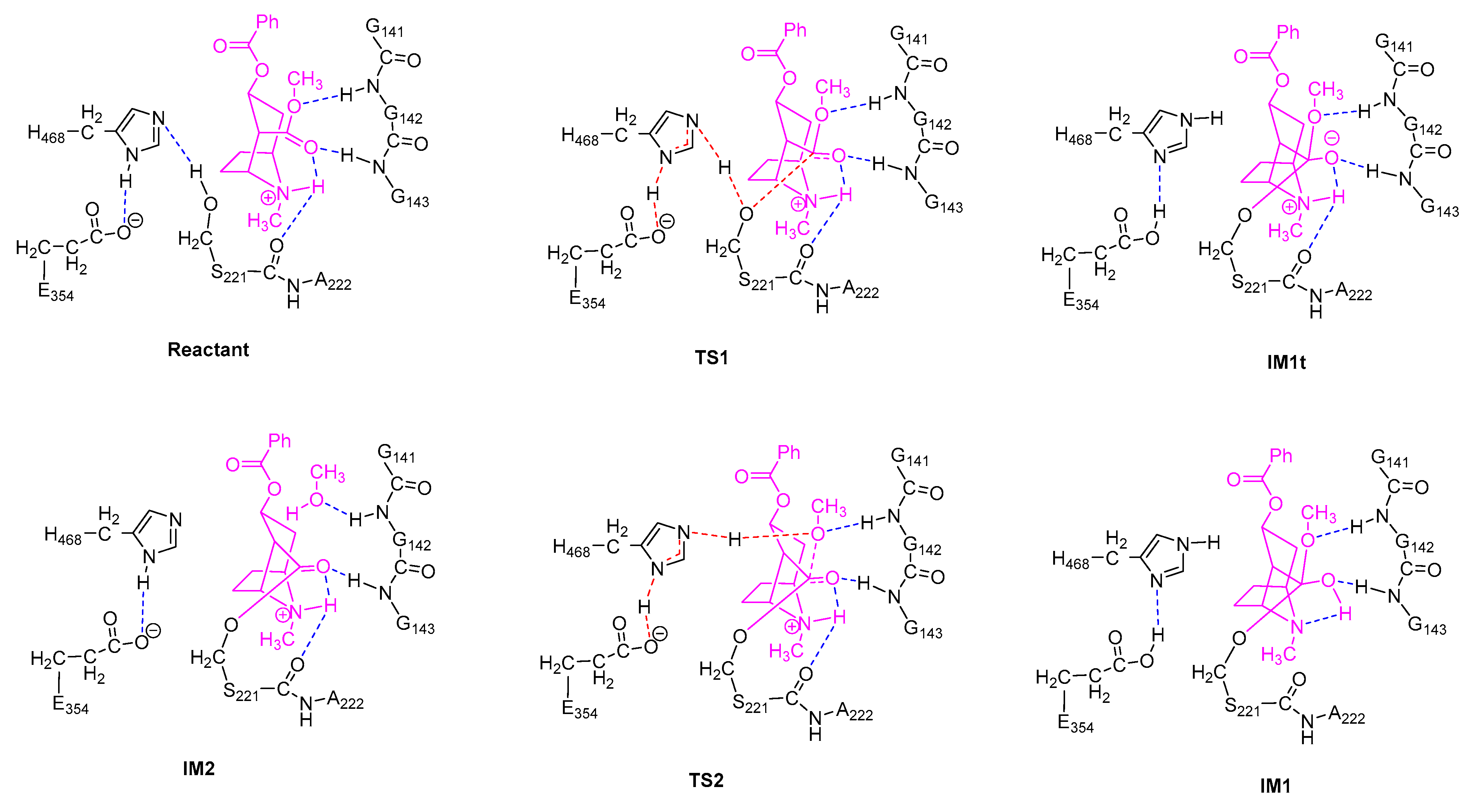
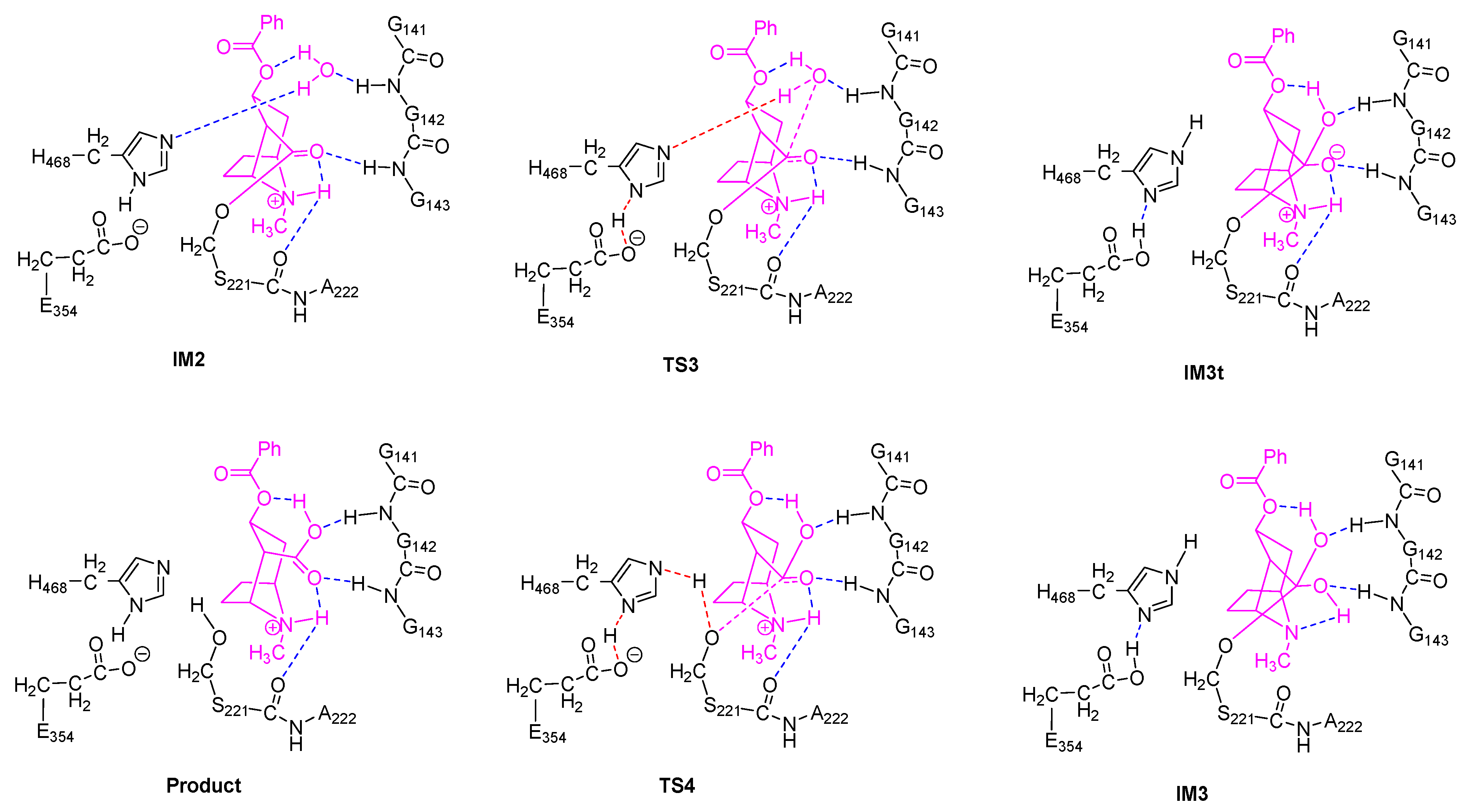
2.2. Acylation Stage of hCES1-Catalyzed Cocaine Hydrolysis
2.3. Deacylation Stage of hCES1-Catalyzed Cocaine Hydrolysis
2.4. Differences Between Our Computational Results and Previous Results
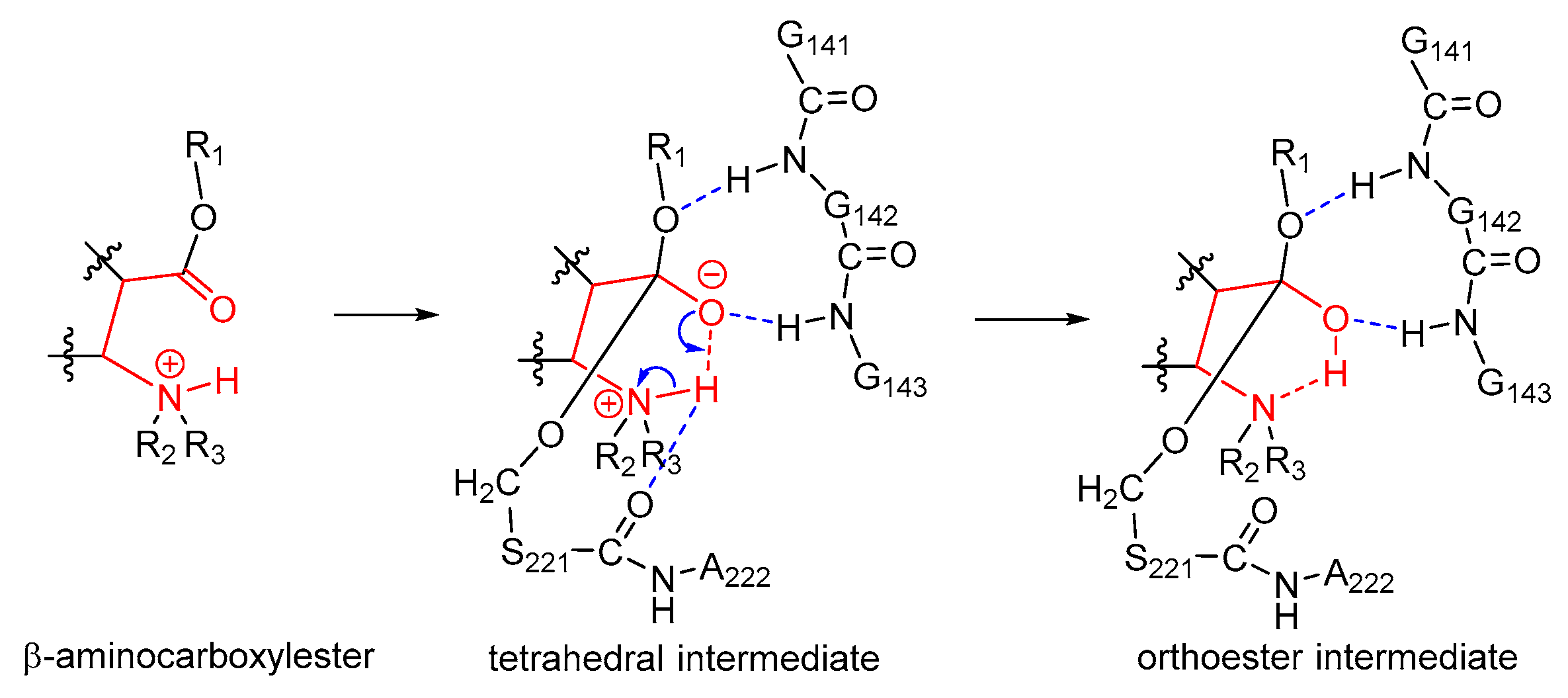
2.5. Orthoester Intermediates in the hCES1-Catalyzed Hydrolysis of Cocaine
3. Materials and Methods
3.1. Construction of Cluster Model of hCES1–Cocaine Complex
3.2. Computation of the Reaction Mechanism and Free Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mentlein, R.; Heymann, E. Hydrolysis of ester- and amide-type drugs by the purified isoenzymes of nonspecific carboxylesterase from rat liver. Biochem. Pharmacol. 1984, 33, 1243–1248. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. The mammalian carboxylesterases: From Molecules to Functions. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 257–288. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M. Structure and Catalytic Properties of Carboxylesterase Isozymes Involved in Metabolic Activation of Prodrugs. Molecules 2008, 13, 412–431. [Google Scholar] [CrossRef]
- Redinbo, M.R.; Potter, P.M. Mammalian carboxylesterases: From drug targets to protein therapeutics. Drug Discovery Today 2005, 10, 313–325. [Google Scholar] [CrossRef]
- Lian, J.; Nelson, R.; Lehner, R. Carboxylesterases in lipid metabolism: From mouse to human. Protein Cell 2018, 9, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zou, L.; Jin, Q.; Hou, J.; Ge, G.; Yang, L. Human carboxylesterases: A comprehensive review. Acta Pharm. Sinica B 2018, 8, 699–712. [Google Scholar] [CrossRef]
- Xu, J.; Qiu, J.C.; Ji, X.; Guo, H.L.; Wang, X.; Zhang, B.; Wang, T.; Chen, F. Potential Pharmacokinetic Herb-Drug Interactions: Have we Overlooked the Importance of Human Carboxylesterases 1 and 2? Curr. Drug Metab. 2019, 20, 130–137. [Google Scholar] [CrossRef]
- Di, L. The Impact of Carboxylesterases in Drug Metabolism and Pharmacokinetics. Curr. Drug Metab. 2019, 20, 91–102. [Google Scholar] [CrossRef]
- Laizure, S.C.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The role of human carboxylesterases in drug metabolism: Have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef]
- Kraut, J. Serine proteases: Structure and mechanism of catalysis. Ann. Rev. Biochem. 1977, 46, 331–358. [Google Scholar] [CrossRef]
- Quinn, D.M. Acetylcholinesterase: Enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Zhang, Y. Catalytic Reaction Mechanism of Acetylcholinesterase Determined by Born-Oppenheimer Ab Initio QM/MM Molecular Dynamics Simulations. J. Phys. Chem. B 2010, 114, 8817–8825. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fang, L.; Liu, J.; Zhan, C.G. Reaction pathway and free energy profile for butyrylcholinesterase-catalyzed hydrolysis of acetylcholine. J. Phys. Chem. B 2011, 115, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamza, A.; Zhan, C.G. Fundamental Reaction Mechanism and Free Energy Profile for (-)-Cocaine Hydrolysis Catalyzed by Cocaine Esterase. J. Am. Chem. Soc. 2009, 131, 11964–11975. [Google Scholar] [CrossRef]
- Sadeghi Googheri, M.S.; Housaindokht, M.R.; Sabzyan, H. Reaction mechanism and free energy profile for acylation of Candida antarctica lipase B with methylcaprylate and acetylcholine: Density functional theory calculations. J. Mol. Graphics. Modell. 2014, 54, 131–140. [Google Scholar] [CrossRef]
- Sadeghi Googheri, M.S.; Housaindokht, M.R.; Sabzyan, H. Theoretical studies on the deacylation step of acylated Candida antarctica lipase B: Structural and reaction pathway analysis. J. Mol. Graphics. Modell. 2015, 57, 9–19. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C.L.; Xue, Y.; Potter, P.M.; Redinbo, M.R. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat. Struc. Biol. 2003, 10, 349–356. [Google Scholar] [CrossRef]
- Fleming, C.D.; Bencharit, S.; Edwards, C.C.; Hyatt, J.L.; Tsurkan, L.; Bai, F.; Fraga, C.; Morton, C.L.; Howard-Williams, E.L.; Potter, P.M.; et al. Structural insights into drug processing by human carboxylesterase 1: Tamoxifen, mevastatin, and inhibition by benzil. J. Mol. Biol. 2005, 352, 165–177. [Google Scholar] [CrossRef]
- Ileperuma, N.R.; Marshall, S.D.; Squire, C.J.; Baker, H.M.; Oakeshott, J.G.; Russell, R.J.; Plummer, K.M.; Newcomb, R.D.; Baker, E.N. High-resolution crystal structure of plant carboxylesterase AeCXE1, from Actinidia eriantha, and its complex with a high-affinity inhibitor paraoxon. Biochemistry 2007, 46, 1851–1859. [Google Scholar] [CrossRef]
- Aranda, J.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Roca, M.; Tuñon, I.; Ramos, M.J. The Catalytic Mechanism of Carboxylesterases: A Computational Study. Biochemistry 2014, 53, 5820–5829. [Google Scholar] [CrossRef]
- Wang, X.; Yao, J. Improvement of the self consistent charge density functional tight binding theory by a modified Mulliken charge. Theor. Chem. Acc. 2017, 136, 124. [Google Scholar] [CrossRef]
- Yao, J.; Yuan, Y.; Zheng, F.; Zhan, C.-G. Unexpected Reaction Pathway for butyrylcholinesterase-catalyzed inactivation of hunger hormone ghrelin. Sci. Rep. 2016, 6, 22322. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chen, X.; Zheng, F.; Zhan, C.-G. Catalytic Reaction Mechanism for Drug Metabolism in Human Carboxylesterase-1: Cocaine Hydrolysis Pathway. Mol. Pharm. 2018, 15, 3871–3880. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Hosokawa, M. Carboxylesterases: Structure, Function and Polymorphism. Biomol. Therap. 2009, 17, 335–347. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. Structure, function and regulation of carboxylesterases. Chem. Bio. Int. 2006, 162, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Wade, L.G. Carboxylic acid derivatives. In Organic Chemistry, 6th ed.; Pearson Education, Inc.: London, UK, 2006. [Google Scholar]
- Himo, F. Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Blomberg, M.R.A. Quantum Chemical Studies of Proton-Coupled Electron Transfer in Metalloenzymes. Chem. Rev. 2010, 110, 7040–7061. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Neves, R.P.P.; Bras, N.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. WIREs Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Swiderek, K.; Tunon, I.; Moliner, V. Predicting enzymatic reactivity: From theory to design. WIREs Comput. Mol. Sci. 2014, 4, 407–421. [Google Scholar] [CrossRef]
- Masgrau, L.; Truhlar, D.G. The Importance of Ensemble Averaging in Enzyme Kinetics. Acc. Chem. Res. 2015, 48, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, S.P.; Sanghani, P.C.; Schiel, M.A.; Bosron, W.F. Human Carboxylesterases: An Update on CES1, CES2 and CES3. Protein Peptide Lett. 2009, 16, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Chladek, S.; Sprinzl, M. The 3’-End of tRNA and Its Role in Protein Biosynthesis. Angew. Chem. Int. Ed. Engl. 1985, 24, 371–391. [Google Scholar] [CrossRef]
- Garrett, B.C.; Truhlar, D.G. Transition State Theory. In Encyclopedia of Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 1998; pp. 3094–3104. [Google Scholar]
- Turner, J.M.; Larsen, N.A.; Basran, A.; Barbas, C.F., III; Bruce, N.C.; Wilson, I.A.; Lerner, R.A. Biochemical Characterization and Structural Analysis of a Highly Proficient Cocaine Esterase. Biochemistry 2002, 41, 12297–12307. [Google Scholar] [CrossRef]
- Sun, H.; Pang, Y.P.; Lockridge, O.; Brimijoin, S. Re-engineering Butyrylcholinesterase as a Cocaine Hydrolase. Mol. Pharmacol. 2002, 62, 220–224. [Google Scholar] [CrossRef]
- Warner, A.; Norman, A.B. Mechanisms of Cocaine Hydrolysis and Metabolism In Vitro and In Vivo: A Clarification. Therapeutic Drug Monit. 2000, 266–270. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent interactions, Excited States, and Transition Elements. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on molecular-orbital hydrogenation energies. Theor. Chem. Acc. 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-Consistent Molecular Orbital Methods. 21. Small Split-Valence Basis Sets for First-Row Elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar]
- Alecu, I.M.; Zheng, J.; Zhao, Y.; Truhlar, D.G. Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries. J. Chem. Theory. Comput. 2010, 6, 2872–2887. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.G.; Scuseria, E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anistropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics, and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B. Analytical derivatives for geometry optimization in solvation continuum models. J. Chem. Phys. 1998, 109, 249–259. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory. Comput. 2011, 7, 291–309. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Eriksson, L.A.; Ramos, M.J. Dehydration of ribonucleotides catalyzed by ribonucleotide reductase: The role of the enzyme. Biophys. J. 2006, 90, 2109–2119. [Google Scholar] [CrossRef][Green Version]
- Ramos, M.J.; Fernandes, P.A. Computational Enzymatic Catalysis. Acc. Chem. Res. 2008, 41, 689–698. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, M.; Zhang, Z.; Liu, Z.; Zhang, C.; Zhang, J.; Fan, S.; Yang, Z. Catalytic Hydrolysis Mechanism of Cocaine by Human Carboxylesterase 1: An Orthoester Intermediate Slows Down the Reaction. Molecules 2019, 24, 4057. https://doi.org/10.3390/molecules24224057
Yan M, Zhang Z, Liu Z, Zhang C, Zhang J, Fan S, Yang Z. Catalytic Hydrolysis Mechanism of Cocaine by Human Carboxylesterase 1: An Orthoester Intermediate Slows Down the Reaction. Molecules. 2019; 24(22):4057. https://doi.org/10.3390/molecules24224057
Chicago/Turabian StyleYan, Maocai, Zhen Zhang, Zhaoming Liu, Chunyan Zhang, Jingchang Zhang, Shuai Fan, and Zhaoyong Yang. 2019. "Catalytic Hydrolysis Mechanism of Cocaine by Human Carboxylesterase 1: An Orthoester Intermediate Slows Down the Reaction" Molecules 24, no. 22: 4057. https://doi.org/10.3390/molecules24224057
APA StyleYan, M., Zhang, Z., Liu, Z., Zhang, C., Zhang, J., Fan, S., & Yang, Z. (2019). Catalytic Hydrolysis Mechanism of Cocaine by Human Carboxylesterase 1: An Orthoester Intermediate Slows Down the Reaction. Molecules, 24(22), 4057. https://doi.org/10.3390/molecules24224057