Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Results and Discussion
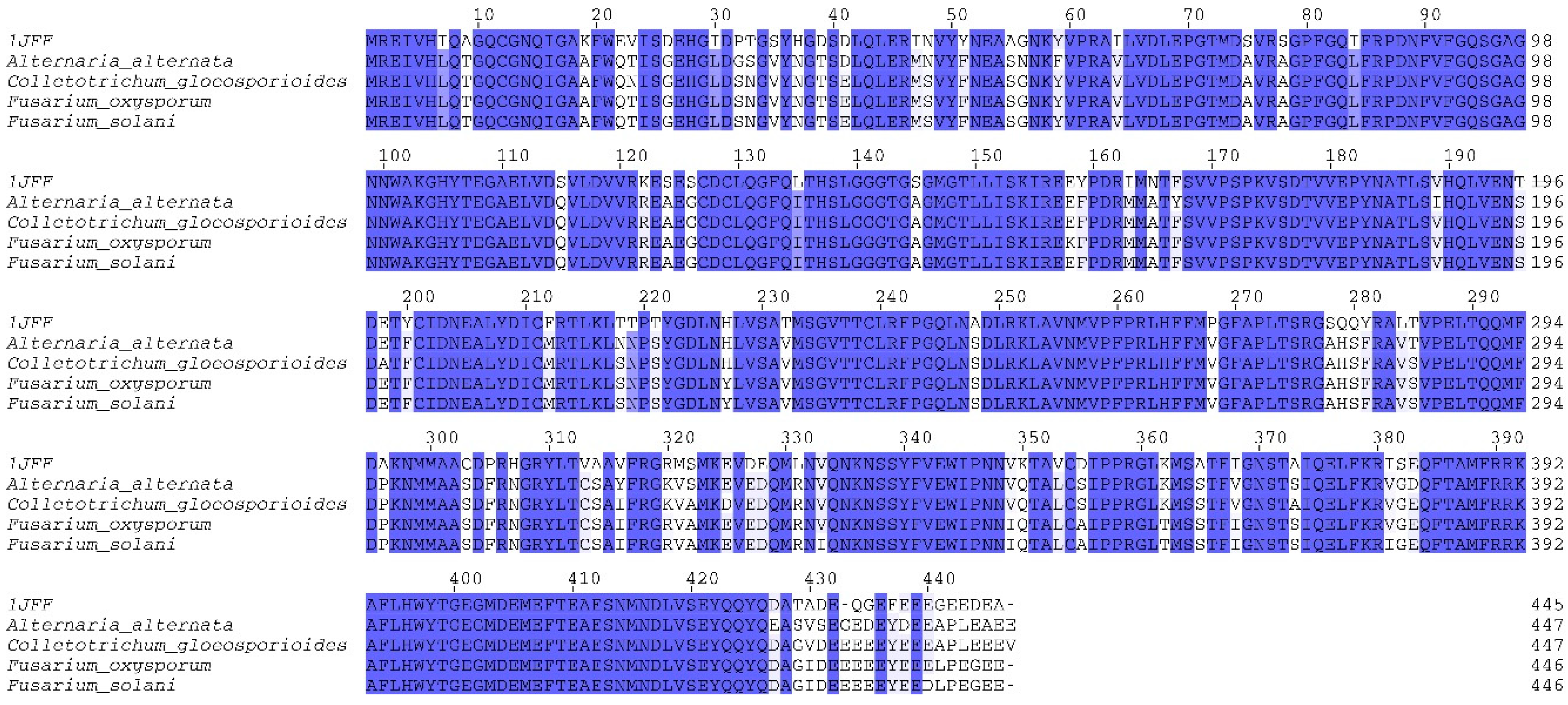
2.1. Homology Modeling of β-Tubulin
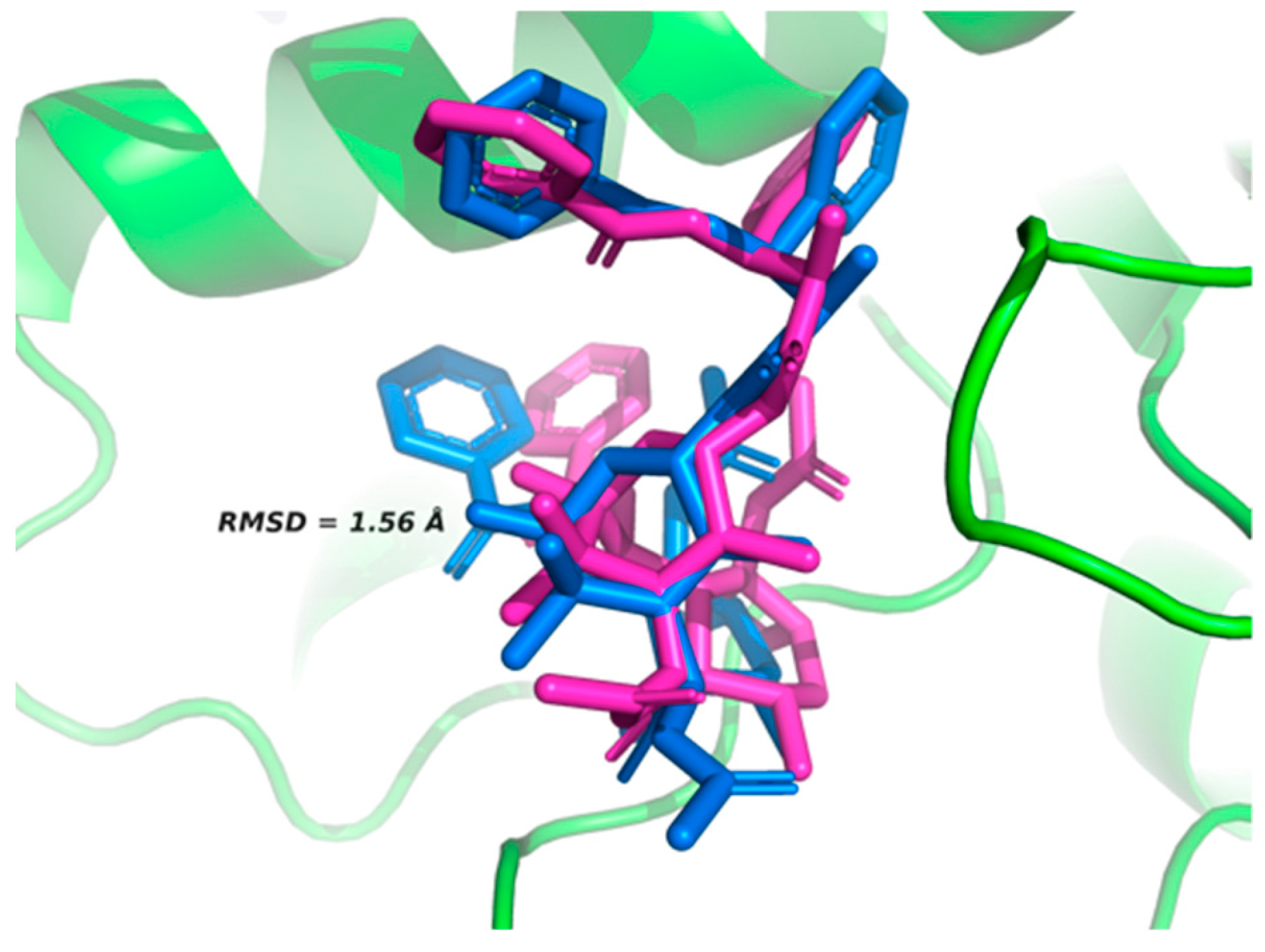
2.2. Active Site Validation
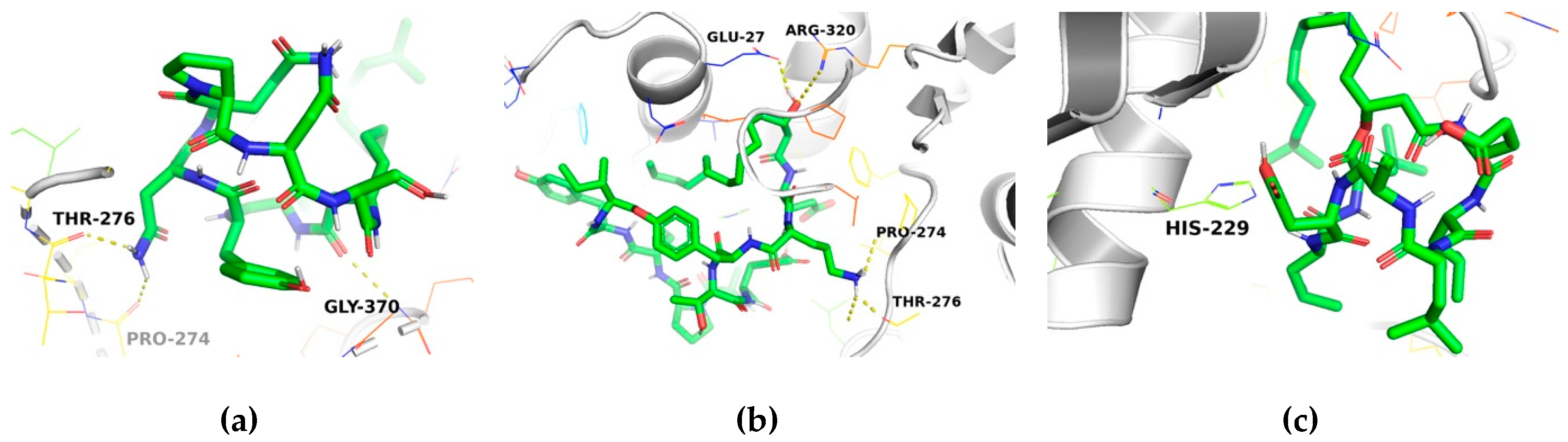
2.3. Molecular Docking Analysis
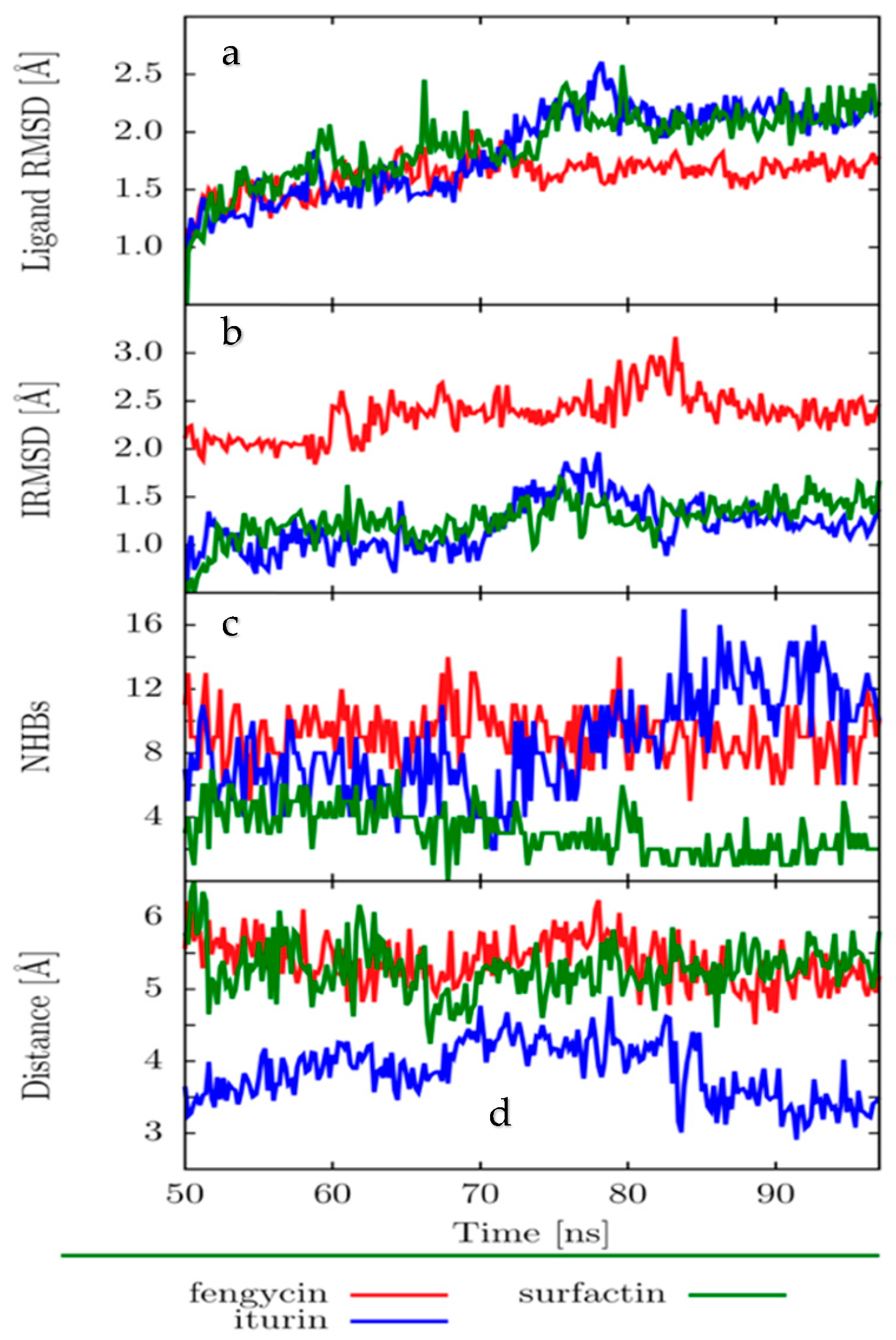
2.4. Molecular Dynamics (MD)
3. Materials and Methods
3.1. Domain Identification and Template Search
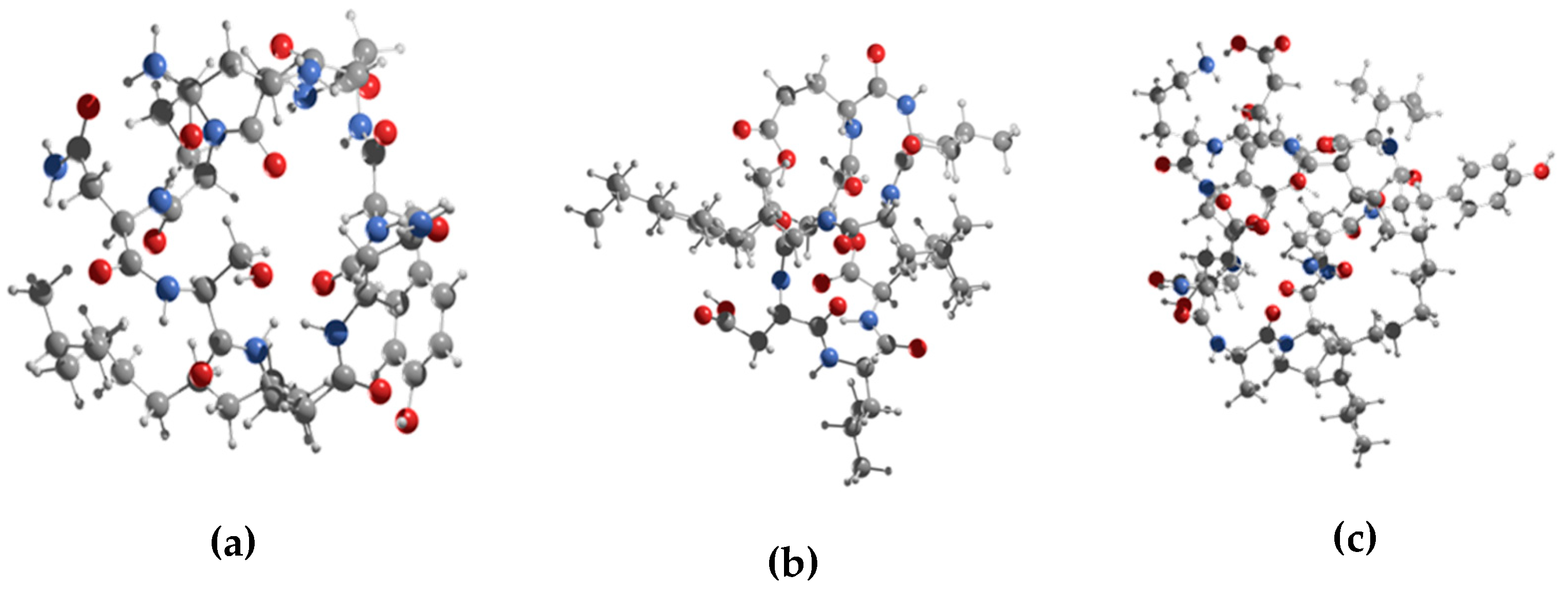
3.2. Conformational Search of the Lipopeptides
3.3. Validation of Active Site
3.4. Molecular Docking
3.5. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ongena, M.; Jacques, P. Bacillus lipopeptides: Versatile weapons for plant disease biocontrol. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef]
- Schneider, T.; Müller, A.; Miess, H.; Gross, H. Cyclic lipopeptides as antibacterial agents—Potent antibiotic activity mediated by intriguing mode of actions. Int. J. Med. Microbiol. 2014, 304, 37–43. [Google Scholar] [CrossRef]
- Eswari, J.S.; Anand, M.; Venkateswarlu, C. Optimum culture medium composition for lipopeptide production by Bacillus subtilis using response surface model-based ant colony optimization. Sadhana 2016, 41, 55–65. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; de Bruijn, I.; de Kock, M.J.D. Cyclic lipopeptide production by plant-associated Pseudomonas spp.: Diversity, activity, biosynthesis, and regulation. Mol. Plant Microbe Interact. 2006, 19, 699–710. [Google Scholar] [CrossRef]
- Ongena, M.; Jacques, P.; Touré, Y.; Destain, J.; Jabrane, A.; Thonart, P. Involvement of fengycin-type lipopeptides in the multifaceted biocontrol potential of Bacillus subtilis. Appl. Microbiol. Biotechnol. 2005, 69, 29–38. [Google Scholar] [CrossRef]
- Chan, Y.K.; Savard, M.E.; Reid, L.M.; Cyr, T.; McCormick, W.A.; Seguin, C. Identification of lipopeptide antibiotics of a Bacillus subtilis isolate and their control of Fusarium graminearum diseases in maize and wheat. BioControl 2009, 54, 567–574. [Google Scholar] [CrossRef]
- Romero, D.; de Vicente, A.; Olmos, J.L.; Dávila, J.C.; Pérez-García, A. Effect of lipopeptides of antagonistic strains of Bacillus subtilis on the morphology and ultrastructure of the cucurbit fungal pathogen Podosphaera fusca. J. Appl. Microbiol. 2007, 103, 969–976. [Google Scholar] [CrossRef]
- Meena, K.R.; Kanwar, S.S. Lipopeptides as the antifungal and antibacterial agents: Applications in food safety and therapeutics. BioMed Res. Int. 2015, 2015, 473050. [Google Scholar] [CrossRef]
- Grau, A.; Gómez Fernández, J.C.; Peypoux, F.; Ortiz, A. A study on the interactions of surfactin with phospholipid vesicles. Biochim. Biophys. Acta Biomembr. 1999, 1418, 307–319. [Google Scholar] [CrossRef]
- Heerklotz, H.; Seelig, J. Leakage and lysis of lipid membranes induced by the lipopeptide surfactin. Eur. Biophys. J. 2007, 36, 305–314. [Google Scholar] [CrossRef]
- Chatterji, B.P.; Jindal, B.; Srivastava, S.; Panda, D. Microtubules as antifungal and antiparasitic drug targets. Expert Opin. 2011, 21, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Downing, K.H. Structural Basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Vindya, N.G.; Sharma, N.; Yadav, M.; Ethiraj, K.R. Tubulins—The target for anticancer therapy. Curr. Top. Med. Chem. 2015, 15, 73. [Google Scholar] [CrossRef]
- St. George, M.; Ayoub, A.T.; Banerjee, A.; Churchill, C.D.M.; Winter, P.; Klobukowski, M.; Cass, C.E.; Ludueña, R.F.; Tuszynski, J.A.; Damaraju, S. Designing and testing of novel Taxanes to probe the highly complex mechanisms by which Taxanes bind to microtubules and cause cytotoxicity to cancer cells. PLoS ONE 2015, 10, e0129168. [Google Scholar] [CrossRef] [PubMed]
- Lone, M.Y.; Athar, M.; Manhas, A.; Jha, P.C.; Bhatt, S.; Shah, A. In Silico exploration of Vinca domain tubulin inhibitors: A combination of 3D-QSAR-Based pharmacophore modeling, docking and molecular dynamics simulations. Chem. Sel. 2017, 2, 10848–10853. [Google Scholar] [CrossRef]
- Oxberry, M.E.; Geary, T.G.; Prichard, R.K. Assessment of benzimidazole binding to individual recombinant tubulin isotypes from Haemonchus contortus. Parasitology 2001, 122, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Dharni, S.; Maurya, A.; Samad, A.; Srivastava, S.K.; Sharma, A.; Patra, D.D. Purification, characterization, and in Vitro activity of 2,4-Di- tert -butylphenol from Pseudomonas monteilii PsF84: Conformational and molecular docking studies. J. Agric. Food Chem. 2014, 62, 6138–6146. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Lee, S.; Jeon, J. Identification of potential nematicidal compounds against the pine wood nematode, Bursaphelenchus xylophilus through an In Silico Approach. Molecules 2018, 23, 1828. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef]
- De Pereda, J.M.; Leynadier, D.; Evangelio, J.A.; Chacón, P.; Andreu, J.M. Tubulin Secondary Structure Analysis, Limited Proteolysis Sites, and Homology to FtsZ. Biochemistry 1996, 35, 14203–14215. [Google Scholar] [CrossRef]
- Carpenter, E.J.; Huzil, J.T.; Ludueña, R.F.; Tuszynski, J.A. Homology modeling of tubulin: Influence predictions for microtubule’s biophysical properties. Eur. Biophys. J. 2006, 36, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Herowati, R.; Widodo, G.P. Molecular Docking studies of chemical constituents of Tinospora cordifolia on glycogen phosphorylase. Procedia Chem. 2014, 13, 63–68. [Google Scholar] [CrossRef]
- Kumar, P.N.; Swapna, T.H.; Khan, M.Y.; Daddam, J.R.; Hameeda, B. Molecular dynamics and protein interaction studies of lipopeptide (Iturin A) on α- amylase of Spodoptera litura. J. Theor. Biol. 2017, 415, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01. Fox; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- González-Trujano, M.E.; Uribe-Figueroa, G.; Hidalgo-Figueroa, S.; Martínez, A.L.; Déciga-Campos, M.; Navarrete-Vazquez, G. Synthesis and antinociceptive evaluation of bioisosteres and hybrids of naproxen, ibuprofen and paracetamol. Biomed. Pharmacother. 2018, 101, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Sharma, O.P.; Pan, A.; Hoti, S.L.; Jadhav, A.; Kannan, M.; Mathur, P.P. Modeling, docking, simulation, and inhibitory activity of the benzimidazole analogue against β-tubulin protein from Brugia malayi for treating lymphatic filariasis. Med. Chem. Res. 2012, 21, 2415–2427. [Google Scholar] [CrossRef]
- Kim, P.I.; Ryu, J.; Kim, Y.H.; Chi, Y.T. Production of biosurfactant lipopeptides Iturin A, fengycin and surfactin A from Bacillus subtilis CMB32 for control of Colletotrichum gloeosporioides. J. Microbiol. Biotechnol. 2010, 20, 138–145. [Google Scholar]
- Li, Y.; Héloir, M.; Zhang, X.; Geissler, M.; Trouvelot, S.; Jacquens, L.; Henkel, M.; Su, X.; Fang, X.; Wang, Q.; et al. Surfactin and fengycin contribute to the protection of a Bacillus subtilis strain against grape downy mildew by both direct effect and defence stimulation. Mol. Plant Pathol. 2019, 20, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Stewart, J.J.P. An investigation into the applicability of the semiempirical method PM7 for modeling the catalytic mechanism in the enzyme chymotrypsin. J. Mol. Model. 2017, 23, 154. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | No. of H-Bonds | Residue Receptor | Ligand | Bond Length (Å) | Docking Score (kcal/mol) | ki (μM) a |
|---|---|---|---|---|---|---|
| Taxol | 1 | Thr276(N) | O06 | 2.92 | −9.1 | 0.202 |
| Iturin A | 2 | Pro274(O) Thr276(O) | (ND2)68 (ND2)66 | 2.38 2.16 | −7.0 | 7.084 |
| Fengycin | 3 | Glu27(OE2) Thr276(O) Thr276(OG1) | (O)114 (N)115 (N)115 | 2.11 2.43 2.09 | −7.0 | 7.084 |
| Surfactin | 1 | His229(NE2) | H | 2.7 | −6.3 | 23.188 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cob-Calan, N.N.; Chi-Uluac, L.A.; Ortiz-Chi, F.; Cerqueda-García, D.; Navarrete-Vázquez, G.; Ruiz-Sánchez, E.; Hernández-Núñez, E. Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides. Molecules 2019, 24, 3387. https://doi.org/10.3390/molecules24183387
Cob-Calan NN, Chi-Uluac LA, Ortiz-Chi F, Cerqueda-García D, Navarrete-Vázquez G, Ruiz-Sánchez E, Hernández-Núñez E. Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides. Molecules. 2019; 24(18):3387. https://doi.org/10.3390/molecules24183387
Chicago/Turabian StyleCob-Calan, Nubia Noemi, Luz America Chi-Uluac, Filiberto Ortiz-Chi, Daniel Cerqueda-García, Gabriel Navarrete-Vázquez, Esaú Ruiz-Sánchez, and Emanuel Hernández-Núñez. 2019. "Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides" Molecules 24, no. 18: 3387. https://doi.org/10.3390/molecules24183387
APA StyleCob-Calan, N. N., Chi-Uluac, L. A., Ortiz-Chi, F., Cerqueda-García, D., Navarrete-Vázquez, G., Ruiz-Sánchez, E., & Hernández-Núñez, E. (2019). Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides. Molecules, 24(18), 3387. https://doi.org/10.3390/molecules24183387