1. Introduction
Ganoderma lucidum Karst is a wood-rotting mushroom generally growing on tree stumps. The fruiting bodies of
G. lucidum Karst are widely used in Northeast Asia as a natural product to treat chronic hepatitis, nephritis, hepatopathy, gastric ulcer and insomnia. Additionally,
G. lucidum extracts (GLE) have been shown to exhibit anticancer properties by inhibiting tumor growth, angiogenesis, metastasis and telomerase activity [
1,
2,
3,
4]. Furthermore, GLE exhibited antiviral activities during various viral lifecycle stages, including viral attachment to host cell receptors and viral penetration into host cells. For example, acidic protein bound polysaccharide (APBP) was isolated from water-soluble GLE and showed direct virucidal effects against Herpes simplex virus (HSV)-1 and HSV-2 [
5]. APBP inhibited up to 50% of the attachment of HSV-1 and HSV-2 to Vero cells and prevented both viruses from penetrating Vero cells. The antiherpetic activity of APBP suggested that APBP could impede the complex interactions of viruses with cell membranes.
The bioactive ingredients of GLE include triterpenes, steroids, polysaccharides and proteoglycans. More than 100 GLE triterpenes have been isolated and designated as ganoderic acid A-T, lucidenic acid A-P, lucidenolactone, methyl ganoderate E-F, and others [
6]. These triterpenes displayed several biological activities, including antitumor activity [
1,
7]. Ganoderic acid A had significant inhibitory effects against both Epstein-Barr virus (EBV) early antigen (EA) and capsid antigen (CA) activation at 16 nmol [
4]. Ganoderic acid B showed significant anti-human immunodeficiency virus (anti-HIV)-1 protease activity with IC
50 values of 20–90 μM [
8]. Lucidenic acids showed inhibitory effects against 12-
O-tetradecanoylphorbol-13-acetate (TPA)-induced EBV EA activation in Raji cells (96–100% inhibition at 1 × 10
3 mol ratio/TPA) [
2]. Lucidenic lactone inhibited HIV-1 reverse transcriptase activity [
9].
EBV is a human herpes virus that causes infectious mononucleosis and is associated with both B cell and epithelial cell malignancies [
10]. EBV can cause two types of infections in cells, latent and lytic [
11]. In the latent state, EBV genome exists in the nucleus as an episome, which is chromatinized with histones and expresses only a few latent genes. As only some gene products that can be targeted by the host immune system are expressed, EBV can escape a host immune attack and indefinitely survive in the host. The stimulation of EBV lytic cascade using chemical or biological agents first leads to the expression of two viral immediately early (IE) genes,
BZLF1 and
BRLF1. These IE genes encode transcriptional activators and coordinately induce the expression of early (E) genes, such as
BALF5 (DNA polymerase catalytic subunit) and
BALF2 (single-stranded DNA-binding protein). By activating these EBV enzymes and proteins, EBV potently replicates its genome in a rolling circular manner.
EBV-associated epithelial cancers (EBVaECs) have received considerable interest for the past two decades because they represent 80% of all EBV-associated malignancies [
12]. Nasopharyngeal carcinomas (NPC) and EBV-associated gastric carcinomas (EBVaGCs) are the most common EBVaECs with 78,000 and 84,000 of new cases, respectively, annually worldwide [
12]. EBVaGCs represent approximately 10% of all gastric cancers. Among EBVaGCs, 16% are conventional gastric adenocarcinomas and 89% are lymphoepithelioma-like gastric carcinomas (LELC) [
13]. LELC is a poorly differentiated carcinoma with dense lymphocytic infiltration. The selective expression of EBV genes (type I latency) contributes to the malignant transformation of epithelial cells by disrupting cellular processes and signaling pathways. EBVaGC genome contains distinct mutations and methylation patterns, indicating that EBV stimulates a unique and alternate tumorigenesis pattern in EBVaGCs [
14].
Flavonoids represent a class of secondary metabolites present in plants and fungi. Over 5000 naturally occurring flavonoids from various plants have been discovered and structurally characterized. They are classified according to their chemical structure as follows: anthoxanthins, flavanones, flavanonols, flavans, and anthocyanidins [
15]. Quercetin (QCT) and isoliquiritigenin (ISL) are present in several herbs and belong to anthoxanthins and flavanones, respectively. QCT has shown anticancer effects owing to its antioxidant activity, the inhibition of enzymes that activate carcinogens, the modification of signal transduction pathways and the interactions with receptors and other proteins [
16]. In fact, QCT contains a polyphenolic structure that acts as a scavenger of free radicals, and is therefore, responsible for its antioxidant effects. In addition, QCT acts as an agonist of the G protein-coupled estrogen receptor that plays a role in breast cancer progression and tamoxifen resistance [
17]. These findings suggest that QCT is a multifunctional compound with potential anticancer properties.
Regarding its anti-EBV and antitumor effects, QCT has been shown to be cytotoxic to SNU719 EBVaGC cells. Additionally, QCT induced apoptosis and a strong EBV lytic reactivation in SUN719 cells, suggesting that QCT could be a promising candidate anti-EBV and antitumor agent [
18]. Triterpenoids, such as ganoderic acid A (GAA) and ganoderic acid B found in GLE, were shown to moderately inhibit the activity of telomerase at 10 μM [
4]. Molecular docking showed that GAA could inhibit telomerase as a ligand. These results suggested that triterpenoids could exhibit antitumor effects in EBVaGC cells. GLE and QCT might exert significant synergistic anti-EBV and antitumor activities against EBVaGC cells. This study aimed to elucidate the synergistic effects of GLE and QCT cotreatment on inhibiting EBV infection and EBVaGC development. This study showed that cotreatment with GLE and QCT at low concentrations could synergistically enhance apoptosis and induce strong EBV lytic reactivation in EBVaGC cells.
3. Discussion
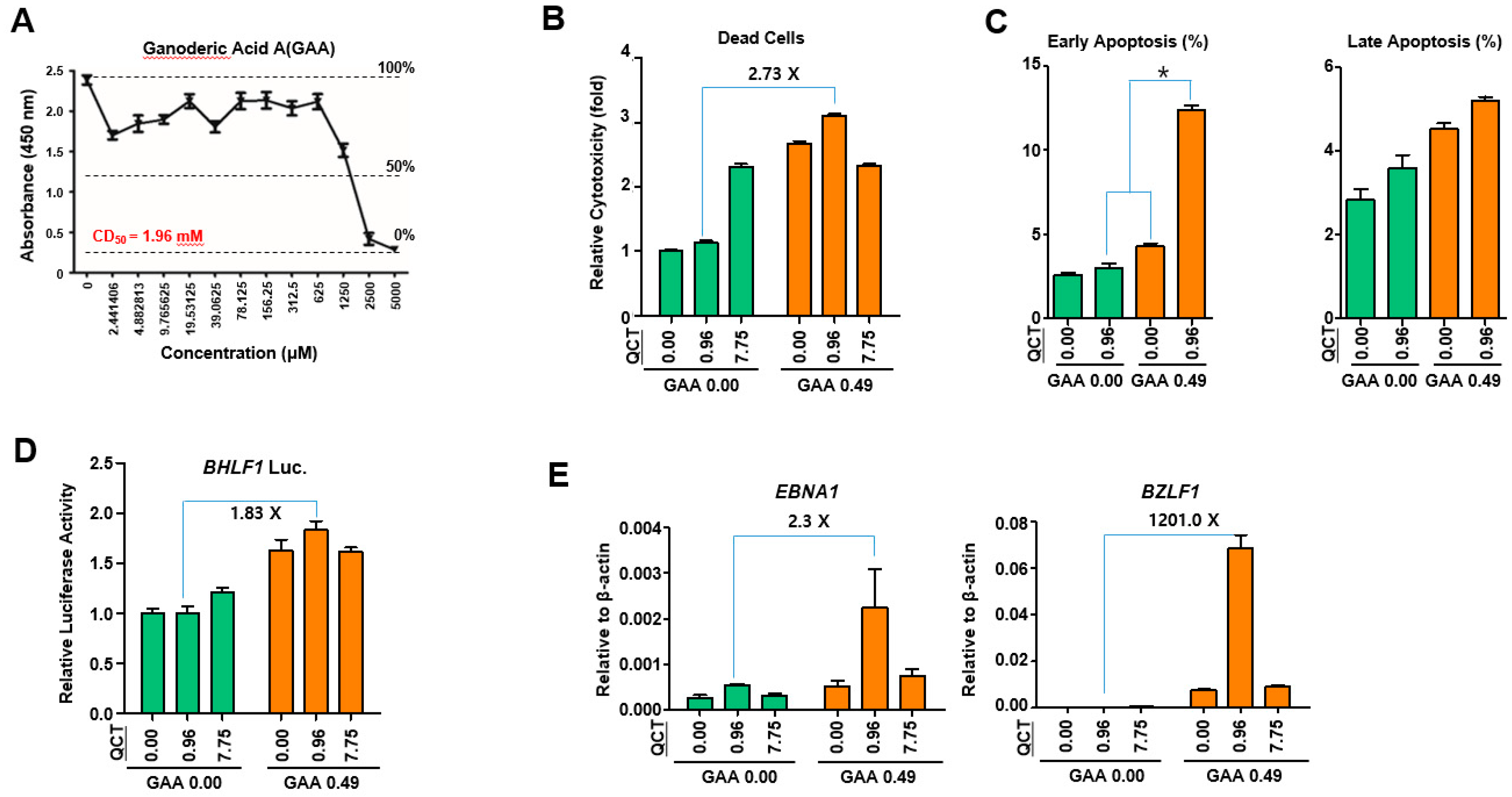
This study showed that the cotreatment of G. lucidum extracts (GLE) and quercetin (QCT) exerted synergistic antitumor and antiviral activities against SNU719 EBVaGC cells. Low concentrations of the cotreatment resulted in both the synergistic activation of cytotoxicity in SNU719 cells and the suppression of tumor development in MKN1-EBV xenograft mice. The addition of a low concentration GLE to QCT reinforced QCT-mediated cytotoxicity and QCT-mediated apoptosis in SNU719 cells. Furthermore, the addition also activated the EBV lytic gene promoter and upregulated EBV genes. Interestingly, ganoderic acid A (GAA), an effective molecule of GLE also showed similar bioactive features with QCT like GLE. The low concentrations of the GAA and QCT cotreatment reinforced QCT-mediated cytotoxicity and QCT-mediated apoptosis in SNU719 cells. Furthermore, the cotreatment activated the EBV lytic gene promoter and upregulated EBV genes, like GLE.
QCT has been shown to exhibit potential anticancer properties owing to its antiproliferative, growth factor suppressive and antioxidant effects [
26]. In addition, QCT can induce apoptosis, where it has been shown to reduce the growth of tumors and inhibit the spread of malignant cells. Moreover, it inhibited chemical carcinogen-induced cell transformation, which was evident as cell viability decreased, ROS generation and microRNA-21 elevation occurred in cancer cells [
27]. Besides its antitumor effects, QCT has been shown to have antiviral activity through its anti-infective and anti-replicative abilities. The replication of herpes viruses, including EBV, was found to be affected by QCT [
18]. EBVaGC exhibits mixed characteristics of both gastric carcinoma and EBV. Apoptosis and EBV lytic reactivation can control the fate of EBVaGC cells, where antitumor agents can induce apoptosis and antiviral agents can stimulate EBV lytic reactivation to further aggravate cell lysis. QCT exhibits both antitumor and antiviral activities against EBVaGC [
18]. This dual function of QCT is likely to be synergistic. GLE, containing polysaccharides and triterpenes, is known to suppress the proliferation and metastatic potential of breast cancer cells by inhibiting Akt, AP-1 and NF-κB [
28]. Moreover, GLE modulated the estrogen receptor signaling and inhibited oxidative stress-induced invasiveness of breast cancer cells [
29]. Consistent with the results of previous studies on breast cancer, our study showed that GLE enhanced both QCT-mediated apoptosis and EBV-lytic reactivation. The effects of GLE were evident at low concentrations of QCT. Furthermore, QCT also enhanced the GLE-mediated upregulation of the EBV gene expression. Similarly, the effects of QCT were evident at low concentrations of GLE. Therefore, these results suggested that GLE enhanced QCT-mediated apoptosis, whereas QCT increased GLE-mediated EBV lytic reactivation.
Mycotherapy has several benefits [
30], where it improves the overall response rate during cancer treatment, enhances immunity owing to the stimulation of T cell proliferation and reduces some chemotherapy-associated adverse events, such as nausea and insomnia. Medicinal mushrooms have mainly been used in Asian countries for hundreds of years for the treatment of infectious diseases. More recently, they have been used for cancer treatment as adjuncts. Importantly, they have an extensive clinical history of safe use as single agents or in combination with chemotherapy. GLE has been intensively combined with mycotherapy for the treatment of broad-spectrum cancers [
30,
31]. GLE has shown antitumor activities against breast cancer, bladder cancer, prostate cancer, colorectal cancer and others. These tumor inhibitory effects of GLE were attributed to the disruption of the proliferation and metastasis of cancer cells. However, the fact that EBV is the initiator of gastric carcinoma should be considered in the treatment of EBVaGC. EBV should be eliminated to reduce the risk of EBVaGC relapse. In this study, QCT strongly induced EBV lytic reactivation. The synergistic effects of the GLE and QCT cotreatment might enhance QCT-mediated apoptosis and GLE-mediated EBV lytic reactivation. Therefore, QCT-supplemented GLE might be used as a medicinal food with a dual function as an antitumor and antiviral agent for EBVaGC treatment.
To the author’s knowledge, this is the first study to report that QCT-supplemented GLE might be effective in treating EBVaGC. The synergistic antitumor and antiviral effects were supported by both in vivo and in vitro studies. Hence, the combined use of GLE and QCT could be beneficial in cancer treatment by alleviating the toxicity of conventional chemotherapy and improving the immune function. However, more studies are still needed to further elucidate the molecular mechanisms of the direct antitumor and antiviral effects of QCT-supplemented GLE.
4. Materials and Methods
4.1. Preparation of GLE
G. lucidum strain CM980736, one of the mushroom culture collections of Chungnam-do Agricultural Technology Institute (Yesan, Korea) was grown into
G. lucidum fruiting bodies by Yeonchen Cheongsan Mushroom Farming Association (YCMFA, Yeonchen, Korea). The
G. lucidum fruiting bodies were obtained from YCMFA who prepared GLE for this study. Small molecules were extracted from 5.0 kg of
G. lucidum fruiting bodies for 24 h at 25 °C using volume of 99% ethanol. This extract was repeated three times with the
G. lucidum fruiting bodies previously extracted. The resultant primary extracts were filtered, concentrated, sterilized and freeze-dried. The final extract was approximately 0.5 g and named as
G. lucidum extracts (GLE). All steps for GEL preparation were described previously [
32]. GLE was dissolved in sterile distilled water to prepare a GLE stock solution (8 mg/mL) and stored at −20 °C until use. GAA was purchased from ChemFaces (Wuhan, China), dissolved in dimethyl sulfoxide (DMSO) to prepare a GAA stock solution (100 mM) and stored at −20 °C until use.
4.2. Cell Lines and Reagents
Both gastric carcinoma cell lines, SNU719 (EBVaGC) and MKN1-EBV (EBVaGC), were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Hyclone, MA, USA) supplemented with 10% fetal bovine serum (Hyclone, MA, USA), antibiotics/antimycotics (Gibco, MD, USA), GlutaMAX® (Gibco, MD, USA), and 25 mmol/mL HEPES (Sigma-Aldrich, MO, USA) at 37 °C in a 5% CO2 and 95% humidity incubator.
4.3. Ethics Statement
The animal experiments were conducted in accordance with the National Research Council’s Guide (IACUC, Seoul, Korea) for the Care and Use of Laboratory Animals. The experimental protocol was approved by the Animal Experiments Committee of Duksung Women’s University (permit number: 2014-016-007).
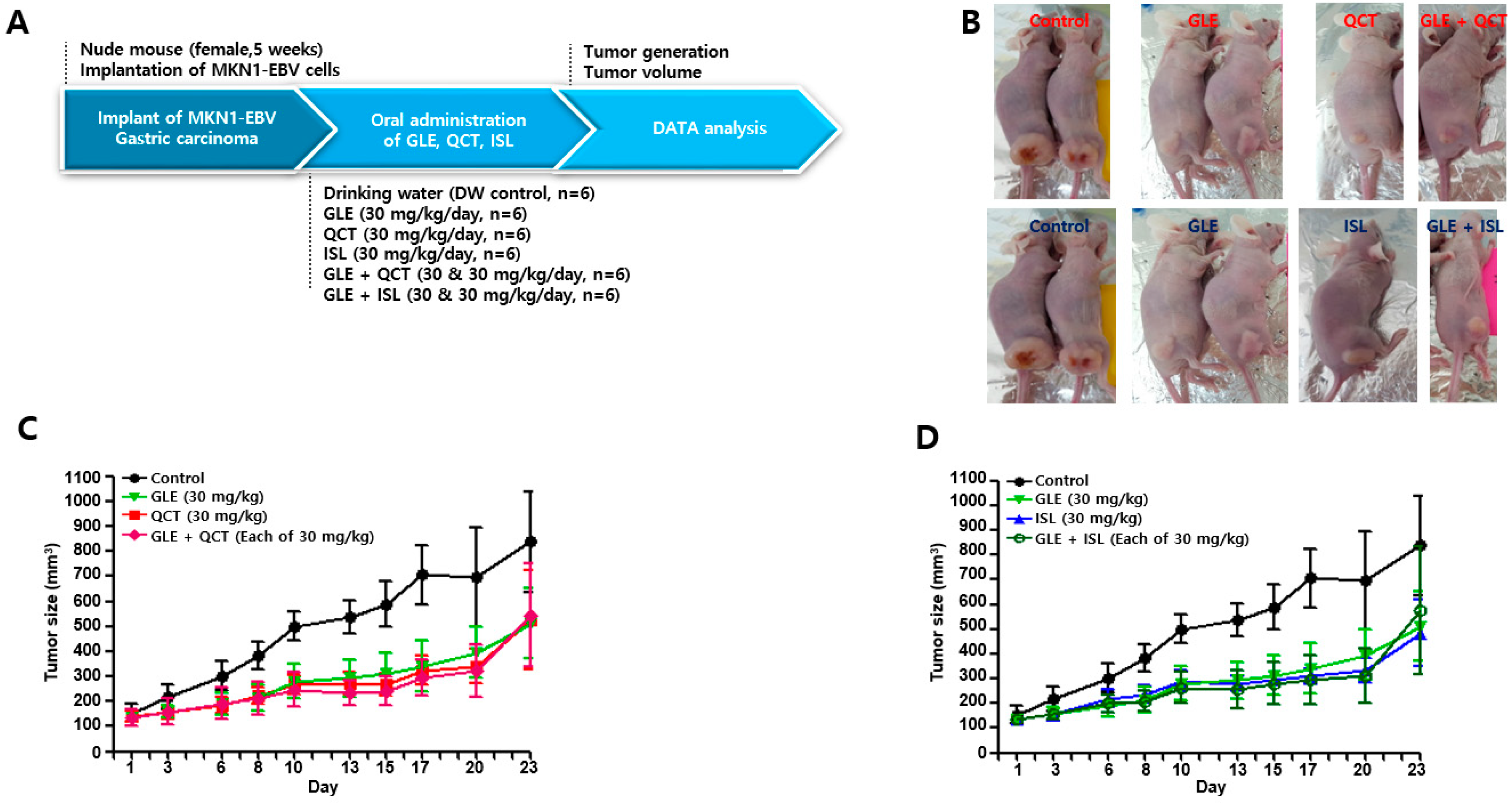
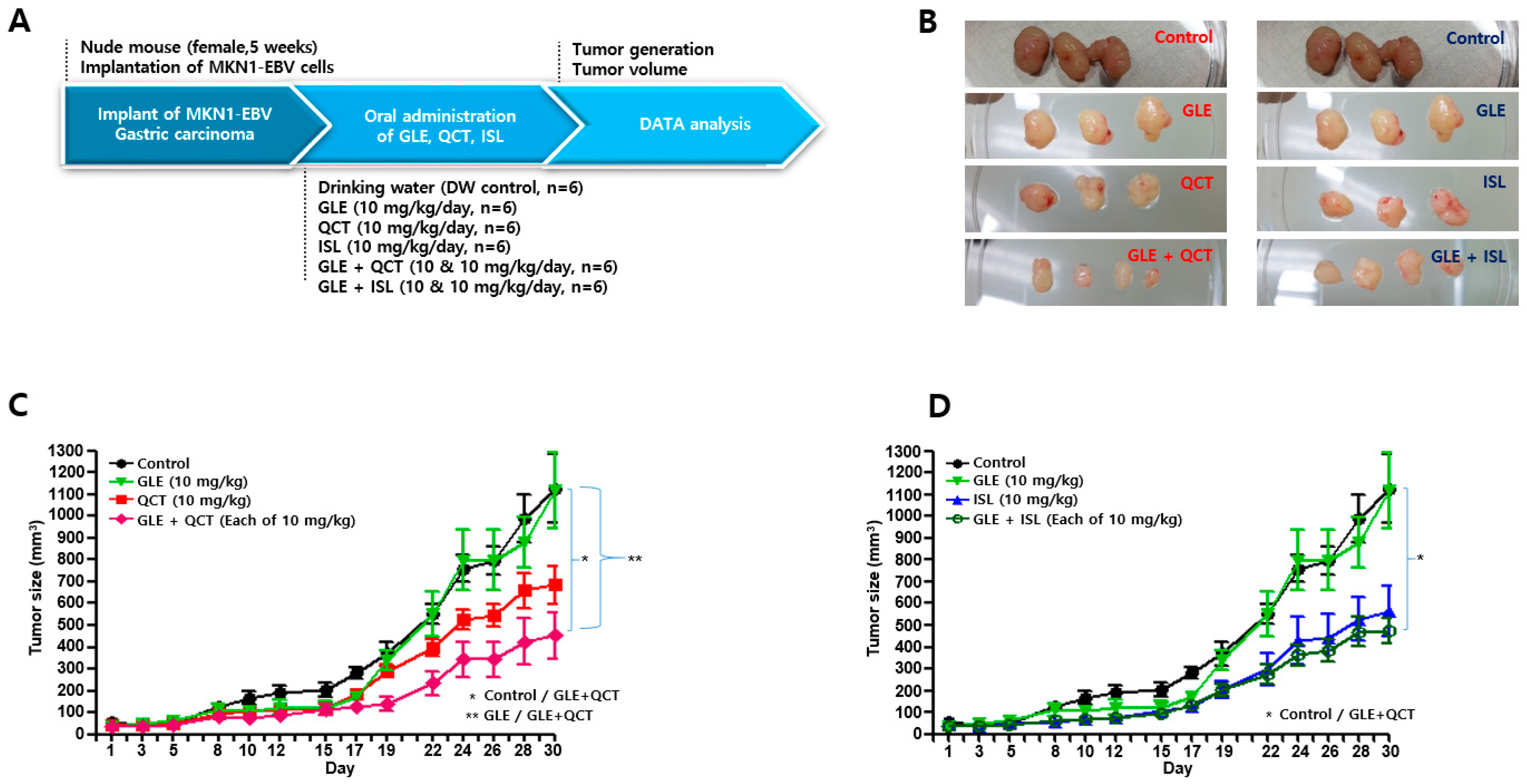
4.4. Antitumor Assay in a Xenograft Mouse Model
Nude mice (female, 5-week-old; Raonbio Co., Ltd., Seoul, Korea) were used as xenograft animal models for the assessment of the antitumor effects. The mice were individually housed in a pathogen-free controlled environment (23–27 °C under a 12 h light/12 h dark cycle) and provided with food and water ad libitum. The mice were first divided randomly into 2 groups (
n = 30) and 5 × 10
6 MKN1-EBV cells were then subcutaneously implanted into the dorsum next to the right hind leg of the mice in both groups. After 14 days, one group was used to test the synergistic antitumor effects of the cotreatment with a relatively high-concentration GLE and QCT or ISL, whereas the other group was used to test the synergistic effects of the cotreatment with a relatively low-concentration GLE and QCT or ISL. Relatively low and high-concentrations were defined based on our previous studies: 100 mg/kg for
Cordyceps militaris extracts [
20], 30 mg/kg quercetin and isoliquiritigenin [
33]. The mice in the group receiving a relatively high-concentration cotreatment were orally administrated drinking water, GLE (30 mg/kg), QCT (30 mg/kg), ISL (30 mg/kg), GLE + QCT (30 mg/kg + 30 mg/kg), or GLE + ISL (30 mg/kg + 30 mg/kg) for 23 days. The mice in the group receiving the low-concentration cotreatment were orally administrated drinking water, GLE (10 mg/kg), QCT (10 mg/kg), ISL (10 mg/kg), GLE + QCT (10 mg/kg + 10 mg/kg), or GLE + ISL (10 mg/kg + 10 mg/kg) for 23 days. The tumors were identified and tumor size was measured every other day using a standard caliper and calculated using the following equation: tumor size = [tumor length (mm) × tumor width (mm)
2]/2, as previously described [
20]. After the tumor size reached approximately 1000 mm
3, the mice were euthanized by an overdose of isoflurane and the euthanasia was confirmed by cervical dislocation. Then, the tumors were harvested surgically.
4.5. Cytotoxicity Assay
The cytotoxic effects of GLE and GAA in SNU719 cells were evaluated using a cytotoxicity assay performed using the cell counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan), according to the manufacturer’s protocol. Briefly, 100 μL of cell suspension (1 × 10
4 cells/well) was seeded into a 96-well plate. On the following day, GLE or GAA was applied at various concentrations (0 ~ 2000 mg/mL for GLE and 0 ~ 5000 μM for GAA). In 48 h post treatment, 10 μL of CCK-8 solution was added to each sample. The samples were incubated for another 3 h and then the absorbance of each cell suspension was measured at 450 nm using an enzyme-linked immunosorbent assay reader. The cytotoxic effects of combined GLE and QCT in SNU719 cells were also evaluated using the CCK-8 cytotoxicity assay mentioned above. Briefly, 100 μL of cell suspension (1 × 10
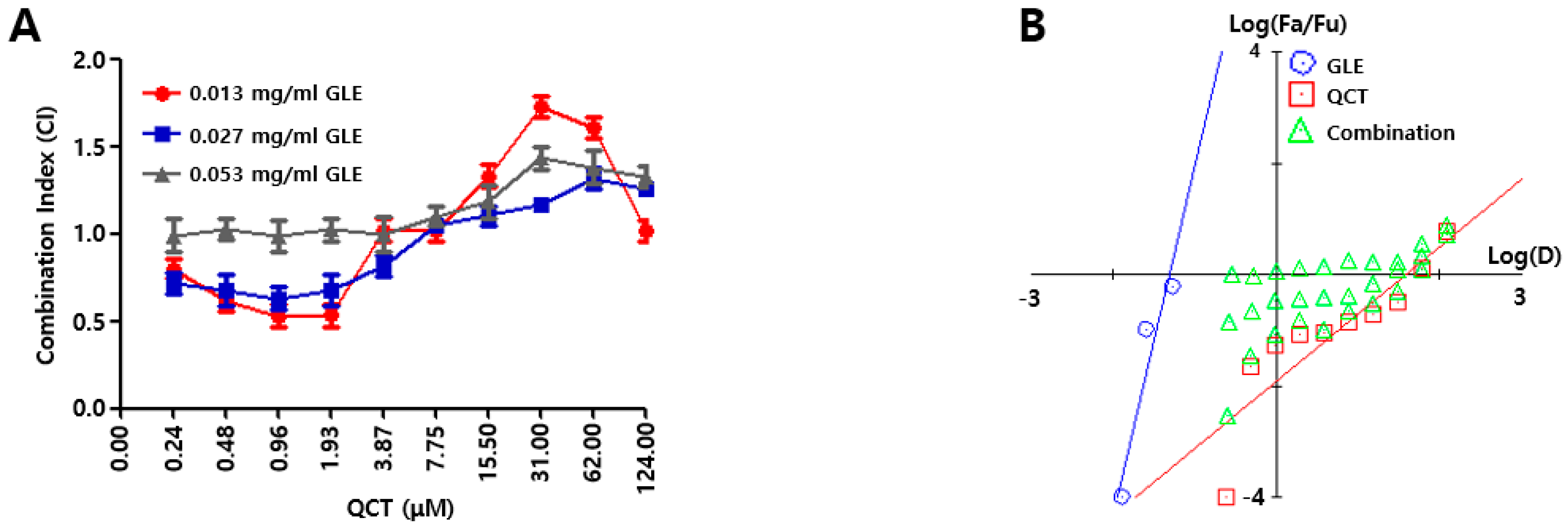
4 cells/well) was seeded into a 96-well plate. On the following day, the combinations of GLE and QCT were made in a series of concentrations (0.000 ~ 0.053 mg/mL for GLE, 0.00 ~ 124.00 μM for QCT) and these combinations were added to the cell suspension in a 96-well plate. Further, 10 μL of CCK-8 solution was added to each sample in 48 h post treatment and the cytotoxicity of the combined compounds were calculated as 450 nm absorbance values of cell suspension. The relative inhibition (RI) of the sample treatment (ST) was evaluated by subtracting RI of the negative control treatment (CT); RI value for ST = 450 nm absorbance value of CT–absorbance value of ST. The measurement of RI values were conducted in triplicate. A synergistic effect was evaluated based on the Chou-Talalay analysis [
19]. First, the intercepts and the slope were assessed from a dose response curve with RI values from GLE and QCT treatments. The RI was used to evaluate the combination index (CI) with a combination of GLE and QCT. The GLE treatments made three data points (0.01 ~ 0.05 mg/mL) and the QCT treatments made 10 data points (0.02 ~ 124.00 μM). Subsequently, both the Fa-CI plot (Fa = fraction affected) and median-effect plot were employed to determine whether the interactions between the two compounds were additive, synergistic or antagonistic. This method involved plotting the dose-effect equation:
In Equation (1), D is the dose, Dm is the required dose for 50% inhibition of cell growth, fa is the faction affected by dose D, fu is the unaffected fraction and m is a coefficient of the sigmoidicity of the dose-effect curve. Both the Fa-CI plot and median-effect plot yielded similar conclusions of synergism or antagonism. Synergism, additivism or antagonism of the combined effects were quantitatively represented by CI. CI < 1.0, 1.0 < CI < 1.1, and CI > 1.1 indicate synergism, additivism and antagonism, respectively.
4.6. Cell Viability Assay
The effects of GLE, QCT, and GAA on SNU719 cell viability were determined using MUSE count and viability kit (Merck Millipore, Darmstadt, Germany), according to the manufacturer’s protocol. The cells (2 × 106) were seeded into a 6-well plate and treated for 48 h with mixtures of GLE and QCT or GLE and GAA at various concentrations (0 ~ 0.027 mg/mL for GLE, 0 ~ 7.75 μM for QCT, and 0 ~ 0.49 mM for GAA). A negative control sample was treated with the same volume of DMSO for 48 h. The treated cells were trypsinized, harvested and resuspended as 106 ~ 107 cells/mL of fresh serum. Cell suspension (20 μL) was mixed with 380 μL of the MUSE count and viability reagent and then incubated at 25 °C for 5 min. Cell viability was measured using MUSE cell analyzer (Millipore, Burlington, MA, USA).
4.7. Apoptosis Assay
The apoptosis effects of QCT and GAA on SNU719 cells were determined using MUSE Annexin V & Dead cell assay kit (Merck Millipore, Darmstadt, Germany), according to the manufacturer’s protocol. The cells (2 × 106) were seeded into a 6-well plate and treated for 48 h with mixtures of QCT and GAA at various concentrations (0 and 0.96 μM for QCT, and 0 and 0.49 mM for GAA). A negative control sample was treated with the same volume of DMSO for 48 h. The treated cells were trypsinized, harvested and resuspended as 105 ~ 106 cells/mL of fresh serum. Cell suspension (180 μL) was mixed with 20 μL of the MUSE Annexin V & Dead Cell reagent and then incubated at 25 °C for 20 min. Cell viability was measured using MUSE cell analyzer (Millipore, Burlington, MA, USA).
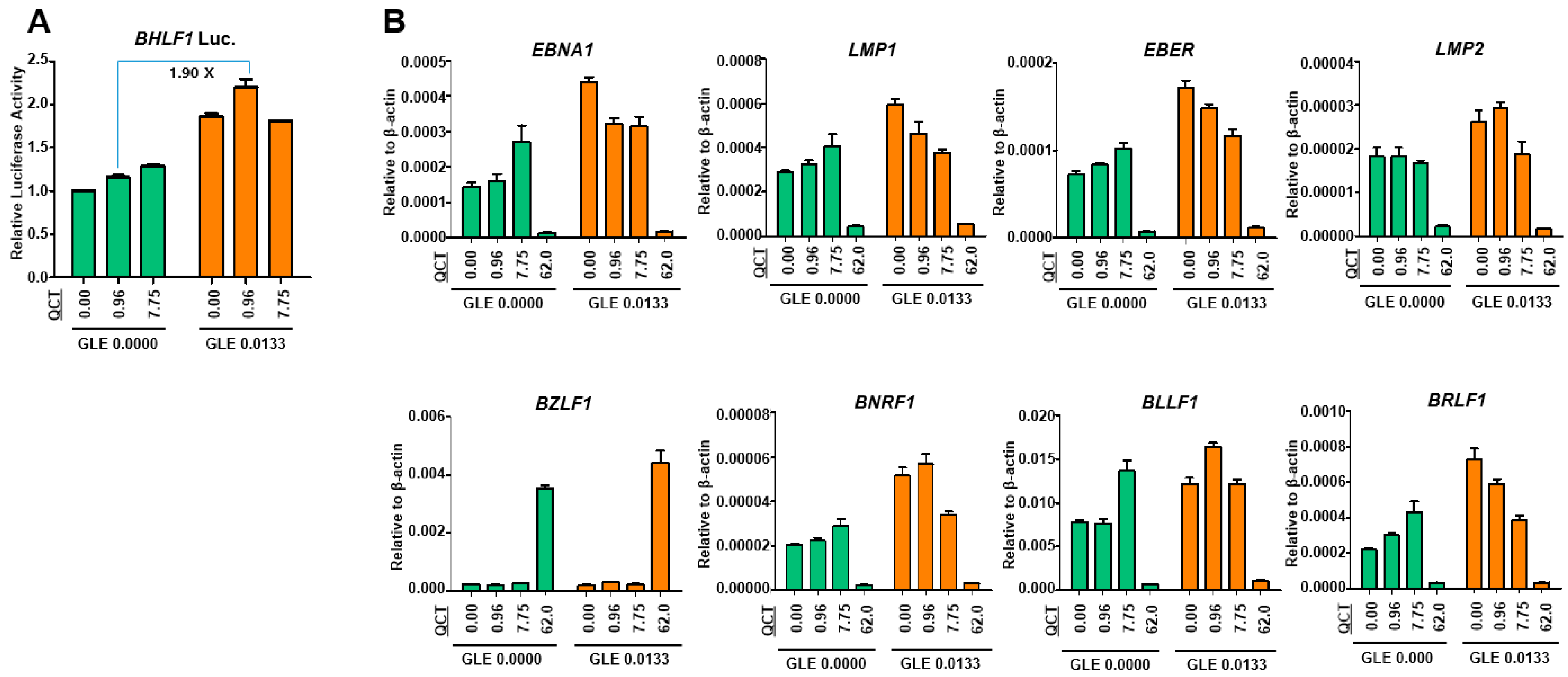
4.8. Luciferase Assay
SNU719 cells were transfected using the Neon system (Invitrogen, Carlsbad, CA, USA) with a luciferase reporter constructs that contained a Renilla luciferase gene transcribed by the promoter of EBV lytic gene, BHLF1. Transfected SNU719 cells were then selected with blasticidin (Invitrogen) for approximately 30 days and established as SNU719-BHLF1 luciferase cells for the luciferase assay. SNU719-BHLF1 luciferase cells were cotreated for 48 h at 37 °C with mixtures of GLE and QCT or GLE and GAA at various concentrations (0 ~ 0.027 mg/mL for GLE, 0 ~ 7.75 μM for QCT, and 0 ~ 0.49 mM for GAA). Luciferase activity was measured using a dual-luciferase reporter assay system (Promega, Madison, WI, USA), according to the manufacturer’s protocol.
4.9. Western Blot Assay
To assess the proapoptotic effects of the cotreatment with GLE and QCT, a western blot analysis was performed using SNU719 cells cotreated with GLE and QCT. The total proteins were harvested using trypsin for 48 h. Cells (107) were lysed using 100 μL of a reporter lysis buffer (Promega, Madison, WI) supplemented with proteinase inhibitor and phenylmethylsulfonyl fluoride. The lysates were further fractionated using a bioruptorsonicator (5 min, 30 s on/off pulses). The total protein content in cell lysates was measured using the Bradford assay. Equivalent amounts of total proteins were subjected to a western blot analysis. The whole proteins were separated using 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to membranes. In addition, cytoplasmic and nuclear proteins of the treated SNU719 cells were recovered using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific, Waltham, MA, USA). The cytoplasmic and nuclear protein expression was determined using a western blot analysis. The membranes were probed with antibodies against the proteins of interest. Anti-poly [ADP-ribose] polymerase 1 (PARP1), anti-Bax, anti-caspase 3, anti-cytochrome C, anti-Bcl-2, anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and anti-proliferating cell nuclear antigen (PCNA) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) and used to detect corresponding proteins. The expression patterns of GAPDH and PCNA were used as internal controls. The antibody-bound proteins were visualized by enhanced chemiluminescence (ECL) detection reagent (GE Healthcare, Chicago, IL, USA). The membranes were stripped and reprobed with other antibodies.
4.10. Quantitative Reverse-Transcriptase PCR Assay
To assess the effects of the cotreatment with GLE and QCT on gene expression, a qRT-PCR assay was performed using SNU719 cells cotreated with GLE and QCT. RNA was isolated from the cells using the RNeasy mini kit (Qiagen, Germantown, MD, USA). Purified RNA was converted into cDNA by using superscript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The primers specific for EBV genes were used. The sequences of primer sets used in this study can be provided upon request.
4.11. LC-MS/MS Analysis
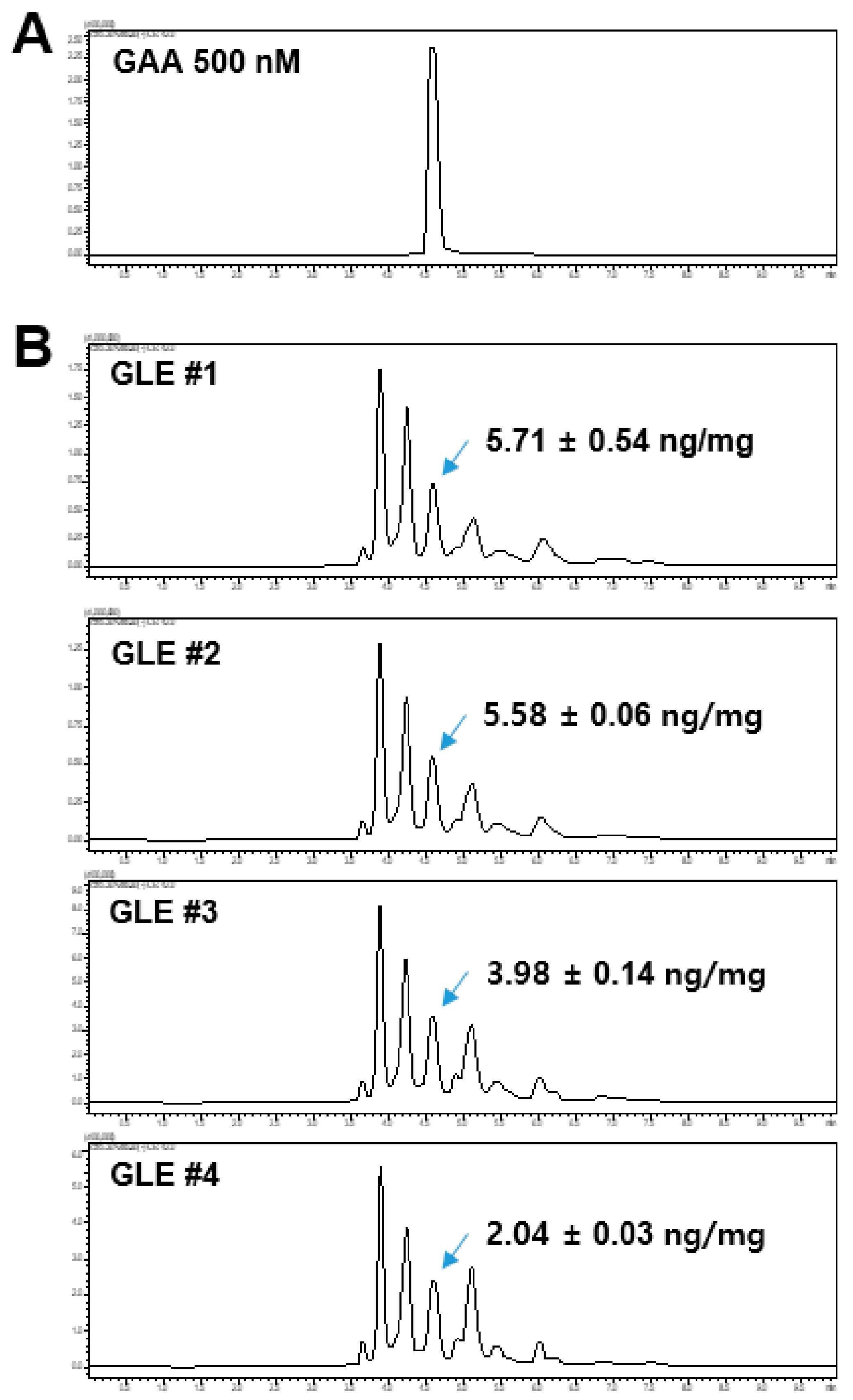
The GLE samples and GAA standard were analyzed using a Shimadzu LCMS-8060 liquid chromatograph-mass spectrometer system (Shimadzu, Tokyo, Japan) equipped with an electrospray ionization (ESI) interface. Analyte separation was performed using the Kinetex C18 column (100 × 2.10 mm, 2.6 μm, 100 Å; Phenomenex, Torrance, CA, USA). The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) and was run on the following gradient: 0–1 min (20% B–40% B), 1–6 min (50% B), and 6.1–10 min (20% B). The total run time was 9 min and the flow rate was 0.2 mL/min. The total run time was 10 min and the flow rate was 0.2 mL/min. Electrospray ionization was performed in negative-ion mode at −3500 V. The optimum operating conditions were determined as follows: capillary temperature, 350 °C; vaporizer temperature, 300 °C; collision gas (argon) pressure, 1.5 mTorr. The quantitation was conducted in multiple reaction monitoring (MRM) mode at 515.3 > 285.3 and the collision energy (CE) value was 40 eV.
4.12. Statistical Analysis
The p-values were calculated by a 2-tailed Student’s t-test using Excel (Microsoft, Redmond, WA, USA). In addition, all statistical analyses were subjected to a one-way analysis of variance (ANOVA) and re-verified by Turkey’s multiple comparison test using GraphPad Prism (San Diego, CA, USA).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






