
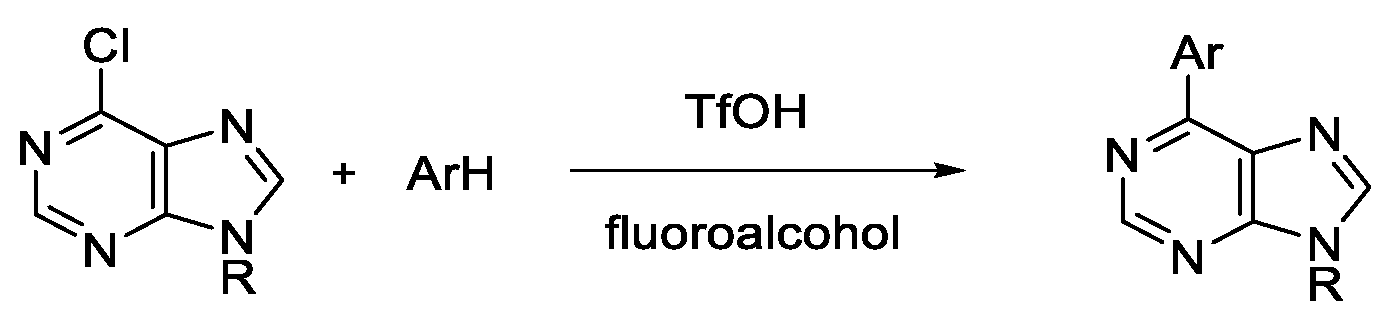
Nucleophilic Arylation of Halopurines Facilitated by Brønsted Acid in Fluoroalcohol

 , and
, and 
Abstract
:1. Introduction
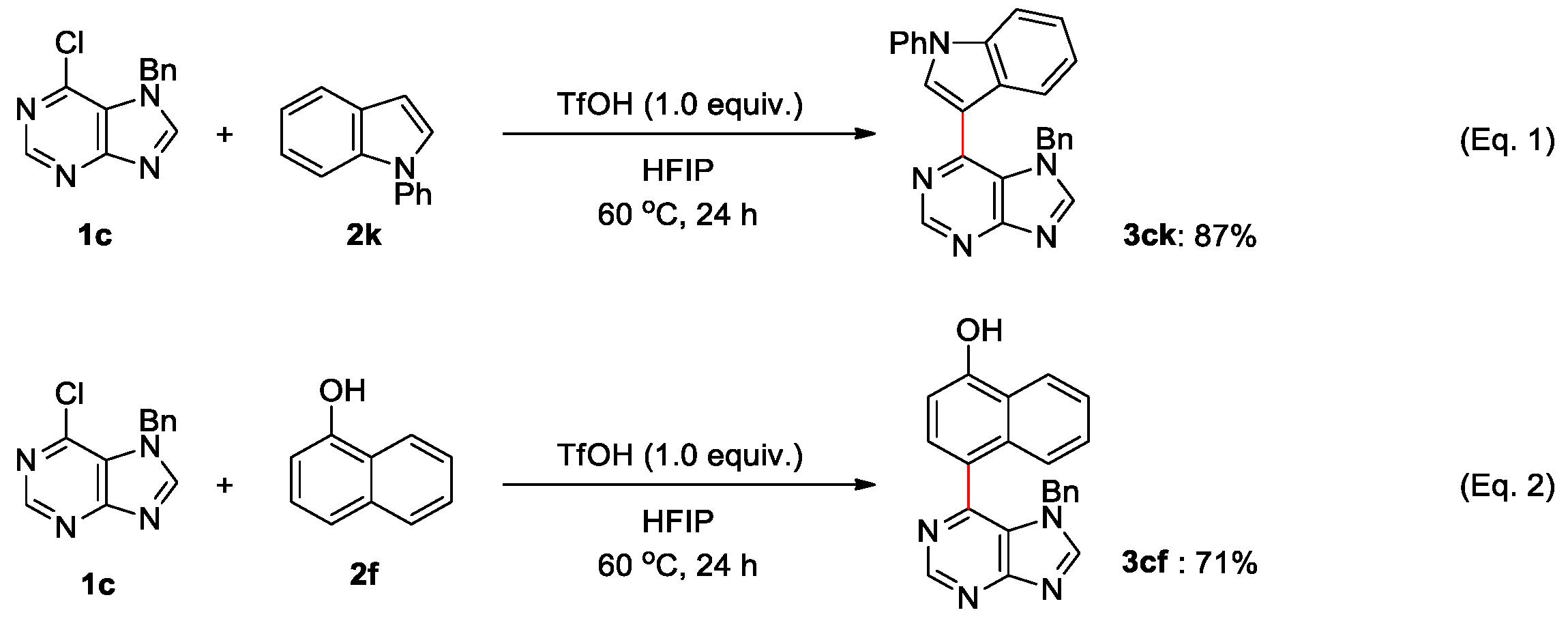
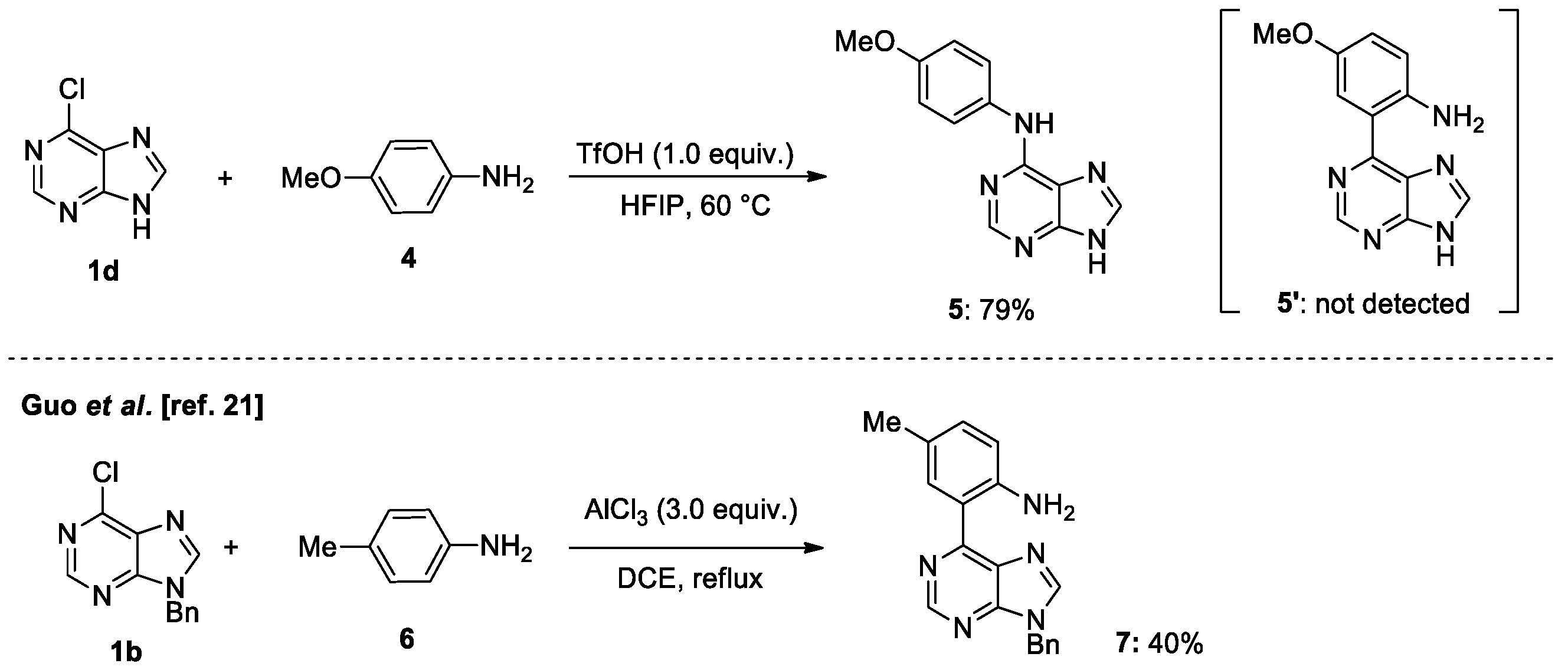
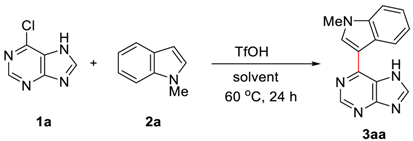
2. Results and Discussions
3. Conclusions
4. Experimental Section
4.1. General Procedure for Brønsted Acid Catalyzed Arylation of Halopurines in Fluoroalcohol (Table 2 and Scheme 3)
4.2. General Procedure for Brønsted Acid Catalyzed N-Coupling of Aniline Derivatives to Halopurines in Fluoroalcohol (Scheme 4)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Legraverend, M.; Grierson, D.S. The purines: Potent and versatile small molecule inhibitors and modulators of key biological targets. Bioorg. Med. Chem. 2006, 14, 3987–4006. [Google Scholar] [CrossRef] [PubMed]
- Bakkestuen, A.K.; Gundersen, L.L.; Utenova, B.T. Synthesis, biological activity, and SAR of antimycobacterial 9-aryl-, 9-arylsulfonyl-, and 9-benzyl-6-(2-furyl)purines. J. Med. Chem. 2005, 48, 2710–2723. [Google Scholar] [CrossRef] [PubMed]
- Hocek, M.; Naus, P.; Pohl, R.; Votruba, I.; Furman, P.A.; Tharnish, P.M.; Otto, M.J. Cytostatic 6-arylpurine nucleosides. 6. SAR in anti-HCV and cytostatic activity of extended series of 6-hetarylpurine ribonucleosides. J. Med. Chem. 2005, 48, 5869–5873. [Google Scholar] [CrossRef] [PubMed]
- Storr, T.E.; Strohmeier, J.A.; Baumann, C.G.; Fairlamb, I.J.S. A sequential direct arylation/Suzuki–Miyaura cross-coupling transformation of unprotected 20-deoxyadenosine affords a novel class of fluorescent analogues. Chem. Commun. 2010, 46, 6470–6472. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G. Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon Press: Oxford, UK, 1984; Volume 5, pp. 501–597. [Google Scholar]
- Agrofoglio, L.A.; Gillaizeau, I.; Saito, Y. Palladium-assisted routes to nucleosides. Chem. Rev. 2003, 103, 1875–1916. [Google Scholar] [CrossRef]
- Hocek, M. Synthesis of purines bearing carbon substituents in positions 2, 6 or 8 by metal- or organometal-mediated C–C bond-forming reaction. Eur. J. Org. Chem. 2003, 245–254. [Google Scholar] [CrossRef]
- Lakshman, M.K.; Hilmer, J.H.; Martin, J.Q.; Keeler, J.C.; Dinh, Y.Q.; Ngassa, F.N.; Russon, L.M. Palladium catalysis for the synthesis of hydrophobic C-6 and C-2 aryl 2′-deoxynucleosides. Comparison of C–C versus C–N bond formation as well as C-6 versus C-2 reactivity. J. Am. Chem. Soc. 2001, 123, 7779–7787. [Google Scholar] [CrossRef]
- Hocek, M.; Holy, A.; Votruba, I.; Dvorakova, H. Synthesis and cytostatic activity of substituted 6-phenylpurine bases and nucleosides: Application of the Suzuki-Miyaura cross-coupling reactions of 6-chloropurine derivatives with phenylboronic acids. J. Med. Chem. 2000, 43, 1817–1825. [Google Scholar] [CrossRef]
- Cerna, I.; Pohl, R.; Klepetarova, B.; Hocek, M. Synthesis of 6,8,9-tri- and 2,6,8,9-tetrasubstituted purines by a combination of the Suzuki cross-coupling, N-arylation, and direct C–H arylation reactions. J. Org. Chem. 2008, 73, 9048–9054. [Google Scholar] [CrossRef]
- Lakshman, M.K.; Thomson, P.F.; Nuqui, M.A.; Hilmer, J.H.; Sevova, N.; Boggess, B. Facile Pd-catalyzed cross-coupling of 2′-deoxyguanosine O6-arylsulfonates with arylboronic acids. Org. Lett. 2002, 4, 1479–1482. [Google Scholar] [CrossRef]
- Gunda, P.; Russon, L.M.; Lakshman, M.K. Pd-catalyzed amination of nucleoside arylsulfonates to yield N6-aryl-2,6-diaminopurine nucleosides. Angew. Chem. Int. Ed. 2004, 43, 6372–6377. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, M.K.; Gunda, P.; Pradhan, P. Mild and room temperature C–C bond forming reactions of nucleoside C-6 arylsulfonates. J. Org. Chem. 2005, 70, 10329–10335. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Robins, M.J. Fluoro, alkylsulfanyl, and alkylsulfonyl leaving groups in suzuki cross-coupling reactions of purine 2′-deoxynucleosides and nucleosides. Org. Lett. 2005, 7, 1149–1151. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Robins, M.J. Azoles as Suzuki cross-coupling leaving groups: Syntheses of 6-arylpurine 2′-deoxynucleosides and nucleosides from 6-(imidazol-1-yl)- and 6-(1,2,4-triazol-4-yl)purine derivatives. Org. Lett. 2004, 6, 3421–3423. [Google Scholar] [CrossRef] [PubMed]
- Kang, F.A.; Sui, Z.; Murray, W.V. Pd-catalyzed direct arylation of tautomerizable heterocycles with aryl boronic acids via C-OH bond activation using phosphonium salts. J. Am. Chem. Soc. 2008, 130, 11300–11302. [Google Scholar] [CrossRef]
- Langli, G.; Gundersen, L.-L.; Rise, F. Regiochemistry in Stille couplings of 2,6-dihalopurines. Tetrahedron 1996, 52, 5625–5638. [Google Scholar] [CrossRef]
- Havelková, M.; Dvořák, D.; Hocek, M. Covalent analogues of DNA base-pairs and triplets. Part 3: Synthesis of 1,4- and 1,3-bis(purin-6-yl)benzenes and 1-(1,3-dimethyluracil-5-yl)-3 or 4-(purin-9-yl)benzenes. Tetrahedron 2002, 58, 7431–7435. [Google Scholar] [CrossRef]
- Gundersen, L.L.; Langli, G.; Rise, F. Regioselective Pd-mediated coupling between 2,6-dichloropurines and organometallic reagents. Tetrahedron Lett. 1995, 36, 1945–1948. [Google Scholar] [CrossRef]
- Furstner, A.; Leitner, A.; Mendez, M.; Krause, H. Iron-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. [Google Scholar] [CrossRef]
- Guo, H.-M.; Li, P.; Niu, H.-Y.; Wang, D.-C.; Qu, G.-R. Direct synthesis of 6-arylpurines by reaction of 6-chloropurines with activated aromatics. J. Org. Chem. 2010, 75, 6016–6018. [Google Scholar] [CrossRef]
- Edenhofer, A. Novel route to imidazoles, and their use for the synthesis of purines and 4,6-dihydro-1,2-dimethyl-8-phenylimidazo[4,5-e]-1,4-diazepin-5(1H)-one. Helv. Chim. Acta 1975, 58, 2192–2209. [Google Scholar] [CrossRef]
- Gundersen, L.-L.; Bakkestuen, A.K.; Aasen, J.; Overas, H.; Rise, F. 6-Halopurines in palladium-catalyzed coupling with organotin and organozinc reagents. Tetrahedron 1994, 50, 9743–9756. [Google Scholar] [CrossRef]
- Havelkova, M.; Hocek, M.; Cesnek, M.; Dvorak, D. The Suzuki-Miyaura cross-coupling reactions of 6-halopurines with boronic acids leading to 6-aryl- and 6-alkenylpurines. Synlett 1999, 1145–1147. [Google Scholar] [CrossRef]
- Takenaga, N.; Ueda, S.; Hayashi, T.; Dohi, T.; Kitagaki, S. Facile synthesis of stable uracil-iodonium(III) salts with various counterions. Heterocycles 2018, 97, 1248–1256. [Google Scholar] [CrossRef]
- Takenaga, N.; Ueda, S.; Hayashi, T.; Dohi, T.; Kitagaki, S. Vicinal functionalization of uracil heterocycles with base activation of iodonium(III) salts. Heterocycles 2019, 99, 865–874. [Google Scholar] [CrossRef]
- Takenaga, N.; Hayashi, T.; Ueda, S.; Satake, H.; Yamada, Y.; Kodama, T.; Dohi, T. Synthesis of uracil-iodonium(III) salts for practical utilization as nucleobase synthetic modules. Molecules 2019, 24, 3034. [Google Scholar] [CrossRef]
- Reichardt, C. Empirical parameters of solvent polarity as linear free-energy relationships. Angew. Chem. Int. Ed. Engl. 1979, 18, 98–110. [Google Scholar] [CrossRef]
- Schadt, F.L.; Bentley, T.W.; Schleyer, P.V.R. The SN2-SN1 spectrum. 2. Quantitative treatments of nucleophilic solvent assistance. A scale of solvent nucleophilicities. J. Am. Chem. Soc. 1976, 98, 7667–7675. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Abbound, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, .pi*., alpha., and .beta., and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Dohi, T.; Yamaoka, N.; Kita, Y. Fluoroalcohols: Versatile solvents in hypervalent iodine chemistry and syntheses of diaryliodonium(III) salts. Tetrahedron 2010, 66, 5775–5785. [Google Scholar] [CrossRef]
- Kamitanaka, T.; Morimoto, K.; Tsuboshima, K.; Koseki, D.; Takamuro, H.; Dohi, T.; Kita, Y. Efficient coupling reaction of quinone monoacetal with phenols leading to phenol biaryls. Angew. Chem. Int. Ed. 2016, 55, 15535–15538. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Ito, M.; Morimoto, K.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Versatile direct dehydrative approach for diaryliodonium(III) salts in fluoroalcohol media. Chem. Commun. 2007, 4152–4154. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Ito, M.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Unusual ipso substitution of diaryliodonium bromides initiated by a single-electron-transfer oxidizing process. Angew. Chem. Int. Ed. 2010, 49, 3334–3337. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, N.; Sumida, K.; Itani, I.; Kubo, H.; Ohnishi, Y.; Sekiguchi, S.; Dohi, T.; Kita, Y. Single-electron-transfer (SET)-induced oxidative biaryl coupling by polyalkoxybenzene-derived diaryliodonium(III) salts. Chem. Eur. J. 2013, 19, 15004–15011. [Google Scholar] [CrossRef]
- Dohi, T.; Ueda, S.; Hirai, A.; Kojima, Y.; Morimoto, K.; Kita, Y. Selective aryl radical transfers into N-heteroaromatics from diaryliodonoium salts with trimethoxybenzene auxiliary. Heterocycles 2017, 95, 1272–1284. [Google Scholar] [CrossRef]
- Carbin, B.; Coxon, C.R.; Lebraud, H.; Elliott, K.J.; Matheson, C.J.; Meschini, E.; Roberts, A.R.; Turner, D.M.; Wong, C.; Cano, C.; et al. Trifluoroacetic acid in 2,2,2-trifluoroethanol facilitates SNAr reactions of heterocycles with arylamines. Chem. Eur. J. 2014, 20, 2311–2317. [Google Scholar] [CrossRef]
- Whitfield, H.J.; Griffin, R.J.; Hardcastle, I.R.; Henderson, A.; Meneyrol, J.; Mesguiche, V.; Sayle, K.L.; Golding, B.T. Facilitation of addition–elimination reactions in pyrimidines and purines using trifluoroacetic acid in trifluoroethanol. Chem. Commun. 2003, 2802–2803. [Google Scholar] [CrossRef]
- Marchetti, F.; Cano, C.; Curtin, N.J.; Golding, B.T.; Griffin, R.J.; Haggerty, K.; Newell, D.R.; Parsons, R.J.; Payne, S.L.; Wang, L.Z.; et al. Synthesis and biological evaluation of 5-substituted O4-alkylpyrimidines as CDK2 inhibitors. Org. Biomol. Chem. 2010, 8, 2397–2407. [Google Scholar] [CrossRef]
- Wong, C.; Griffin, R.J.; Hardcastle, I.R.; Northen, J.S.; Wang, L.Z.; Golding, B.T. Synthesis of sulfonamide-based kinase inhibitors from sulfonates by exploiting the abrogated SN2 reactivity of 2,2,2-trifluoroethoxysulfonates. Org. Biomol. Chem. 2010, 8, 2457–2464. [Google Scholar] [CrossRef]
- Bégué, J.; Bonnet-delpon, D.; Crousse, B. Fluorinated alcohols: A new medium for selective and clean reaction. Synlett 2004, 18–29. [Google Scholar] [CrossRef]
- Shuklov, I.A.; Dubrovina, N.V.; Boerner, A. Fluorinated alcohols as solvents, cosolvents and additives in homogeneous catalysis. Synthesis 2007, 2925–2943. [Google Scholar] [CrossRef]
- Baeza, A.; Najera, C. Recent advances in the direct nucleophilic substitution of allylic alcohols through SN1-type reactions. Synthesis 2014, 46, 25–34. [Google Scholar] [CrossRef]
- Berkessel, A.; Adrio, J.A. Dramatic acceleration of olefin epoxidation in fluorinated alcohols: Activation of hydrogen peroxide by multiple H-bond networks. J. Am. Chem. Soc. 2006, 128, 13412–13420. [Google Scholar] [CrossRef] [PubMed]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar] [CrossRef]
- Zhou, Z.; Cheng, Q.-Q.; Kürti, L. Aza-Rubottom oxidation: Synthetic access to primary α-aminoketones. J. Am. Chem. Soc. 2019, 141, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Robins, M.J. SNAr displacements with 6-(fluoro, chloro, bromo, iodo, and alkylsulfonyl)purine nucleosides: Synthesis, kinetics, and mechanism. J. Am. Chem. Soc. 2007, 129, 5962–5968. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, V.D.; Richmond, E.; Wolf, E.; Moran, J. Catalytic Friedel-Crafts reactions of highly electronically deactivated benzylic alcohols. Angew. Chem. Int. Ed. 2017, 56, 3085–3089. [Google Scholar] [CrossRef]
- Liu, W.; Wang, H.; Li, C.-J. Metal-free markovnikov-type alkyne hydration under mild conditions. Org. Lett. 2016, 18, 2184–2187. [Google Scholar] [CrossRef]
- Dohi, T.; Hu, Y.; Kamitanaka, T.; Washimi, N.; Kita, Y. [3 + 2] Coupling of quinone monoacetals by combined acid-hydrogen bond donor. Org. Lett. 2011, 13, 4814–4817. [Google Scholar] [CrossRef]
- Lee, J.W.; Oliveira, M.T.; Jang, H.B.; Lee, S.; Chi, D.Y.; Kim, D.W.; Song, C.E. Hydrogen-bond promoted nucleophilic fluorination: Concept, mechanism and applications in positron emission tomography. Chem. Soc. Rev. 2016, 45, 4638–4650. [Google Scholar] [CrossRef]
- Braendvang, M.; Gundersen, L.-L. Selective anti-tubercular purines: Synthesis and chemotherapeutic properties of 6-aryl- and 6-heteroaryl-9-benzylpurines. Bioorg. Med. Chem. 2005, 13, 6360–6373. [Google Scholar] [CrossRef]
- Wang, X.; Han, C.; Wu, K.; Luo, L.; Wang, Y.; Du, X.; He, Q.; Ye, F. Design, synthesis and ability of non-gold complexed substituted purine derivatives to inhibit LPS-induced inflammatory response. Eur. J. Med. Chem. 2018, 149, 10–21. [Google Scholar] [CrossRef]
Sample Availability: Samples of the products are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | TfOH | Yield (%) b |
|---|---|---|---|
| 1 | HFIP | 0.1 equiv. | 8 |
| 2 | HFIP | 0.2 equiv. | 54 |
| 3 | HFIP | 0.5 equiv. | 89 |
| 4 | HFIP | 1.0 equiv. | quant. |
| 5 | HFIP/DCE = 9:1 | 1.0 equiv. | 60 |

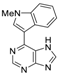 | 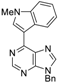 | 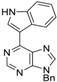 |  |
| 3aa: quant. | 3ba: 82% | 3bb: quant. | 3bc: 96% |
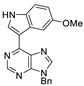 | 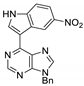 | 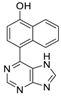 | 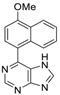 |
| 3bd: 90% | 3be: n.r | 3af: 97% | 3ag: 85% |
 | 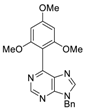 | 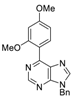 | 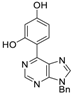 |
| 3bg: 92% | 3bh: quant. | 3bi: quant. | 3bj: quant. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takenaga, N.; Shoji, T.; Menjo, T.; Hirai, A.; Ueda, S.; Kikushima, K.; Hanasaki, T.; Dohi, T. Nucleophilic Arylation of Halopurines Facilitated by Brønsted Acid in Fluoroalcohol. Molecules 2019, 24, 3812. https://doi.org/10.3390/molecules24213812
Takenaga N, Shoji T, Menjo T, Hirai A, Ueda S, Kikushima K, Hanasaki T, Dohi T. Nucleophilic Arylation of Halopurines Facilitated by Brønsted Acid in Fluoroalcohol. Molecules. 2019; 24(21):3812. https://doi.org/10.3390/molecules24213812
Chicago/Turabian StyleTakenaga, Naoko, Toshitaka Shoji, Takayuki Menjo, Akiko Hirai, Shohei Ueda, Kotaro Kikushima, Tomonori Hanasaki, and Toshifumi Dohi. 2019. "Nucleophilic Arylation of Halopurines Facilitated by Brønsted Acid in Fluoroalcohol" Molecules 24, no. 21: 3812. https://doi.org/10.3390/molecules24213812
APA StyleTakenaga, N., Shoji, T., Menjo, T., Hirai, A., Ueda, S., Kikushima, K., Hanasaki, T., & Dohi, T. (2019). Nucleophilic Arylation of Halopurines Facilitated by Brønsted Acid in Fluoroalcohol. Molecules, 24(21), 3812. https://doi.org/10.3390/molecules24213812