
Auriculatone Sulfate Effectively Protects Mice Against Acetaminophen-Induced Liver Injury

 ,
, 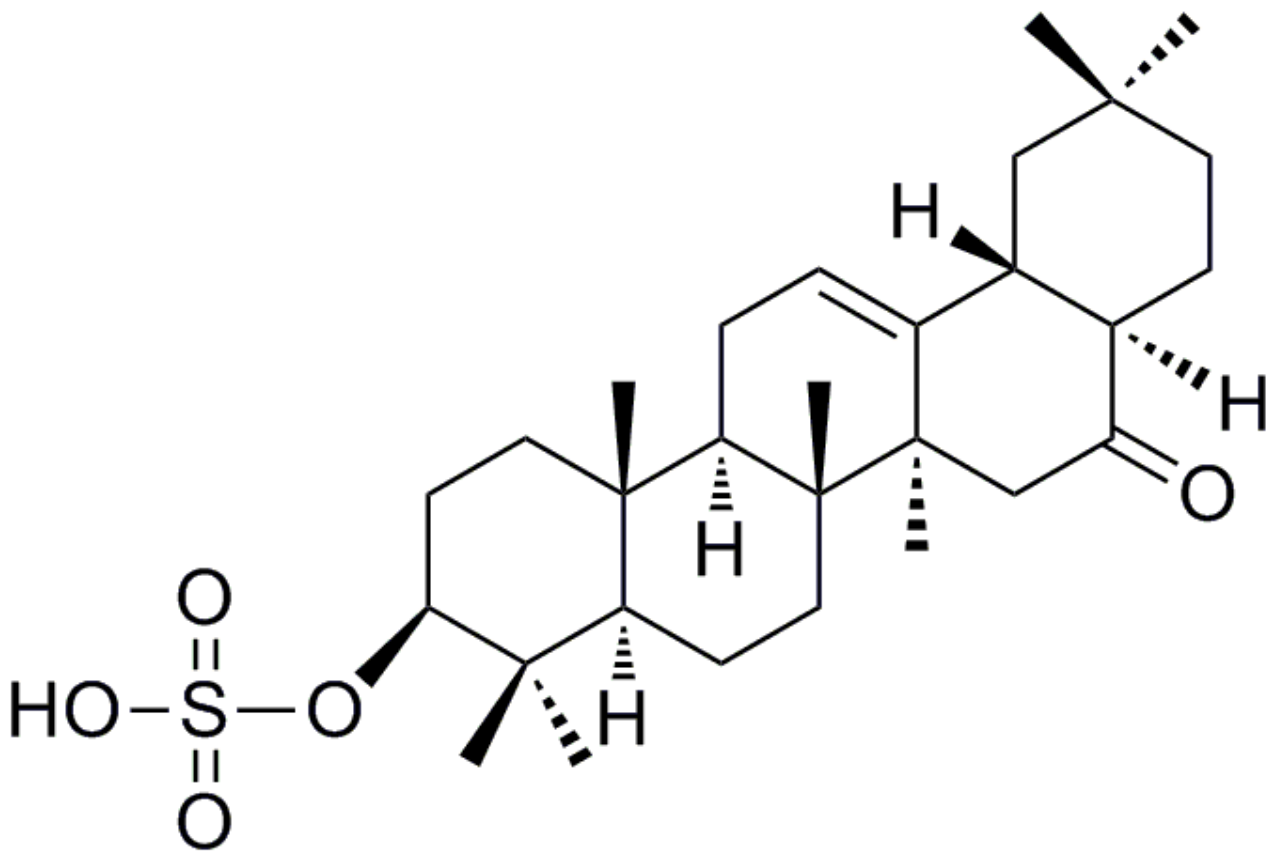{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
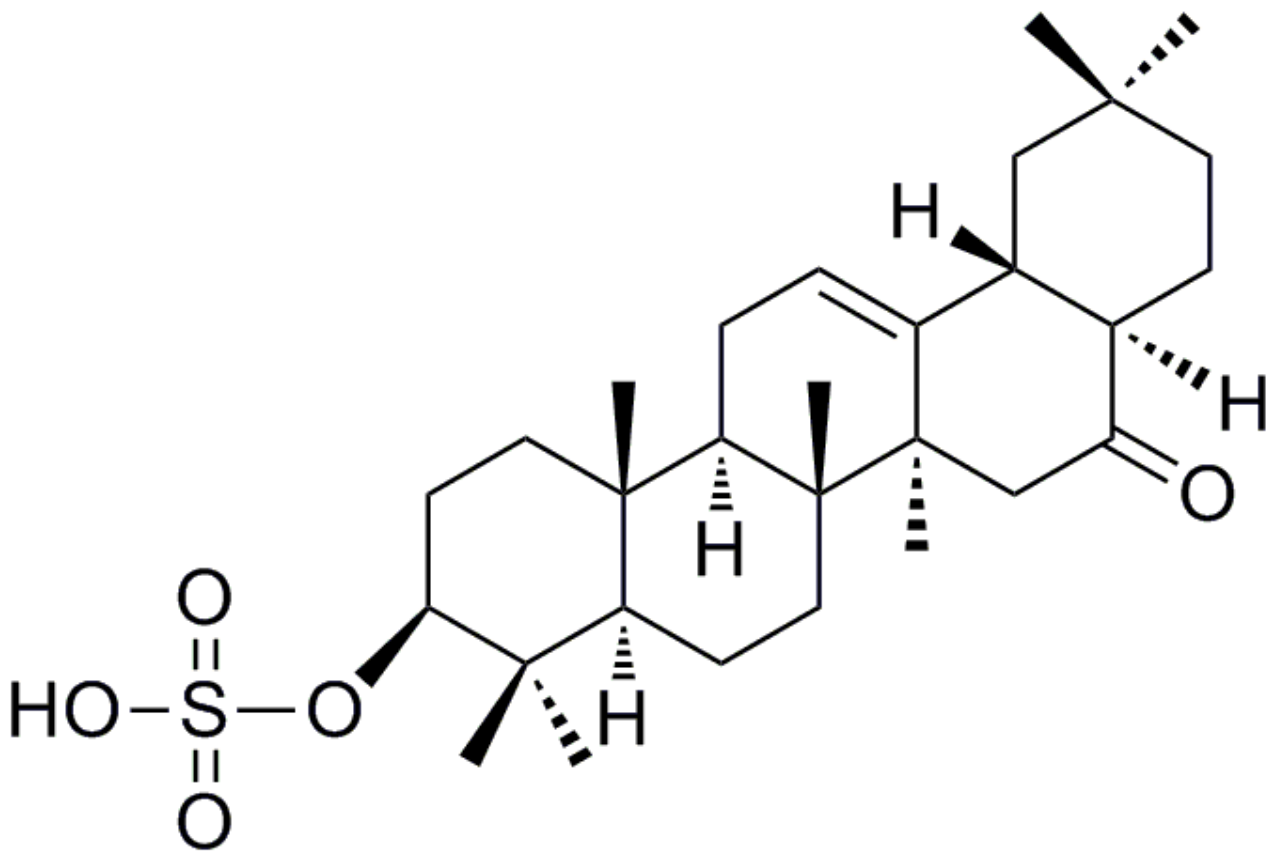
2.1. Protection of the Liver by AS against the APAP-Induced Injury in Mice
2.2. Effects of AS on Liver Histopathology
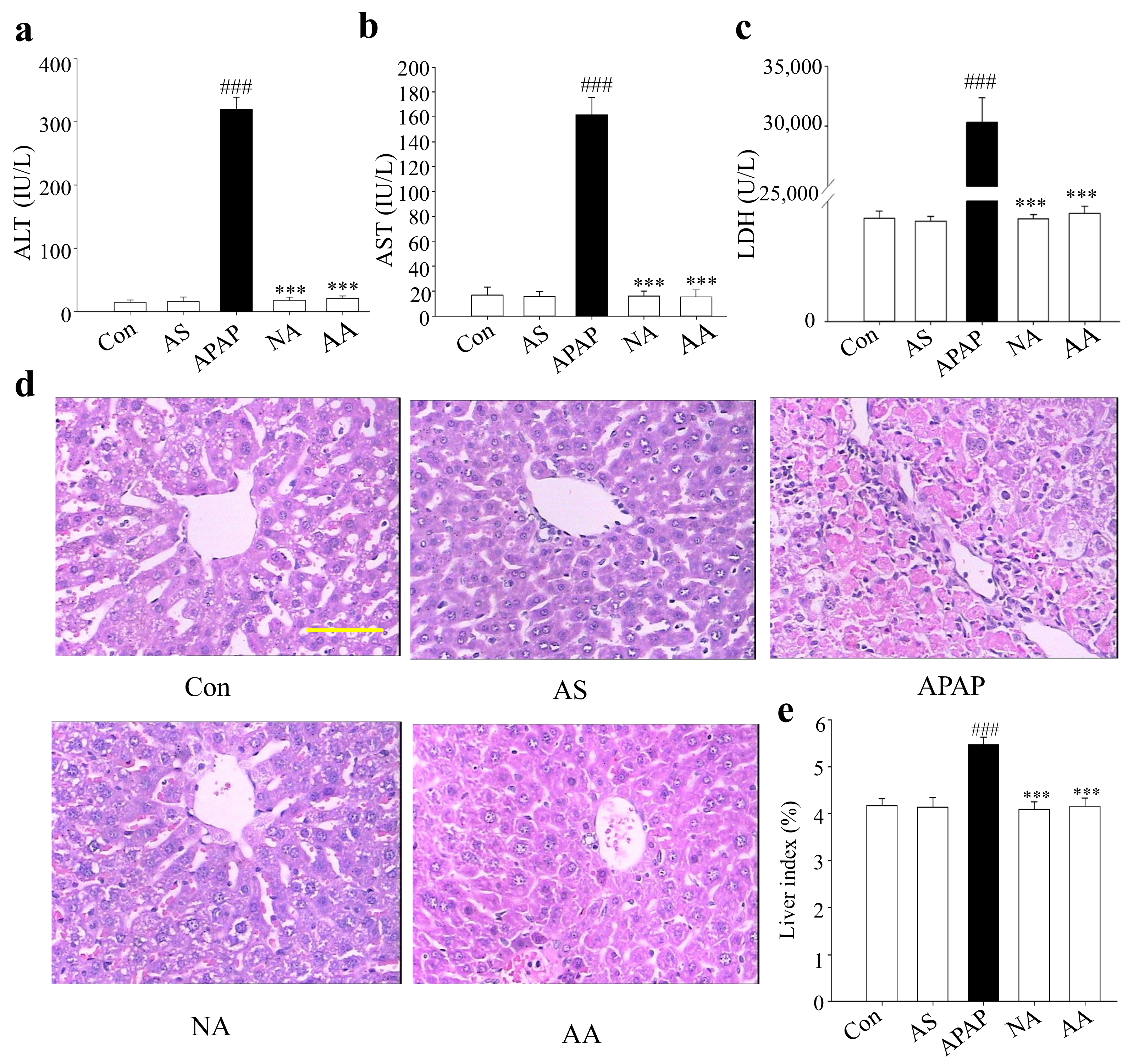
2.3. AS Inhibits APAP-Induced Hepatic Mitochondrial Injury
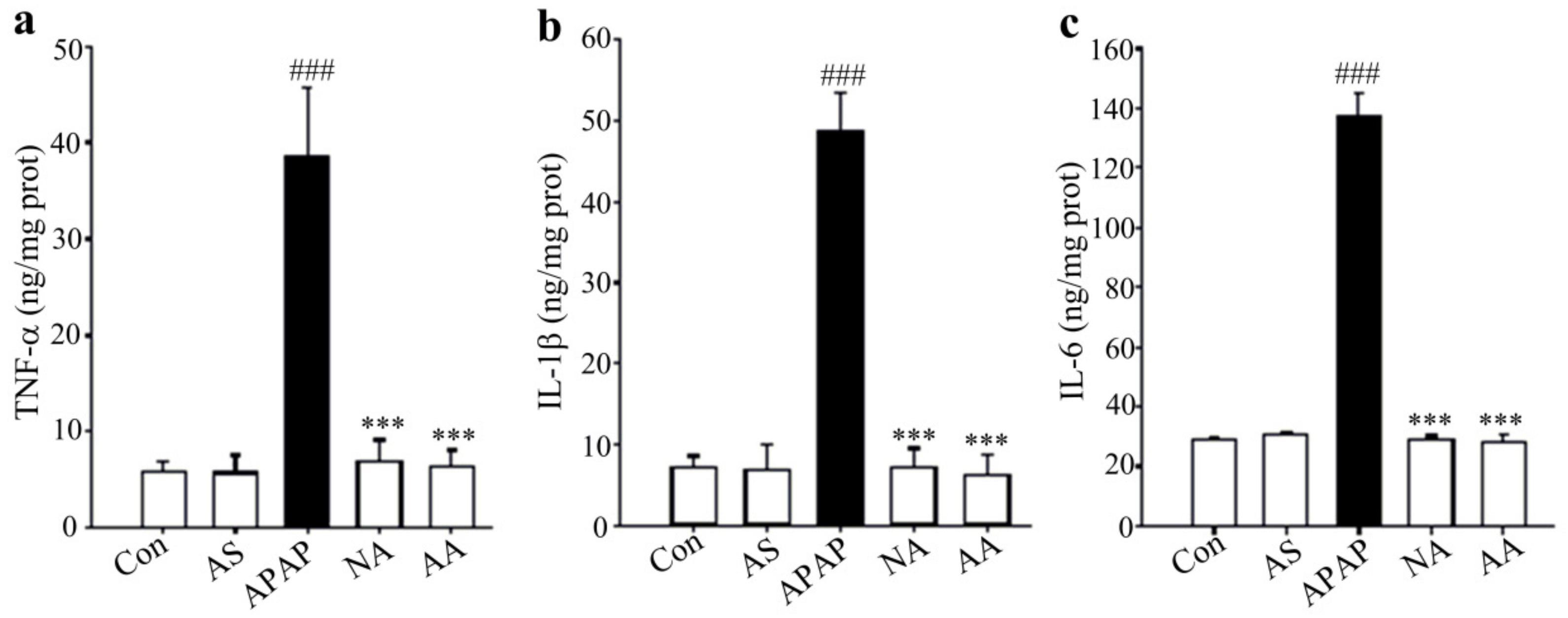
2.4. Effects of AS on TNF-α, IL-1β, and IL-6 Levels in the Liver
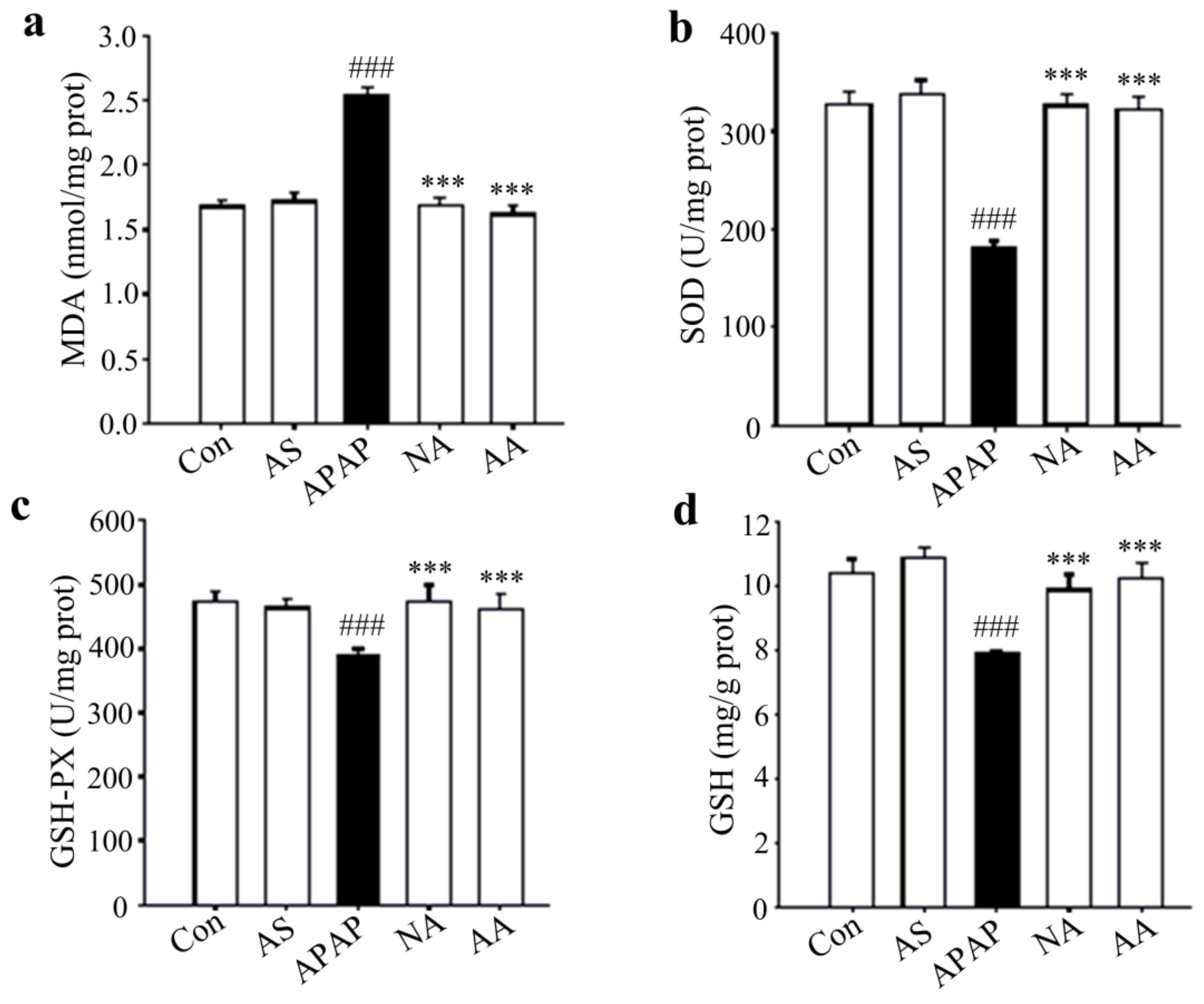
2.5. Effects of AS on MDA, SOD, GSH and GSH-PX Levels of Liver Tissues
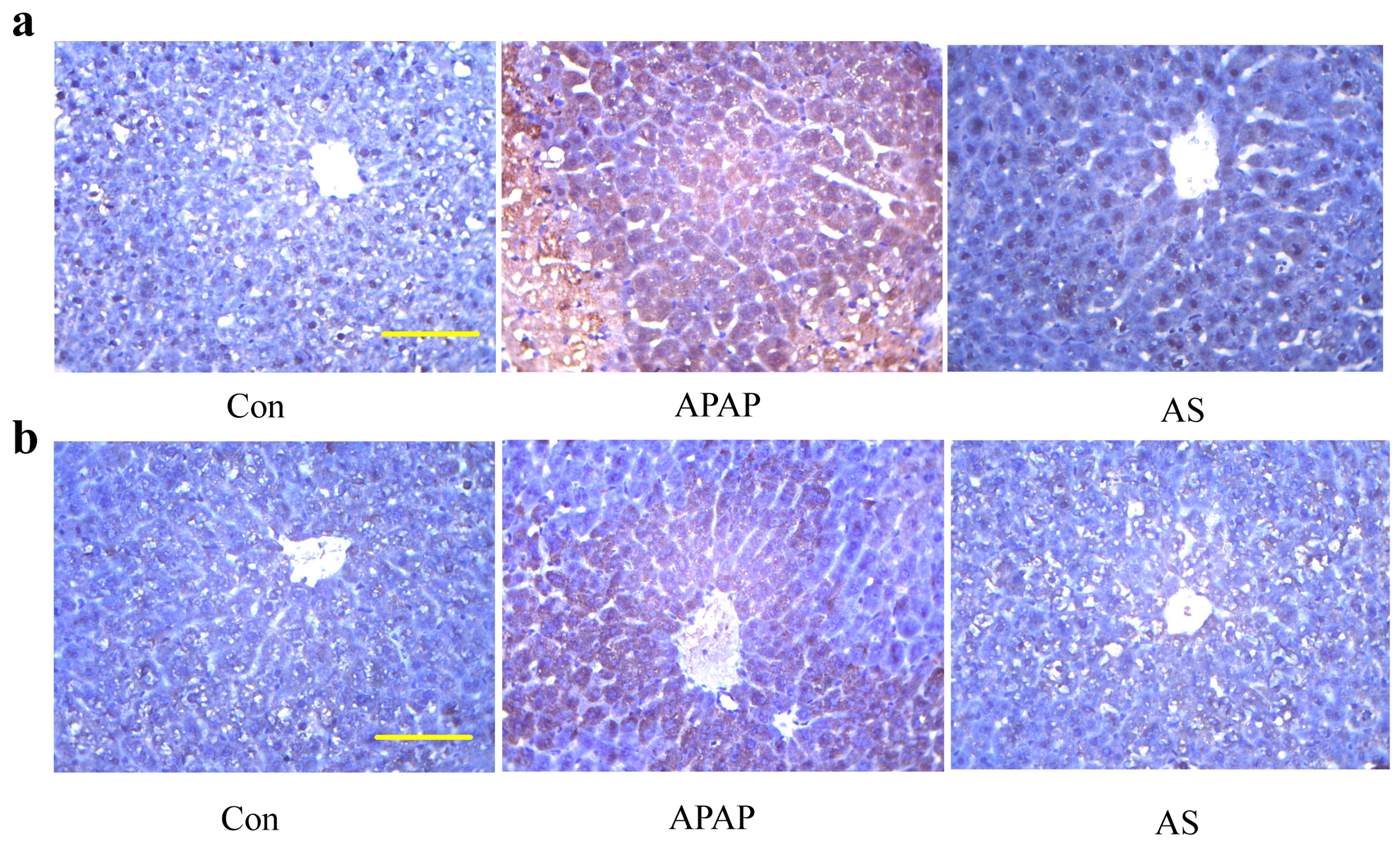
2.6. Immunohistochemical Examination
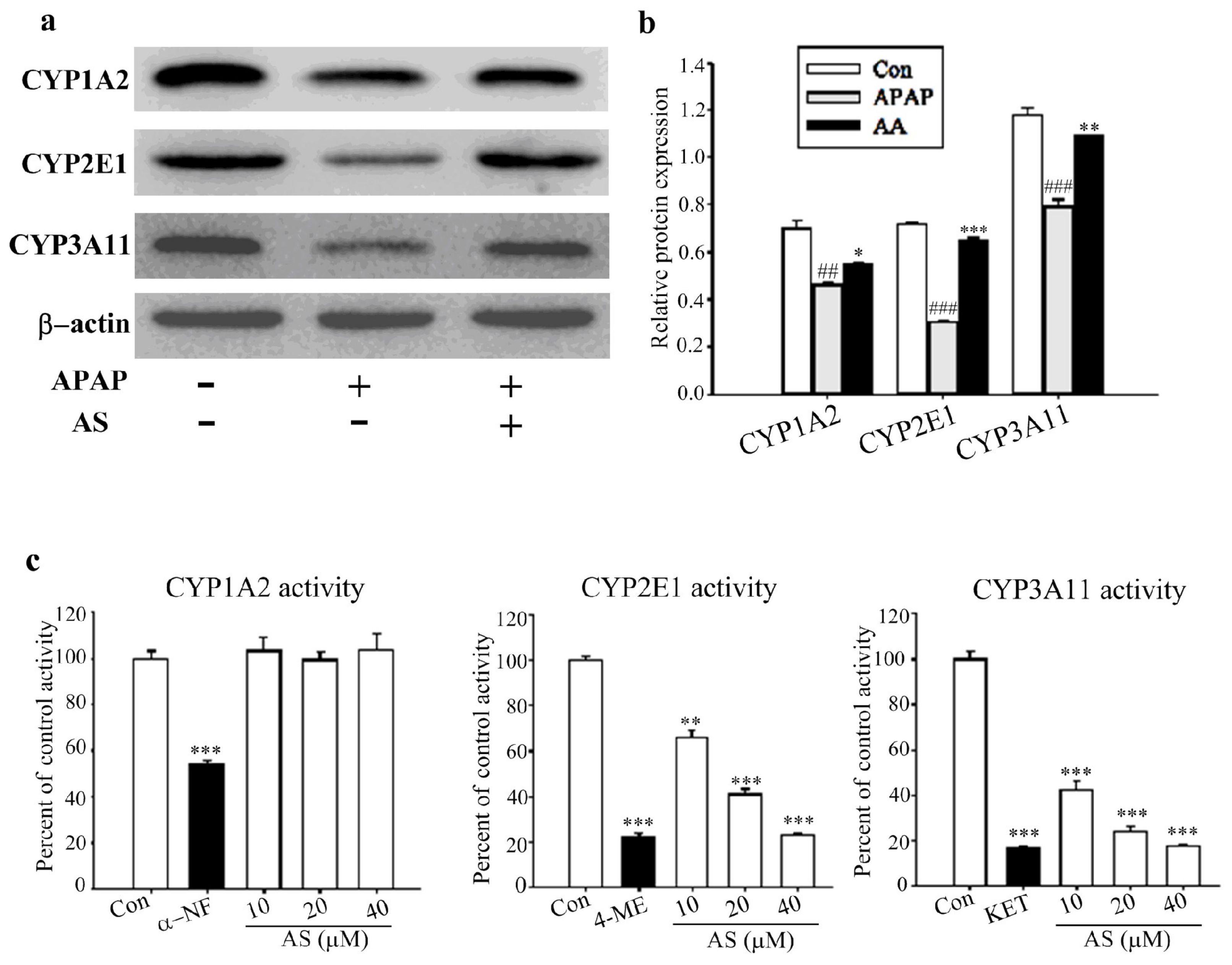
2.7. Effects of AS on the Expressions and Activities of CYPs
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals and Treatments
4.3. Assays of Serum ALT, AST and LDH
4.4. Determinations of Hepatic MDA, SOD, GSH and GSH-PX
4.5. Determination of Cytokines
4.6. Preparation of Mitochondria and Measurement of Mitochondrial ATPase Activity
4.7. Histological Studies
4.8. Immunohistochemical Staining
4.9. Measurement of Liver CYP Activity
4.10. Western Blot Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blazer, D.G.; Wu, L.T. Nonprescription use of pain relievers by middle-aged and elderly community-living adults: National Survey on Drug Use and Health. J. Am. Geriatr. Soc. 2009, 57, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. Arjunolic acid, a triterpenoid saponin, prevents acetaminophen (APAP)-induced liver and hepatocyte injury via the inhibition of APAP bioactivation and JNK-mediated mitochondrial protection. Free Radic. Biol. Med. 2010, 48, 535–553. [Google Scholar] [CrossRef]
- Nelson, S.D. Metabolic activation and drug toxicity. J. Med. Chem. 1982, 25, 753–765. [Google Scholar] [CrossRef]
- Wang, E.J.; Li, Y.; Lin, M.; Chen, L.; Stein, A.P.; Reuhl, K.R.; Yang, C.S. Protective effects of garlic and related organosulfur compounds on acetaminophen-induced hepatotoxicity in mice. Toxicol. Appl. Pharmacol. 1996, 136, 146–154. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; McCullough, S.S.; Knight, T.R.; Jaeschke, H.; Hinson, J.A. Acetaminophen toxicity in mice lacking NADPH oxidase activity: Role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic. Res. 2003, 37, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Sun, J.; Zhang, X.; Melzig, M.F. Necrosis factor-alpha (TNF-alpha) response in human hepatoma HepG2 cells treated with hepatotoxic agents. Pharmazie 2014, 69, 379–384. [Google Scholar] [PubMed]
- Roberts, D.W.; Bucci, T.J.; Benson, R.W.; Warbritton, A.R.; McRae, T.A.; Pumford, N.R.; Hinson, J.A. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am. J. Pathol. 1991, 138, 359–371. [Google Scholar] [PubMed]
- Bajt, M.L.; Knight, T.R.; Lemasters, J.J.; Jaeschke, H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: Protection by N-acetyl cysteine. Toxicol. Sci. 2004, 80, 343–349. [Google Scholar] [CrossRef]
- Terneus, M.V.; Kiningham, K.K.; Carpenter, A.B.; Sullivan, S.B.; Valentovic, M.A. Comparison of S-Adenosyl-L-methionine and N-acetylcysteine protective effects on acetaminophen hepatic toxicity. J. Pharmacol. Exp. Ther. 2007, 320, 99–107. [Google Scholar] [CrossRef]
- Mullins, M.E.; Schmidt, R.U.; Jang, T.B. What is the rate of adverse events with intravenous versus oral N-acetylcysteine in pediatric patients? Ann. Emerg. Med. 2004, 44, 548–549. [Google Scholar] [CrossRef]
- Schmidt, L.E.; Dalhoff, K. Risk factors in the development of adverse reactions to N-acetylcysteine in patients with paracetamol poisoning. Br. J. Clin. Pharmacol. 2001, 51, 87–91. [Google Scholar] [PubMed]
- Zhang, A.; Sun, H.; Wang, X. Recent advances in natural products from plants for treatment of liver diseases. Eur. J. Med. Chem. 2013, 63, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.B.; Chai, D.W.; Jin, X.J.; Bi, Y.R.; Yao, X.J.; Wu, W.S.; Zhu, Y. Triterpenes and neolignans from the roots of Nannoglottis carpesioides. Phytochemistry 2011, 72, 1804–1813. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, M.; Zhong, R.F.; Liao, X.M.; Deng, L.L.; Xu, G.B.; He, X.; Li, J.; Li, Y.J.; Liu, T.; et al. Discovery and structure-activity relationship of auriculatone: A potent hepatoprotective agent against acetaminophen-induced liver injury. Bioorg. Med. Chem. Lett. 2017, 27, 3636–3642. [Google Scholar] [PubMed]
- Guo, Q.; Shen, Z.; Yu, H.; Lu, G.; Yu, Y.; Liu, X.; Zheng, P. Carnosic acid protects against acetaminophen-induced hepatotoxicity by potentiating Nrf2-mediated antioxidant capacity in mice. Korean J. Physiol Pharmacol 2016, 20, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Guan, C.; Sun, X.; Zhao, Z.; Li, J.; Fu, X.; Qiu, Y.; Huang, M.; Jin, J.; Huang, Z. Tanshinone IIA protects against acetaminophen-induced hepatotoxicity via activating the Nrf2 pathway. Phytomedicine 2016, 23, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.D.; Pumford, N.R.; Khairallah, E.A.; Boekelheide, K.; Pohl, L.R.; Amouzadeh, H.R.; Hinson, J.A. Selective protein covalent binding and target organ toxicity. Toxicol. Appl. Pharmacol. 1997, 143, 1–12. [Google Scholar] [CrossRef]
- Qiu, Y.; Benet, L.Z.; Burlingame, A.L. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J. Biol. Chem. 1998, 273, 17940–17953. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef]
- Moore, M.; Thor, H.; Moore, G.; Nelson, S.; Moldeus, P.; Orrenius, S. The toxicity of acetaminophen and N-acetyl-p-benzoquinone imine in isolated hepatocytes is associated with thiol depletion and increased cytosolic Ca2+. J. Biol. Chem. 1985, 260, 13035–13040. [Google Scholar]
- Burcham, P.C.; Harman, A.W. Acetaminophen toxicity results in site-specific mitochondrial damage in isolated mouse hepatocytes. J. Biol. Chem. 1991, 266, 5049–5054. [Google Scholar] [PubMed]
- Holt, M.P.; Ju, C. Mechanisms of drug-induced liver injury. AAPS J. 2006, 8, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Blazka, M.E.; Wilmer, J.L.; Holladay, S.D.; Wilson, R.E.; Luster, M.I. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 1995, 133, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Bourdi, M.; Masubuchi, Y.; Reilly, T.P.; Amouzadeh, H.R.; Martin, J.L.; George, J.W.; Shah, A.G.; Pohl, L.R. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: Role of inducible nitric oxide synthase. Hepatology 2002, 35, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Wang, M.; Chen, C.; Zhang, X.; Melzig, M.F. Hepatoprotective effect of isoquercitrin against acetaminophen-induced liver injury. Life Sci. 2016, 152, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, F.; Wang, K.; Liu, G.; Yang, M.; Luan, Y.; Zhao, Z. Protective effect of allyl methyl disulfide on acetaminophen-induced hepatotoxicity in mice. Chem. Biol. Interact. 2016, 249, 71–77. [Google Scholar] [CrossRef]
- Potter, W.Z.; Davis, D.C.; Mitchell, J.R.; Jollow, D.J.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. 3. Cytochrome P-450-mediated covalent binding in vitro. J. Pharmacol. Exp. Ther. 1973, 187, 203–210. [Google Scholar]
- Jollow, D.J.; Mitchell, J.R.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [Google Scholar]
- Jollow, D.J.; Thorgeirsson, S.S.; Potter, W.Z.; Hashimoto, M.; Mitchell, J.R. Acetaminophen-induced hepatic necrosis. VI. Metabolic disposition of toxic and nontoxic doses of acetaminophen. Pharmacology 1974, 12, 251–271. [Google Scholar] [CrossRef]
- Jaeschke, H.; Knight, T.R.; Bajt, M.L. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol. Lett. 2003, 144, 279–288. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Malondialdehyde as biomarker of oxidative damage to lipids caused by smoking. Clin. Chim. Acta 2007, 380, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Moon, K.H.; Olsson, N.U.; Salem, N. Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J. Hepatol. 2008, 49, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, I.; Saha, T. Effect of garlic on lipid peroxidation and antioxidation enzymes in DMBA-induced skin carcinoma. Nutrition 2009, 25, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. 4-Hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Xie, W.; Jiang, Z.; Wang, J.; Zhang, X.; Melzig, M.F. Protective effect of hyperoside against acetaminophen (APAP) induced liver injury through enhancement of APAP clearance. Chem. Biol. Interact. 2016, 246, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Mbimba, T.; Awale, P.; Bhatia, D.; Geldenhuys, W.J.; Darvesh, A.S.; Carroll, R.T.; Bishayee, A. Alteration of hepatic proinflammatory cytokines is involved in the resveratrol-mediated chemoprevention of chemically-induced hepatocarcinogenesis. Curr. Pharm. Biotechnol. 2012, 13, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.B.; Kurten, R.C.; McCullough, S.S.; Brock, R.W.; Hinson, J.A. Mechanisms of acetaminophen-induced hepatotoxicity: Role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J. Pharmacol. Exp. Ther. 2005, 312, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Laine, J.E.; Auriola, S.; Pasanen, M.; Juvonen, R.O. Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica 2009, 39, 11–21. [Google Scholar] [CrossRef]
- Fan, X.; Jiang, Y.; Wang, Y.; Tan, H.; Zeng, H.; Wang, Y.; Chen, P.; Qu, A.; Gonzalez, F.J.; Huang, M.; et al. Wuzhi tablet (Schisandra Sphenanthera extract) protects against acetaminophen-induced hepatotoxicity by inhibition of CYP-mediated bioactivation and regulation of NRF2-ARE and p53/p21 pathways. Drug Metab. Dispos. 2014, 42, 1982–1990. [Google Scholar] [CrossRef]
- Graham, N.M.; Burrell, C.J.; Douglas, R.M.; Debelle, P.; Davies, L. Adverse effects of aspirin, acetaminophen, and ibuprofen on immune function, viral shedding, and clinical status in rhinovirus-infected volunteers. J. Infect. Dis. 1990, 162, 1277–1282. [Google Scholar] [CrossRef]
- McGregor, A.H.; More, L.J.; Simpson, K.J.; Harrison, D.J. Liver death and regeneration in paracetamol toxicity. Hum. Exp. Toxicol. 2003, 22, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Subramanya, S.; Venkataraman, B.; Meeran, M.; Goyal, S.; Patil, C.; Ojha, S. Therapeutic Potential of Plants and Plant Derived Phytochemicals against Acetaminophen-Induced Liver Injury. Int. J. Mol. Sci. 2018, 19, 3776. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fan, X.; Wang, Y.; Tan, H.; Chen, P.; Zeng, H.; Huang, M.; Bi, H. Hepato-protective effects of six schisandra lignans on acetaminophen-induced liver injury are partially associated with the inhibition of CYP-mediated bioactivation. Chem. Biol. Interact. 2015, 231, 83–89. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Bi, H.C.; Xie, Z.Y.; Zuo, Z.; Li, J.K.; Li, X.; Zhao, L.Z.; Chen, X.; Huang, M. Rapid determination of six metabolites from multiple cytochrome P450 probe substrates in human liver microsome by liquid chromatography/mass spectrometry: Application to high-throughput inhibition screening of terpenoids. Rapid Commun. Mass Spectrom. 2007, 21, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fan, X.; Wang, Y.; Chen, P.; Zeng, H.; Tan, H.; Gonzalez, F.J.; Huang, M.; Bi, H. Schisandrol B protects against acetaminophen-induced hepatotoxicity by inhibition of CYP-mediated bioactivation and regulation of liver regeneration. Toxicol. Sci. 2015, 143, 107–115. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, L.; Guan, H.; Li, R.; Liao, X.; Zhao, F.; Wang, M.; Li, J.; Xu, G.; He, X.; Zhang, J.; et al. Auriculatone Sulfate Effectively Protects Mice Against Acetaminophen-Induced Liver Injury. Molecules 2019, 24, 3642. https://doi.org/10.3390/molecules24203642
Lin L, Guan H, Li R, Liao X, Zhao F, Wang M, Li J, Xu G, He X, Zhang J, et al. Auriculatone Sulfate Effectively Protects Mice Against Acetaminophen-Induced Liver Injury. Molecules. 2019; 24(20):3642. https://doi.org/10.3390/molecules24203642
Chicago/Turabian StyleLin, Liangcai, Huanyu Guan, Rui Li, Xiangming Liao, Feifei Zhao, Min Wang, Jing Li, Guobo Xu, Xun He, Jinjuan Zhang, and et al. 2019. "Auriculatone Sulfate Effectively Protects Mice Against Acetaminophen-Induced Liver Injury" Molecules 24, no. 20: 3642. https://doi.org/10.3390/molecules24203642
APA StyleLin, L., Guan, H., Li, R., Liao, X., Zhao, F., Wang, M., Li, J., Xu, G., He, X., Zhang, J., Li, Y., Wang, Y., Zhou, M., & Liao, S. (2019). Auriculatone Sulfate Effectively Protects Mice Against Acetaminophen-Induced Liver Injury. Molecules, 24(20), 3642. https://doi.org/10.3390/molecules24203642