Abstract
7-Oxo-1,2,4-benzotriazines (benzo[1,2,4]triazin-7-ones) are reversible thioredoxin reductase inhibitors that exhibit very strong correlations to pleurotin. In this article, we provide the first synthesis of fluorinated derivatives. Fluorination using Selectfluor of benzo[1,2,4]triazin-7-ones occurs regioselectively and in high yield at the enamine-activated position. This electron N-lone pair activation overrides the activation/deactivation effects of some other substituents. The reaction time was significantly reduced with the use of microwave irradiation at 120 °C and 7 bar. The cytotoxicity and cyclic voltammetry measurements for 8-fluoro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (2) are presented and compared with its synthetic precursor, 1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (1a).
1. Introduction
The remarkable expansion in the use of fluorinated chemicals has attracted the attention of organic, agricultural, medicinal, and materials scientists. Uniquely, the incorporation of fluorine atoms into organic molecules introduces polar hydrophobicity. This has been significantly utilized in medicinal chemistry as a means to increase efficacy [1,2], with some 30% of blockbuster drugs containing fluorine atoms [3]. Selectfluor is perhaps the most versatile, stable, cheap, and effective commercial electrophilic fluorinating reagent [4], and in this article, we demonstrate its use in the selective fluorination of some 7-oxo-1,2,4-benzotriazines (benzo[1,2,4]triazin-7-ones) (Figure 1). Selectfluor is also a strong oxidant, and mediator or catalyst of several “fluorine-free” transformations [5]. Selectfluor is known to directly substitute fluorine into activated positions on anilines, benzamides, and phenols [6,7]; however, to date, examples of fluorinated quinones are few and low-yielding (<20%) [8,9]. Typically, in such cases, the fluorine was introduced via a halogen exchange with nucleophilic fluoride reagents, such as KF [10,11,12]. Interestingly, 2-hydroxymethylindole can undergo simultaneous electrophilic aromatic substitution with fluorine at the activated enamine C-3 position with oxidation of the alcohol to the aldehyde by using Selectfluor [13].
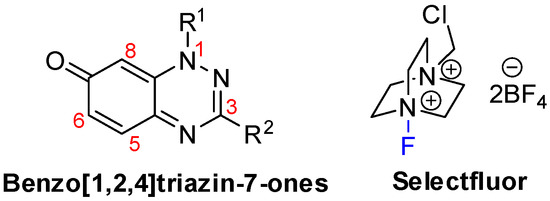
Figure 1.
Key chemical structures.
Benzo[1,2,4]triazin-7-ones are reversible thioredoxin reductase inhibitors with significant anti-cancer activities, which strongly correlate to pleurotin [14]. Derivatives also exhibit anti-Alzheimer’s disease activity [15], while the cytotoxicity is far greater than that of the derived Blatter-type (benzotriazin-4-yl) radicals [16]. Herein, we present the first fluorination of several benzo[1,2,4]triazin-7-one derivatives (that were available to us). Additionally, the cytotoxicity and cyclic voltammetry of 8-fluoro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (2) is compared to that of the non-fluorinated scaffold 1a.
2. Results and Discussion
2.1. Fluorinations
2.1.1. Optimizing the Fluorination and Confirming Selectivity
Selectfluor was found to fluorinate at the 8-position of the parent 1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (1a), presumably due to enamine conjugation with the N-1 atom (Scheme 1). Initially, low yields of 8-fluoro derivative 2 were obtained, when using less than 2 equivalents of Selectfluor at room temperature and at reflux in acetonitrile, with recovery of 1a (Table 1). The complete conversion of 1a to 2 was observed by TLC after 1 h when increasing the reaction temperature to 120 °C, which was facilitated by using a sealed Ace pressure tube with 2 isolated in 94% yield after column chromatography. The reaction time was reduced to 20 min when performing the transformation in a microwave reactor (150 W, 7 bar), with 2 isolated at 97% yield. Applying the latter optimized conditions on 8-chloro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (1b), a halogen exchange proceeds efficiently yielding 2 in 96% yield, although after a longer reaction time of 1 h, as monitored by TLC. Jiang et al. [13] reported a similar aromatic substitution of bromine by fluorine at the C-3 position of indole, using Selectfluor. In these cases, the defluorinated Selectfluor by-product (1-chloromethyl-1,4-diazobicyclo[2.2.2]octane) may assist with the elimination of the halogen atom.
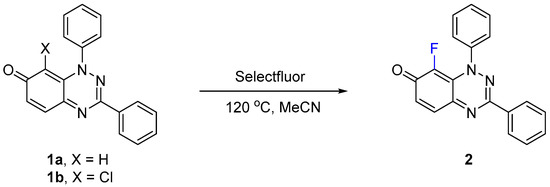
Scheme 1.
Synthesis of 8-fluoro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (2).

Table 1.
Optimization of reaction conditions a.
The position of the fluorination on 2 is discernible by comparing its 1H-NMR spectrum with that of substrate 1a (Figure 2) with the disappearance of the H-8 signal of 1a at 6.10 ppm being clearly visible, and 1H-19F (meta) coupling for H-6 of J = 7.2 Hz. The 13C-NMR spectrum of 2 gives the expected 13C-19F couplings, including 1JCF of 247.5 Hz for C-8, and 2JCF of 15.7 Hz for C=O.
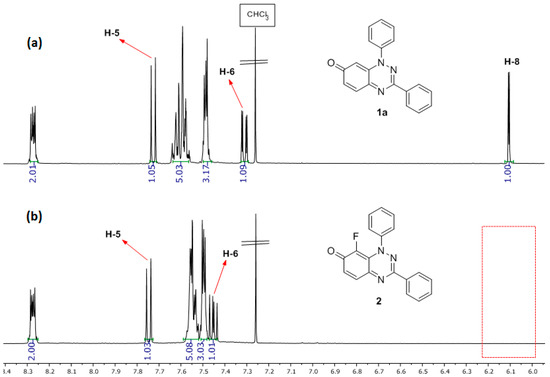
Figure 2.
1H-NMR spectrum: (a) of 1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (1a) and (b) of 8-fluoro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (2).
2.1.2. Fluorination of C-6-Substituted Benzo[1,2,4]triazin-7-ones
The optimized conditions in Table 1 of treatment with Selectfluor (2 equiv, MW at 120 °C, 20 min) were applied to benzo[1,2,4]triazin-7-ones 3a and 3b containing phenyl and benzylthio substituents at C-6 (Scheme 2). C-8 Fluorinated derivatives 4a and 4b were isolated in 59% and 86% yield, respectively, with 4a accompanied by significant recovery of 3a (31%). Interestingly, fluorination at C-8 remained the only path for substitution despite activation at C-5 by the benzylthio substituent of 3b.

Scheme 2.
Synthesis of 8-fluoro-1,3,6-triphenylbenzo[e][1,2,4]triazin-7(1H)-one (4a) and 6-(benzylthio)-8-fluoro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (4b).
2.1.3. Fluorination of 1,2,5-Thiadiazolo-Fused Benzotriazinones
The preparation of 1,2,5-thiadiazolo-fused benzotriazinones 5a and 5b has been recently reported by using the reaction of S4N4 with 1a and the 3-trifluoromethyl analogue [17]. The reaction of thiadiazoles 5a and 5b with Selectfluor using the optimized conditions in Table 1 gave the fluorinated adducts 6a (93%) and 6b (89%) in excellent yields, where the reaction occurred at the only available CH of the benzotriazinone scaffold (Scheme 3). The regioselective formation of 6b in high yield demonstrated that the electrophilic fluorination remained facile, despite the strongly deactivating inductive effect of the CF3 substituent of 5b.

Scheme 3.
Synthesis of 5-fluoro-6,8-diphenyl[1,2,5]thiadiazolo[3′,4′:5,6]benzo[1,2-e][1,2,4]triazin-4(6H)-one (6a) and 5-fluoro-6-phenyl-8-(trifluoromethyl)[1,2,5]thiadiazolo[3′,4′:5,6]benzo[1,2-e][1,2,4]triazin-4(6H)-one (6b).
The NMR data for fluorinated adducts revealed a “through space” coupling with the ortho-CH on the N-1-Ph, supported by distance measurements using Spartan (Figure S16).
2.2. Cytotoxicity against MCF-7 using the MTT Assay
Having the fluorinated benzotriazin-7-one 2, we investigated the effect of the fluorine substituent on cytotoxicity. The breast cancer cell line MCF-7, and the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay were employed by using the conditions previously described on the parent compound 1a [14]. The fluorinated derivative 2 proved to be approximately five times more cytotoxic than 1a towards the MCF-7 cell line (Table 2).
Table 2.
Cytotoxicity evaluation using the MTT colorimetric assay.
2.3. Cyclic Voltammetry
Cyclic voltammetry studies were carried out on compounds 1a and 2 (Figure 3). Redox response experiments showed that both 1a and 2 undergo two characteristic quasi-reversible one-electron redox processes corresponding to the 0/−1 redox transition (I and I′) and the −1/−2 redox transition (II and II′). The fluorinated derivative 2 produced a similar redox response to 1a, with surprisingly similar formal potentials (E0′) (Table 3), despite the presence of the electronegative fluorine at the C-8 position. This indicates that factors other than bioreduction may account for the differences in cytotoxicity between 1a and 2 against the MCF-7 cell line.
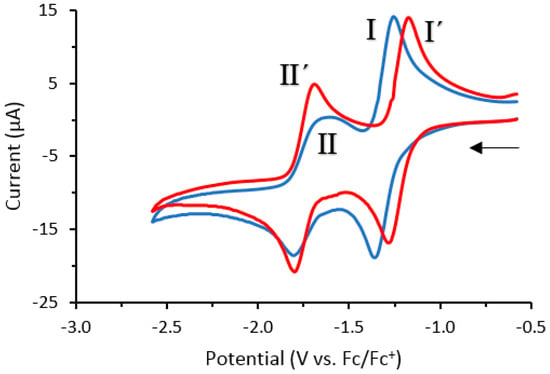
Figure 3.
Cyclic voltammograms of 1a (blue line) and 2 (red line) recorded in CH2Cl2 (0.1 M n-Bu4NPF6) at a glassy carbon electrode (scan rate: 0.1 V·s−1). Arrow indicates the direction of the scan.
Table 3.
Formal potentials (E0′) (±0.010 V) calculated as (Epc + Epa)/2 from cyclic voltammograms recorded at 100 mV·s−1.
3. Experimental Section
3.1. General Materials and Methods
1,3-Diphenylbenzo[e][1,2,4]triazin-7(1H)-one (1a) [18,19] and 1,3,6-triphenylbenzo[e][1,2,4]-triazin-7(1H)-one (3a) [18] were prepared according to literature procedures. 8-Chloro-1,3-diphenyl-benzo[e][1,2,4]triazin-7(1H)-one (1b) [18], 6-(benzylthio)-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (3b) [18], 6,8-diphenyl[1,2,5]thiadiazolo[3′,4′:5,6]benzo[1,2-e][1,2,4]triazin-4(6H)-one (5a) [17] and 6-phenyl-8-(trifluoromethyl)-[1,2,5]thiadiazolo[3′,4′:5,6]benzo[1,2-e][1,2,4]triazin-4(6H)-one (5b) [17] were provided by Koutentis Research Laboratory (University of Cyprus), and used as received. All other solvents and reagents were used as received from Sigma-Aldrich (Gillingham, Dorset, SP8 4XT, UK). Acetonitrile (MeCN, Sigma-Aldrich, ≥99.9%) was freshly distilled over 3 Å molecular sieves and then over CaH2 (Sigma-Aldrich, 95%). Thin-layer chromatography (TLC) was performed on Merck TLC Silica gel 60 F254 plates using a UV lamp for visualization. The technique of dry flash chromatography [20] was used throughout for all non-TLC-scale chromatographic separations using silica gel 60 (<0.063 mm). Microwave irradiation was conducted in a CEM Discover SP Microwave Reactor using 150 watts of microwave power. The pressure was controlled by a load cell connected to the vessel via the cap on top of the sealed pressure vessel. The temperature of the content of the vessel was monitored by an infrared temperature control system, which uses a non-contact, infrared sensor mounted under the vessel. All reactions were performed in Pyrex pressure vessels (capacity 10 mL) sealed with silicone caps. All reaction mixtures were stirred with a Teflon-coated, magnetic stirring bar in the vessel. A ramp temperature of 2 min was set for each experiment. Ultraviolet spectra were obtained on a Varian (Cary 100) UV-Vis spectrometer, where inf = inflection. Infrared spectra were recorded using a PerkinElmer Spec 1 with attenuated total reflection (ATR) attached. NMR spectra were recorded using Varian 500 MHz, and an Agilent DD2 600 MHz instrument was used to be obtain the 13C-NMR spectrum of compound 6b. The chemical shifts were recorded in ppm relative to SiMe4. 13C-NMR data were collected at 125 MHz and 150 MHz for compound 6b with complete proton decoupling. NMR assignments were supported by distortionless enhancement by polarization transfer (DEPT). 19F-NMR spectra were obtained at 470 MHz. Deuterated solvents were used for the homonuclear lock, and the signals were referenced to the deuterated solvent peaks. High resolution mass spectra (HRMS) was carried out using an ESI time-of light mass spectrometer (TOFMS) in positive or negative mode, using a Waters LCT Mass Spectrometry instrument. The precision of all accurate mass measurements was better than 5 ppm. Melting points were determined by using differential scanning calorimetry (DSC), which was performed on a Mettler Toledo Simultaneous Thermal Analyzer using standard aluminium pans.
3.2. Synthetic Procedures and Characterization

Method A: Selectfluor (238.2 mg, 0.8 mmol) was added to the solution of 1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (1a) (120 mg, 0.4 mmol) in dry MeCN (2 mL) in an Ace pressure tube (15 mL). The reaction mixture was immersed in a preheated oil bath at ca. 120 °C and left to stir for 1 h, under monitoring by TLC. The reaction mixture was cooled to ca. 20 °C, diluted with EtOAc (30 mL), and washed with brine (3 × 30 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered, and evaporated to dryness. The residue was purified by column chromatography using EtOAc and petroleum ether to give 8-fluoro-1,3-diphenylbenzo[e][1,2,4]-triazin-7(1H)-one (2) (119.3 mg, 94%) as blue fine needles; m.p. (DSC) onset 202.3 °C, peak max 205.2 °C (from cyclohexane/CH2Cl2, 90:10); Rf 0.42 (EtOAc/Petroleum ether, 60:40); λmax(CH2Cl2)/nm 300 (log ε 5.12), 350 inf (4.69), 360 inf (4.65), 575 (4.29), 615 inf (4.25), 680 inf (3.84); νmax (neat, cm−1) 3057, 1608 (C=O), 1545, 1491, 1436, 1377, 1344, 1211, 1160, 955; 1H-NMR (500 MHz, CDCl3) δH 7.45 (1H, dd, J = 7.2 Hz, J = 9.8 Hz, H-6), 7.48–7.51 (3H, m), 7.52–7.57 (5H, m), 7.75 (1H, d, J = 9.8 Hz, H-5), 8.26–8.29 (2H, m); 13C-NMR (125 MHz, CDCl3) δC 121.2 (d, J = 3.8 Hz, C), 124.8 (d, J = 4.2 Hz, CH), 126.8, 128.9 (×2), 129.6, 130.7, 130.9 (all CH), 133.4 (C), 136.1 (d, 1JCF = 247.5 Hz, CF), 141.3 (CH), 143.6 (d, J = 2.7 Hz, C), 150.9, 153.9 (both C), 172.7 (d, J = 15.7 Hz, C=O); 19F-NMR (470 MHz, CDCl3) δF −145.8 (1F, s); HRMS (ESI) m/z [M + H]+, C19H13FN3O calcd. 318.1043, observed 318.1054.
Method B (General Procedure): Selectfluor (70.8 mg, 0.2 mmol) was added to the solution of the benzo[e][1,2,4]triazin-7(1H)-one 1a–1b, 3a–3b, 5a–5b (0.1 mmol) in dry MeCN (0.5 mL). The reaction mixture was stirred under microwave irradiation (150 W, ca. 120 °C, 7 bar) for 20 min (1 h in the case of 1b). EtOAc (10 mL) was added and the mixture was extracted with brine (3 × 10 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered, and evaporated to dryness. The residue was dissolved in CH2Cl2, poured onto a short pad of silica, and washed with EtOAc and petroleum ether to give the desired products 2, 4a–4b, and 6a–6b.

8-Fluoro-1,3,6-triphenylbenzo[e][1,2,4]triazin-7(1H)-one (4a) (23.4 mg, 59%) as blue fine needles; m.p. (DSC) onset 252.0 °C, peak max 252.8 °C (from cyclohexane/CH2Cl2, 90:10); Rf 0.50 (EtOAc/petroleum ether, 30:70); λmax(CH2Cl2)/nm 260 (log ε 5.03), 330 (4.98), 410 inf (4.13), 595 (4.06), 670 inf (3.76); νmax (neat, cm−1) 3068, 1605 (C=O), 1542, 1491, 1424, 1312, 1281, 1210, 1160, 1108, 962; 1H-NMR (500 MHz, CDCl3) δH 7.47–7.62 (11H, m), 7.79–7.82 (2H, m), 7.88 (1H, s, H-5), 8.29–8.31 (2H, m); 13C-NMR (125 MHz, CDCl3) δC 120.5 (d, J = 4.2 Hz, C), 125.1 (d, J = 4.3 Hz, 5-CH), 126.9, 127.8, 128.6, 129.0 (×2), 129.6, 129.7, 130.2, 130.8 (all CH), 134.0 (C), 134.9 (d, J = 2.3 Hz, C), 136.3 (d, 1JCF = 244.6 Hz, CF), 143.8 (C), 150.6 (d, J = 3.2 Hz, C), 151.2, 153.4 (both C), 171.9 (d, J = 15.7 Hz, C=O); 19F-NMR (470 MHz, CDCl3) δF −145.0 (1F, s); HRMS (ESI) m/z [M + H]+, C25H17FN3O calcd. 394.1356, observed 394.1349. Further elution with EtOAc and petroleum ether (30:70), gave the recovered starting material 3a (11.6 mg, 31%).

6-(Benzylthio)-8-fluoro-1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one (4b) (38.0 mg, 86%) as olive green fine needles; m.p. (DSC) onset 217.5 °C, peak max 219.2 °C (from cyclohexane/CH2Cl2, 90:10); Rf 0.43 (EtOAc/petroleum ether, 30:70); λmax(CH2Cl2)/nm 260 (log ε 4.95), 325 (4.82), 410 inf (4.27), 425 (4.31), 575 (3.95), 635 inf (3.72); νmax (neat, cm−1) 3061 (Ar CH), 1610 (C=O), 1574, 1523, 1491, 1430, 1311, 1281, 1214, 1112, 1072, 974; 1H-NMR (500 MHz, CDCl3) δH 4.29 (2H, s, CH2), 7.33–7.40 (3H, m), 7.46–7.52 (5H, m), 7.53–7.60 (6H, m), 8.26–8.32 (2H, m); 13C-NMR (125 MHz, CDCl3) δC 36.2 (CH2), 118.7 (CH), 120.8 (d, J = 3.1 Hz, C), 125.1 (d, J = 4.2 Hz, 5-CH), 127.0, 128.1, 128.9, 129.0, 129.1 (×2), 129.7, 130.8 (all CH), 133.6, 134.1 (both C), 134.6 (d, 1JCF = 245.0 Hz, CF), 143.9 (d, J = 2.9 Hz, C), 150.5, 151.6 (both C), 158.4 (d, J = 4.6 Hz, C), 168.7 (d, J = 16.4 Hz, C=O); 19F-NMR (470 MHz, CDCl3) δF −147.5 (1F, s); HRMS (ESI) m/z [M + H]+, C26H19FN3OS calcd. 440.1233, observed 440.1220.

5-Fluoro-6,8-diphenyl[1,2,5]thiadiazolo[3′,4′:5,6]benzo[1,2-e][1,2,4]triazin-4(6H)-one (6a) (35.1 mg, 93%) as brown fine needles; m.p. (DSC) onset 315.7 °C, peak max 316.7 °C (from cyclohexane/CH2Cl2, 90:10); Rf 0.38 (EtOAc/petroleum ether, 40:60); λmax(CH2Cl2)/nm 275 (log ε 4.94), 310 (5.09), 430 (4.74), 515 inf (3.95), 555 (4.04), 600 inf (3.97), 660 inf (3.54); νmax (neat, cm−1) 3068 (Ar CH), 1624 (C=O), 1573, 1534, 1464, 1434, 1345, 1238, 1198, 1161, 1061, 922; 1H-NMR (500 MHz, CDCl3) δH 7.50–7.58 (8H, m), 8.36–8.38 (2H, m); 13C-NMR (125 MHz, CDCl3) δC 124.7 (d, J = 4.6 Hz, CH), 125.0 (d, J = 5.3 Hz, C), 127.2, 129.1, 129.2, 129.9, 131.6 (all CH), 132.8 (C), 138.1 (d, 1JCF = 249.3 Hz, CF), 143.6 (d, J = 2.9 Hz, C), 147.8, 151.2, 151.6 (all C), 156.6 (d, J = 7.8 Hz, C), 165.1 (d, J = 18.4 Hz, C=O); 19F-NMR (470 MHz, CDCl3) δF −145.5 (1F, s); HRMS (ESI) m/z [M + H]+, C19H11FN5OS calcd. 376.0668, observed 376.0654.

5-Fluoro-6-phenyl-8-(trifluoromethyl)[1,2,5]thiadiazolo[3′,4′:5,6]benzo[1,2-e][1,2,4]triazin-4(6H)-one (6b) (32.5 mg, 89%) as purple fine needles; m.p. (DSC) onset 279.0 °C, peak max 281.5 °C (from cyclohexane/CH2Cl2, 90:10); Rf 0.59 (EtOAc/petroleum ether, 40:60); λmax(CH2Cl2)/nm 260 (log ε 4.93), 300 (4.96), 315 inf (4.90), 325 inf (4.76), 390 inf (4.52), 405 (4.53), 500 inf (4.02), 540 (4.09), 590 inf (3.97), 645 inf (3.57); νmax (neat, cm−1) 3072 (Ar CH), 1652 (C=O), 1587, 1567, 1487, 1414, 1387, 1294, 1204, 1131, 1068, 928; 1H-NMR (500 MHz, CDCl3) δH 7.49–7.53 (2H, m), 7.54–7.59 (3H, m); 13C-NMR (150 MHz, CDCl3) δC 119.0 (q, 1JCF = 272.4 Hz, F3C), 124.1 (d, J = 5.8 Hz, C), 124.4 (d, J = 4.5 Hz, CH), 129.5, 130.4 (both CH), 138.4 (d, 1JCF = 255.7 Hz, C-8), 142.6 (d, J = 2.7 Hz, C), 142.9 (q, J = 39.4 Hz, F3CC), 149.3, 150.8 (both C), 156.0 (d, J = 7.5 Hz, C), 165.9 (d, J = 19.5 Hz, C=O); 19F-NMR (470 MHz, CDCl3) δF −70.1 (3F, s, CF3), −141.3 (1F, s); HRMS (ESI) m/z [M + H]+, C14H6F4N5OS calcd. 368.0229, observed 368.0230.
3.3. Cell Culture and Cytotoxicity Evaluation
3.3.1. Materials and Cell Lines
MCF-7 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose (4.5 g/mL) and supplemented with 1% penicillin-streptomycin and 10% heat-inactivated foetal bovine serum (FBS). Cells grew as adherent cultures. Cell culture reagents were obtained from Sigma-Aldrich. Disposable sterile plasticware was obtained from Sarstedt (Numbrecht, Germany).
3.3.2. Cytotoxicity Measurements Using the MTT Assay
The MTT colorimetric assay was used to determine cell viability. MCF-7 cells were added to 96-well plates at a cell density of 1000 cells per well (200 µL per well) and allowed to adhere over 24 h. Compound solutions in DMSO were added after 24 h (1% v/v final concentration in the well). The control cells were exposed to the same concentration of the vehicle control alone (DMSO). All cells were incubated at 37 °C and 5% CO2 (humidified atmosphere) for 72 h. MTT (20 µL, 5 mg/mL solution) was added after 72 h and the cells were incubated for a further 3 h. The supernatant was then removed by using a multi-transfer pipette, and DMSO (100 µL) was added to dissolve the MTT formazan crystals. The absorbance was determined by using a plate reader at 550 nm with a reference at 690 nm. Cell viability is expressed as a percentage of the vehicle-only treated control (DMSO). Dose-response curves were analysed by non-linear regression analysis, and IC50 values were determined by using GraphPad Prism software, v 8.0 (GraphPad Inc., San Diego, CA, USA). The in vitro activity of the drugs towards all cell lines is expressed as IC50 (i.e., the concentration required for the reduction of the mean cell viability to 50%).
3.4. Electrochemistry
Cyclic voltammograms were recorded using a PalmSens3+ potentiostat. The concentrations of all studied compounds were 0.001 mol·L−1 in dry (over CaH2) HPLC grade CH2Cl2 (5.0 mL) containing n-Bu4NPF6 (0.1 M) as a supporting electrolyte. A three-electrode electrochemical cell was employed with glassy carbon, Pt wire, and Ag/AgCl (1 M NaCl) as the working, counter, and reference electrodes, respectively. The ferrocene/ferrocenium (Fc/Fc+) couple was used as an internal reference, and all redox couples are referenced against it (EFc/Fc+ 0.0 V). The scan rate was 0.1 V·s−1 and the temperature was 20 °C.
4. Conclusions
Selectfluor regioselectively fluorinates benzo[1,2,4]triazin-7-ones and derivatives in high yield. One of the fluorinated iminoquinones is shown to be cytotoxic, offering the potential for further investigation of fluorinated adducts as bioreductive antibiotics and anti-cancer agents.
Supplementary Materials
Supplementary materials are available online. Figures S1–S15: 1H, 13C and 19F-NMR of compounds 2, 4a–4b and 6a–6b, Figure S16: 1H-13C HSQC NMR and the optimized geometry of compound 6b, Figure S17: Viability of the MCF-7 cell line as determined by using the MTT assay for compounds 1a and 2.
Author Contributions
S.I.M. carried out all of the experimental work and data analysis. S.I.M. drafted the manuscript in consultation with P.A.K. and F.A. All authors approved the manuscript.
Funding
F.A. thanks the Irish Research Council (IRC) for a Government of Ireland Postdoctoral Fellowship for S.I.M.
Acknowledgments
The authors thank Seán Hennessy and Pau Farràs (of the NUI Galway) for use of the cyclic voltammetry equipment, and Daniele Lo Re and Georgia Zissimou (of the University of Cyprus) for preparing compounds 1b and 3b, and 5a–5b, respectively. We thank Stephen Rea (of the NUI Galway) for the MCF-7 cell line.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; DurÓn, S.G.; Burkart, M.D.; Vincent, S.P.; Wong, C.-H. Selectfluor: Mechanistic insight and applications. Angew. Chem. Int. Ed. 2005, 44, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Stavber, S. Recent Advances in the Application of SelectfluorTM F-TEDA-BF4 as a Versatile Mediator or Catalyst in Organic Synthesis. Molecules 2011, 6, 6432–6464. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Li, Y.; Gao, S.; Li, R.; Li, X.; Wang, B.; Yang, H. Amide-assisted radical strategy: Metal-free direct fluorination of arenes in aqueous media. Green Chem. 2017, 19, 3344–3349. [Google Scholar] [CrossRef]
- Heravi, M.R.P. Fluorination of activated aromatic systems with Selectfluor™ F-TEDA-BF4 in ionic liquids. J. Fluor. Chem. 2008, 129, 217–221. [Google Scholar] [CrossRef]
- Keinan, S.; Paquette, W.D.; Skoko, J.J.; Beratan, D.N.; Yang, W.; Shinde, S.; Johnston, P.A.; Lazo, J.S.; Wipf, P. Computational design, synthesis and biological evaluation of para-quinone-based inhibitors for redox regulation of the dual-specificity phosphatase Cdc25B. Org. Biomol. Chem. 2008, 6, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.-T.; Zhou, D.-C.; Mai, Y.-W.; Huo, L.; Yao, P.-F.; Huang, S.-L.; Wang, H.-G.; Huang, Z.-S.; Gu, L.-Q. Construction of the oxaphenalene skeletons of mansonone F derivatives through C-H bond functionalization and their evaluation for anti-proliferative activities. RSC Adv. 2017, 7, 20919–20928. [Google Scholar] [CrossRef]
- Cameron, D.W.; Feutrill, G.I.; Griffiths, P.G.; Richards, K.R. Synthesis of fluoronaphthoquinones: Halide displacement by naked fluoride. Aust. J. Chem. 1982, 35, 1509–1512. [Google Scholar] [CrossRef]
- Cameron, D.W.; Chalmers, P.J.; Feutrill, G.I. Regiochemistry of nucleophilic displacements in chloroquinones. Tetrahedron Lett. 1984, 25, 6031–6032. [Google Scholar] [CrossRef]
- Kim, B.G.; Chun, T.G.; Lee, H.-Y.; Snapper, M.L. A new structural class of S-adenosylhomocysteine hydrolase inhibitors. Bioorg. Med. Chem. 2009, 17, 6707–6714. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, F.; Yang, J.; Yu, P.; Yi, P.; Sun, Y.; Wang, Y. Fluorination-oxidation of 2-hydroxymethylindole using Selectfluor. Adv. Synth. Catal. 2017, 359, 853–858. [Google Scholar] [CrossRef]
- Sweeney, M.; Coyle, R.; Kavanagh, P.; Berezin, A.A.; Lo Re, D.; Zissimou, G.A.; Koutentis, P.A.; Carty, M.P.; Aldabbagh, F. Discovery of anti-cancer for benzo[1,2,4]triazin-7-ones: Very strong correlation to pleurotin and thioredoxin reductase inhibition. Bioorg. Med. Chem. 2016, 24, 3565–3570. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Berezin, A.A.; Lo Re, D.; Loizou, G.; Demetriades, M.; De Stradis, A.; Campagna, F.; Koutentis, P.A.; Carotti, A. Design, synthesis and biological evaluation of benzo[e][1,2,4]triazin-7(1H)-one and [1,2,4]-triazino[5,6,1-jk]carbazol-6-one derivatives as dual inhibitors of beta-amyloid aggregation and acetyl/butyryl cholinesterase. Eur. J. Med. Chem. 2012, 58, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Keane, L.-A.J.; Mirallai, S.I.; Sweeney, M.; Carty, M.P.; Zissimou, G.A.; Berezin, A.A.; Koutentis, P.A.; Aldabbagh, F. Anti-cancer activity of phenyl and pyrid-2-yl 1,3-substituted benzo[1,2,4]triazin-7-ones and stable free radical precursors. Molecules 2018, 23, 574. [Google Scholar] [CrossRef] [PubMed]
- Zissimou, G.A.; Kourtellaris, A.; Manoli, M.; Koutentis, P.A. Redox active quinoidal 1,2,4-Benzotriazines. J. Org. Chem. 2018, 83, 9391–9402. [Google Scholar] [CrossRef] [PubMed]
- Koutentis, P.A.; Lo Re, D. Catalytic oxidation of N-phenylamidrazones to 1,3-diphenyl-1,4-dihydro-1,2,4-benzotriazin-4-yls: An improved synthesis of Blatter’s radical. Synthesis 2010, 2075–2079. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Krassos, H.; Lo Re, D. 1,3-Diphenylbenzo[e][1,2,4]-7(1H)-one: Selected chemistry at the C-6, C-7 and C-8 positions. Org. Biomol. Chem. 2011, 9, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Harwood, L.M. Dry-column flash chromatography. Aldrichim. Acta 1985, 18, 25. [Google Scholar]
Sample Availability: Samples of the compounds 2, 4a–4b, 6a–6b are available from the authors. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).