
Ferrocene-Based Compounds with Antimalaria/Anticancer Activity



Abstract
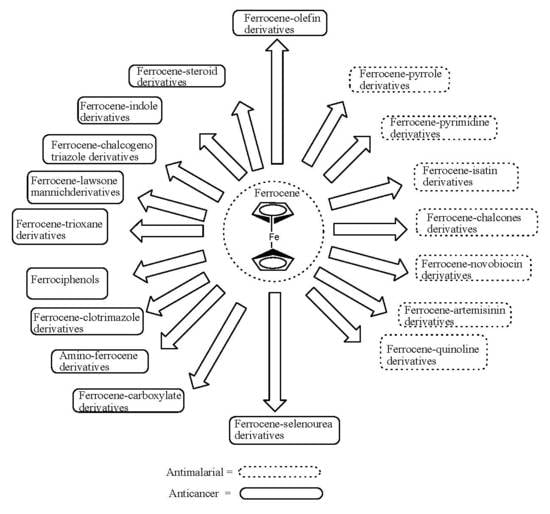
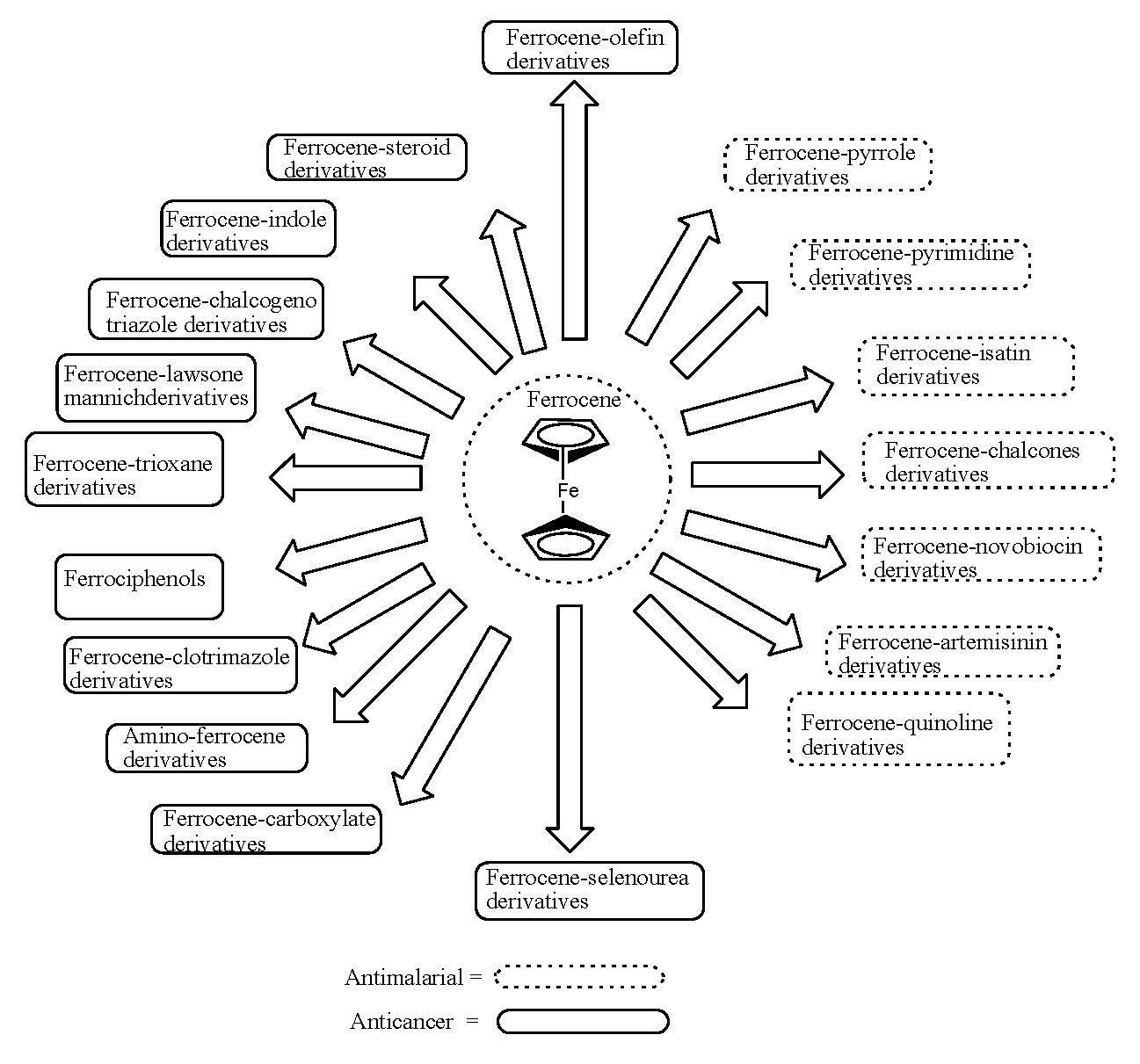
1. Introduction
2. Ferrocene (Biological Activity)
3. Ferrocene-Based Compounds with Antimalaria Activity
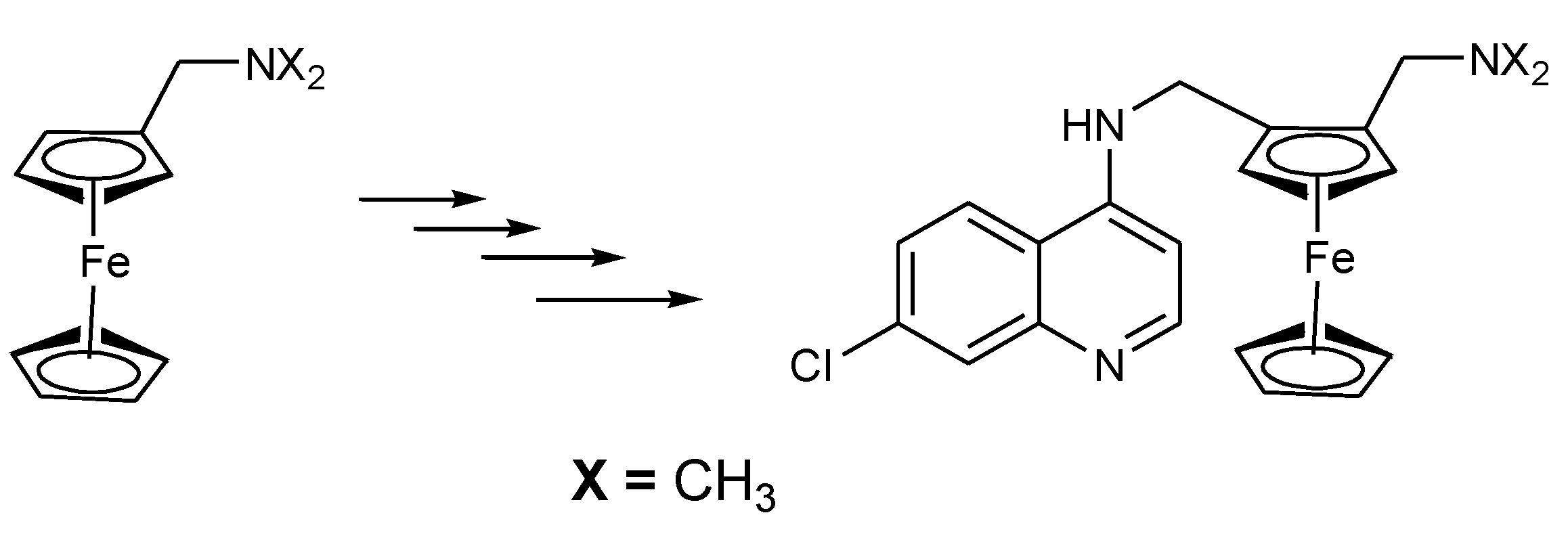
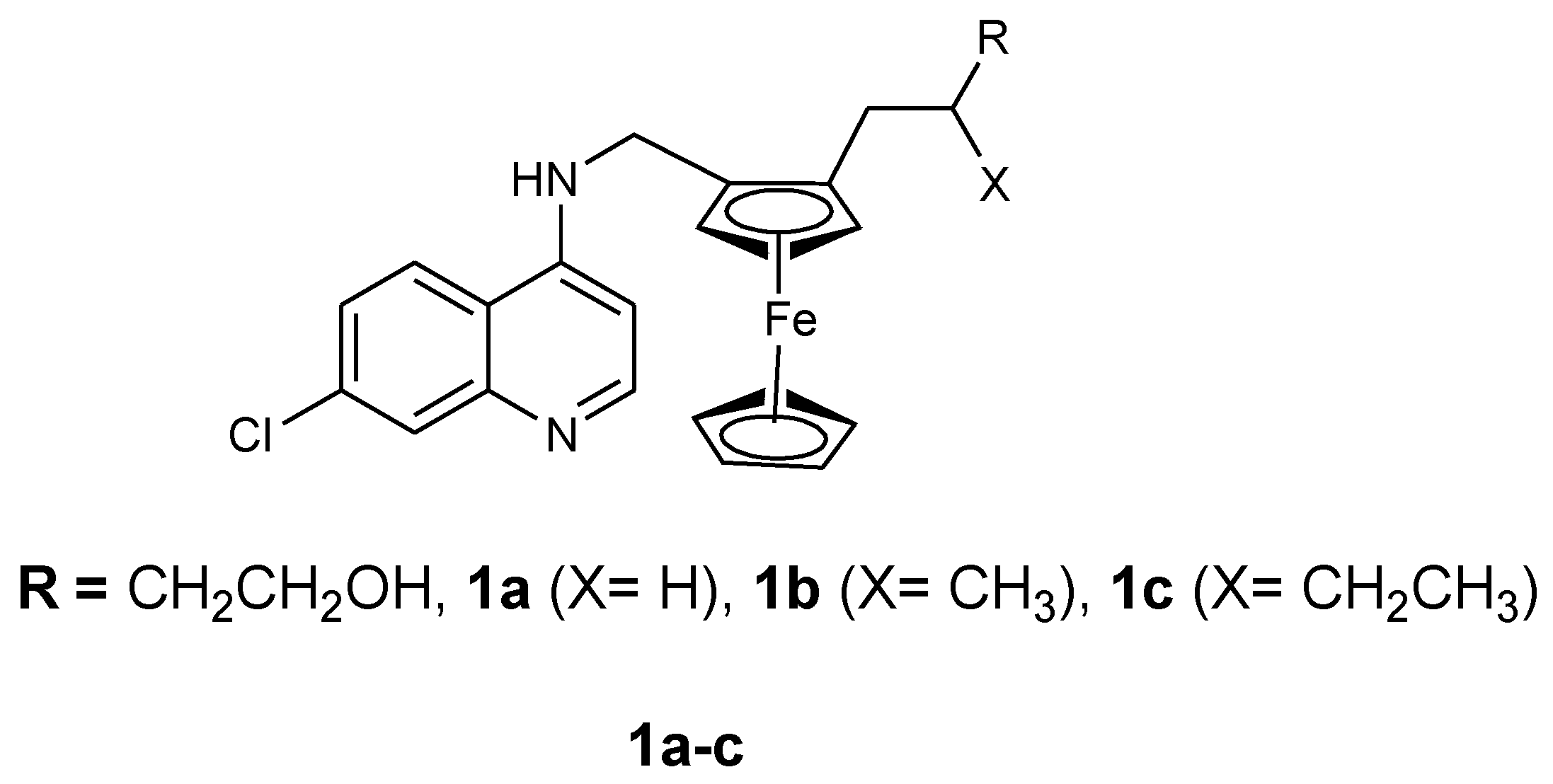
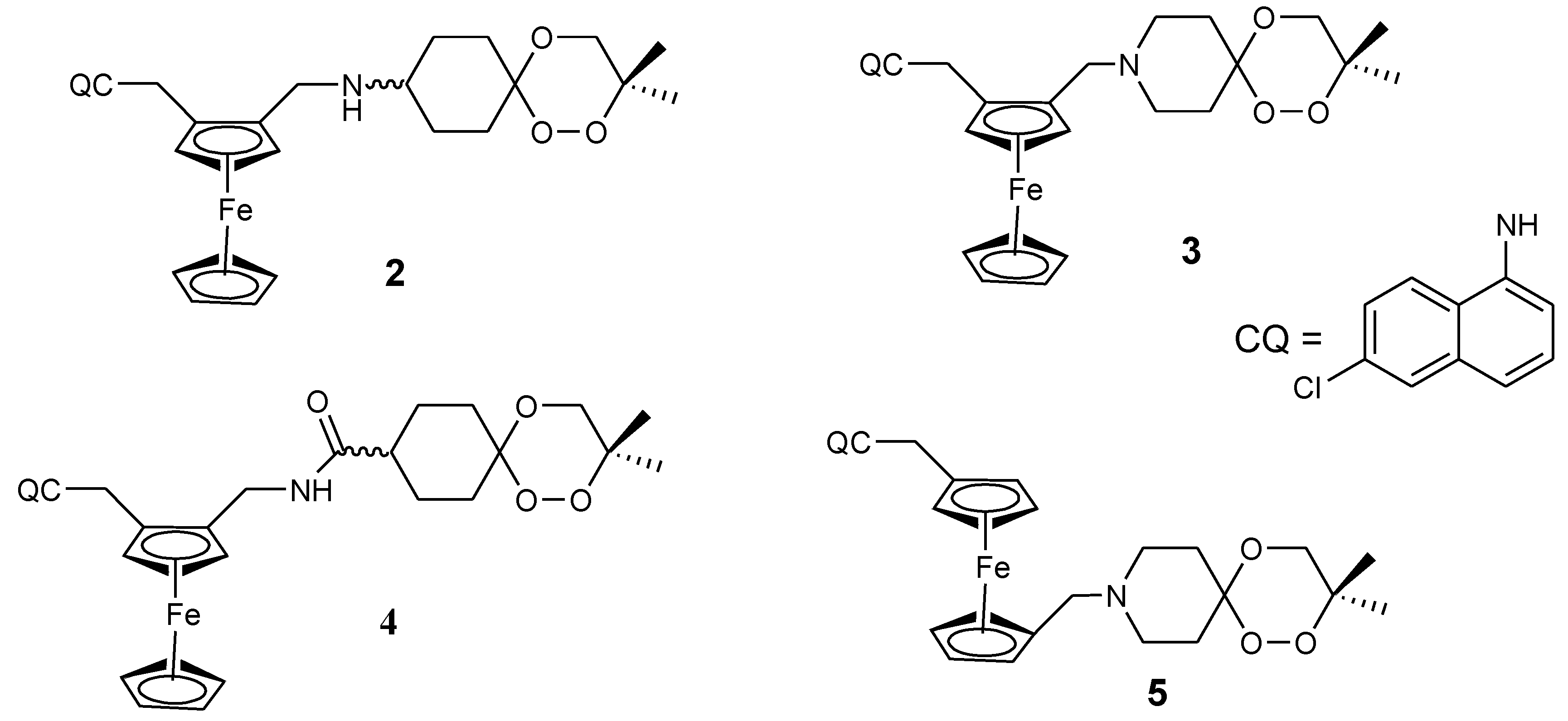
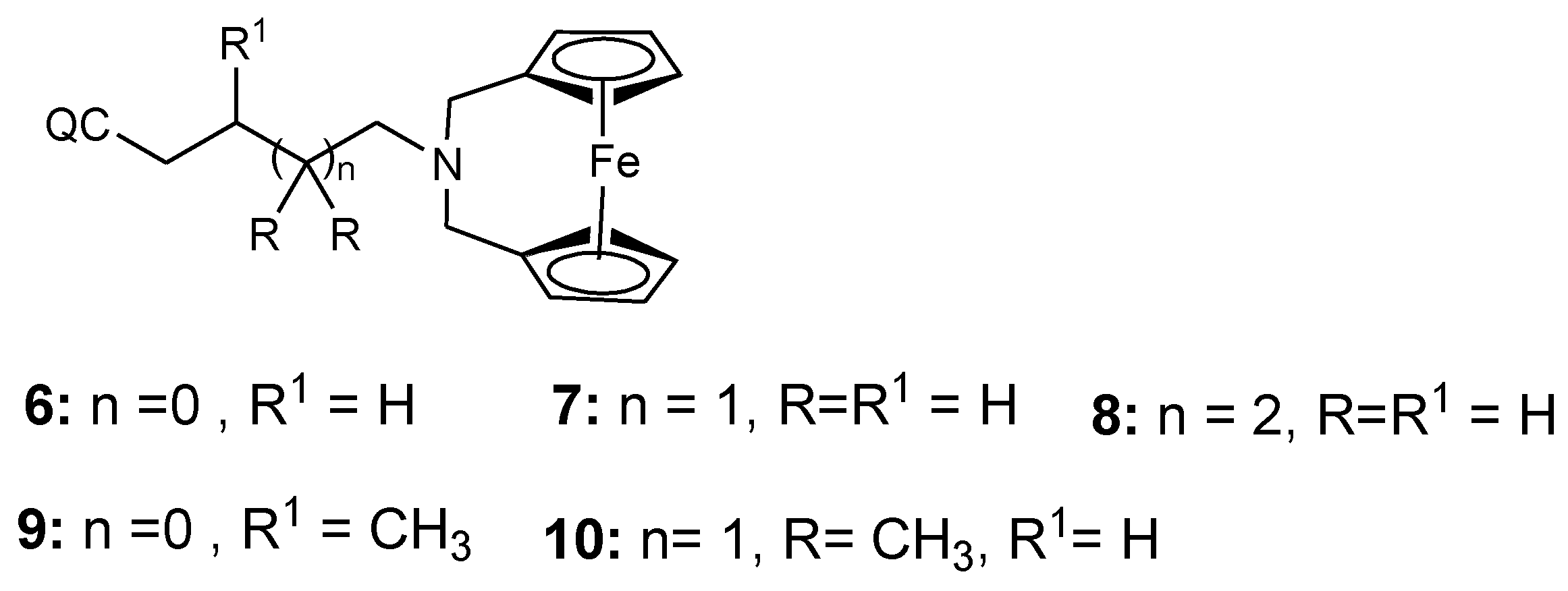
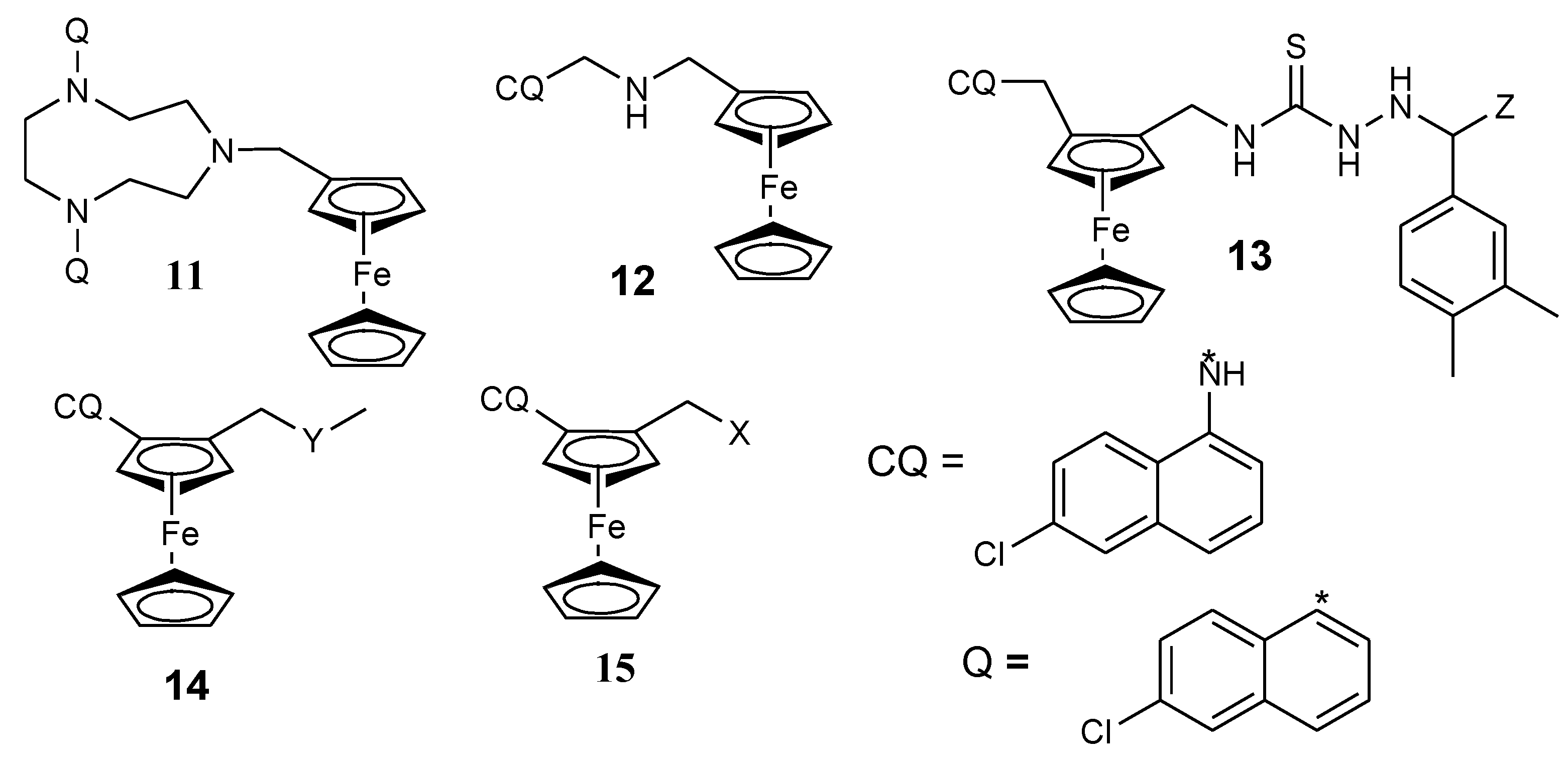
3.1. Ferrocene-Quinoline Derivatives
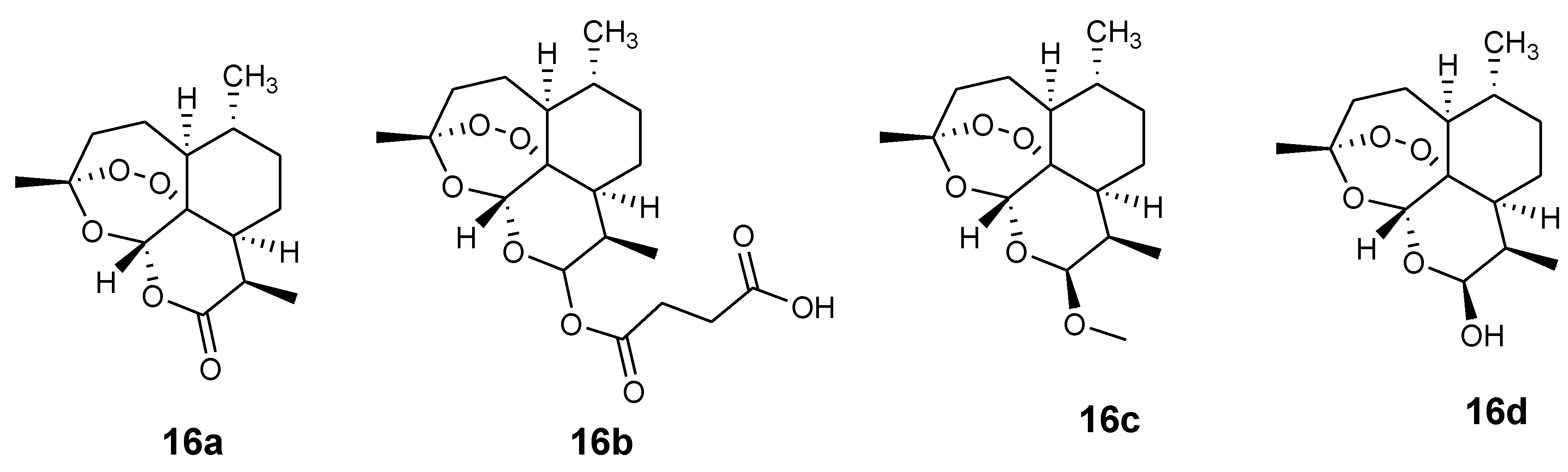
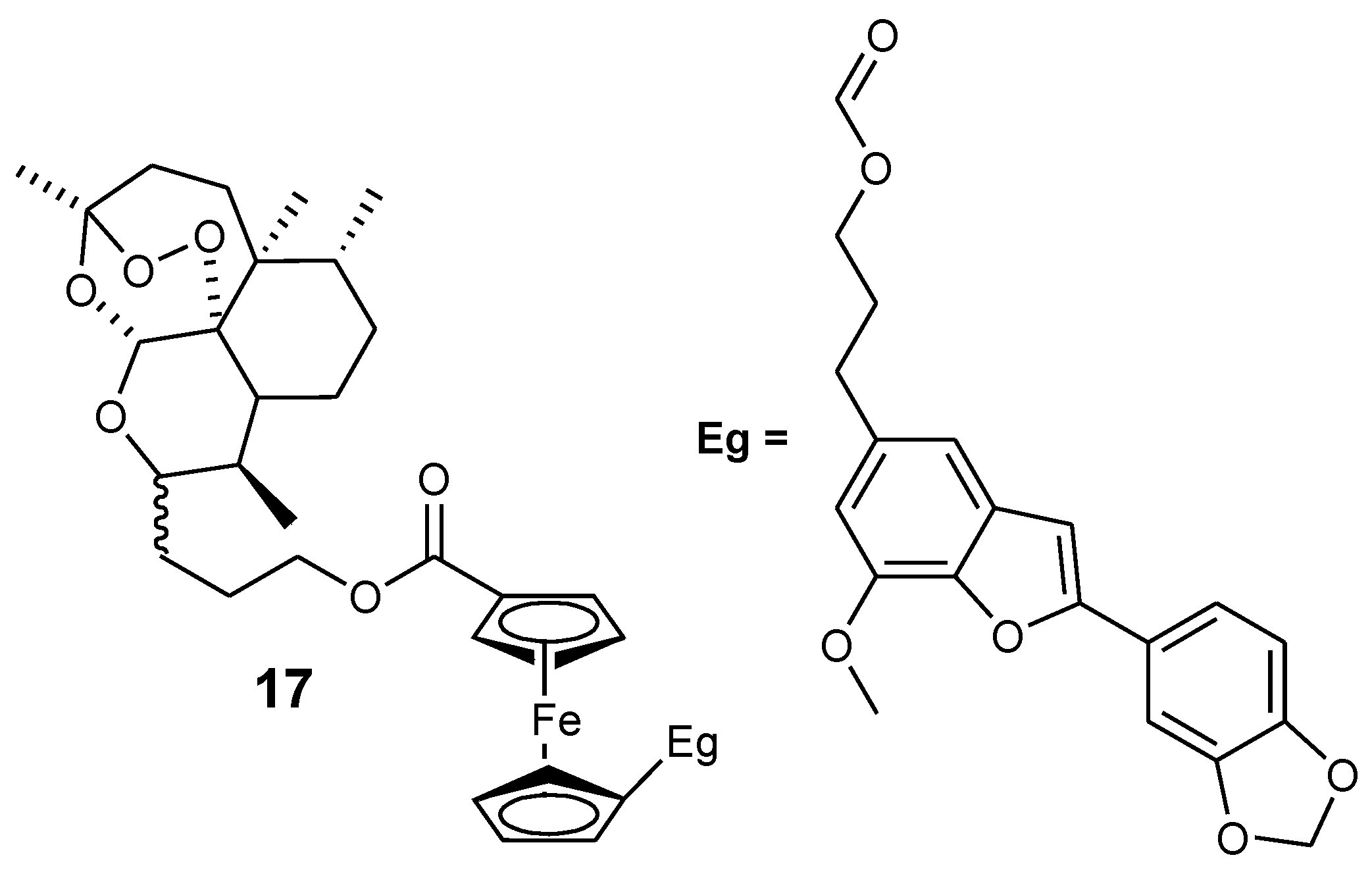
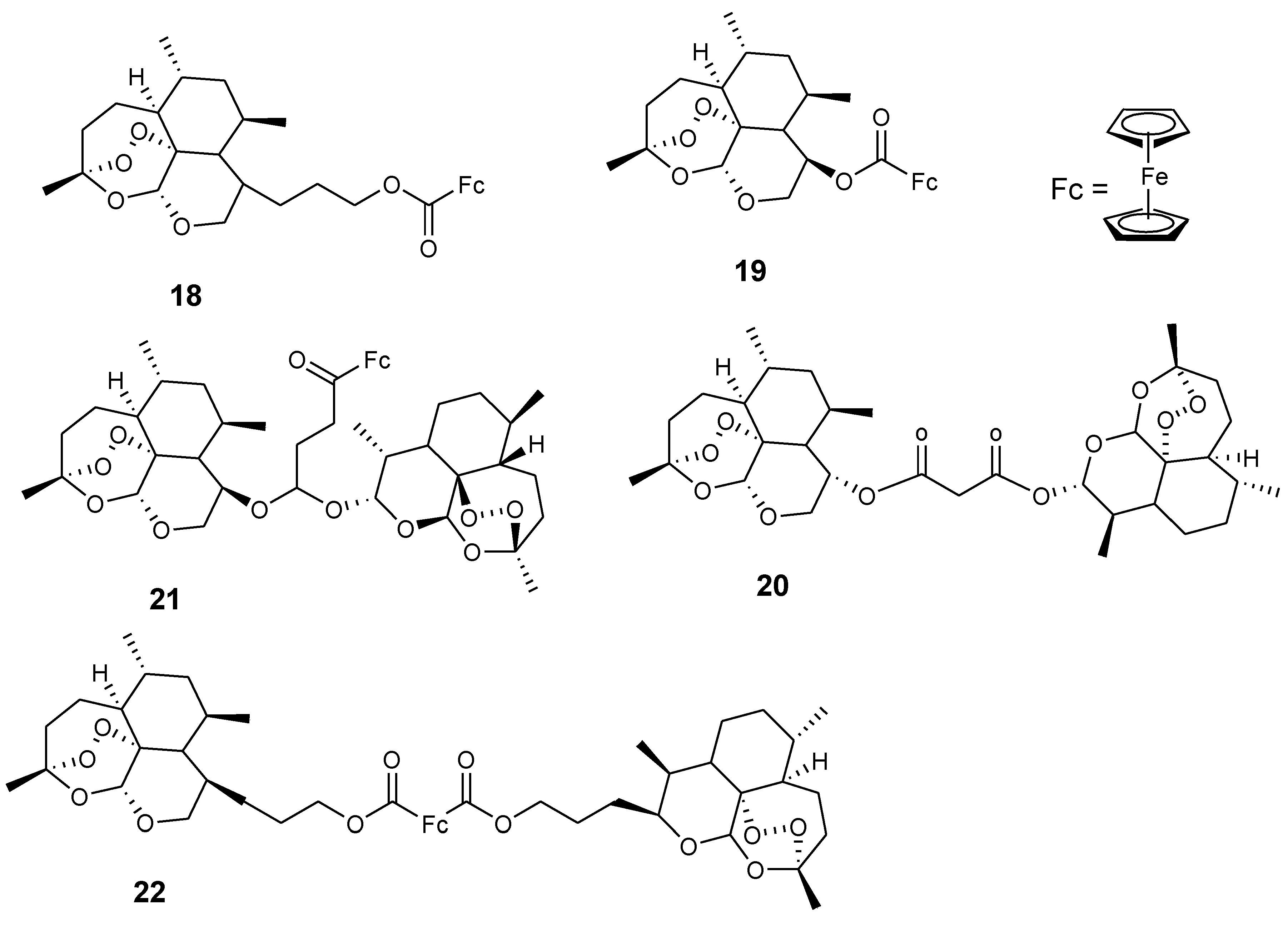
3.2. Artemisinin-Ferrocene Derivative
3.3. Ferrocene–Novobiocin Derivatives
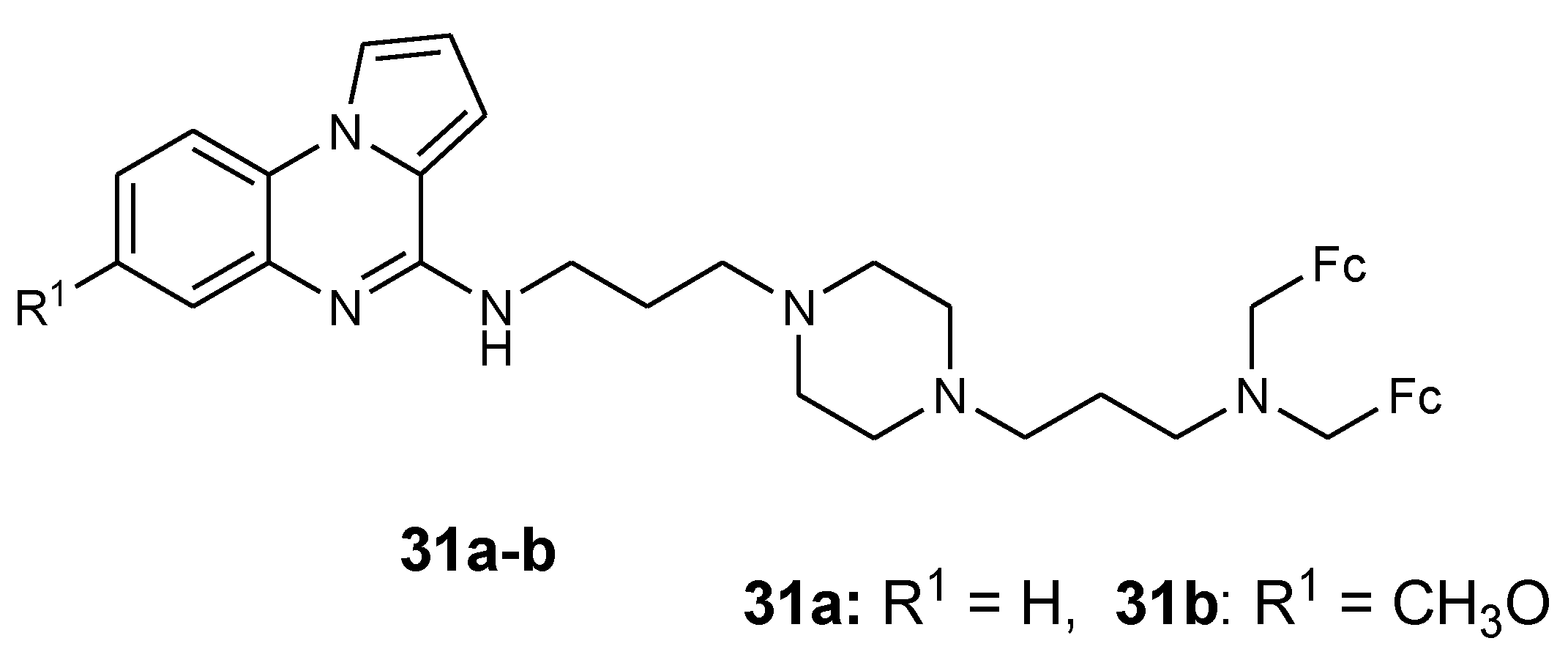
3.4. Ferrocene- Pyrrole Derivatives
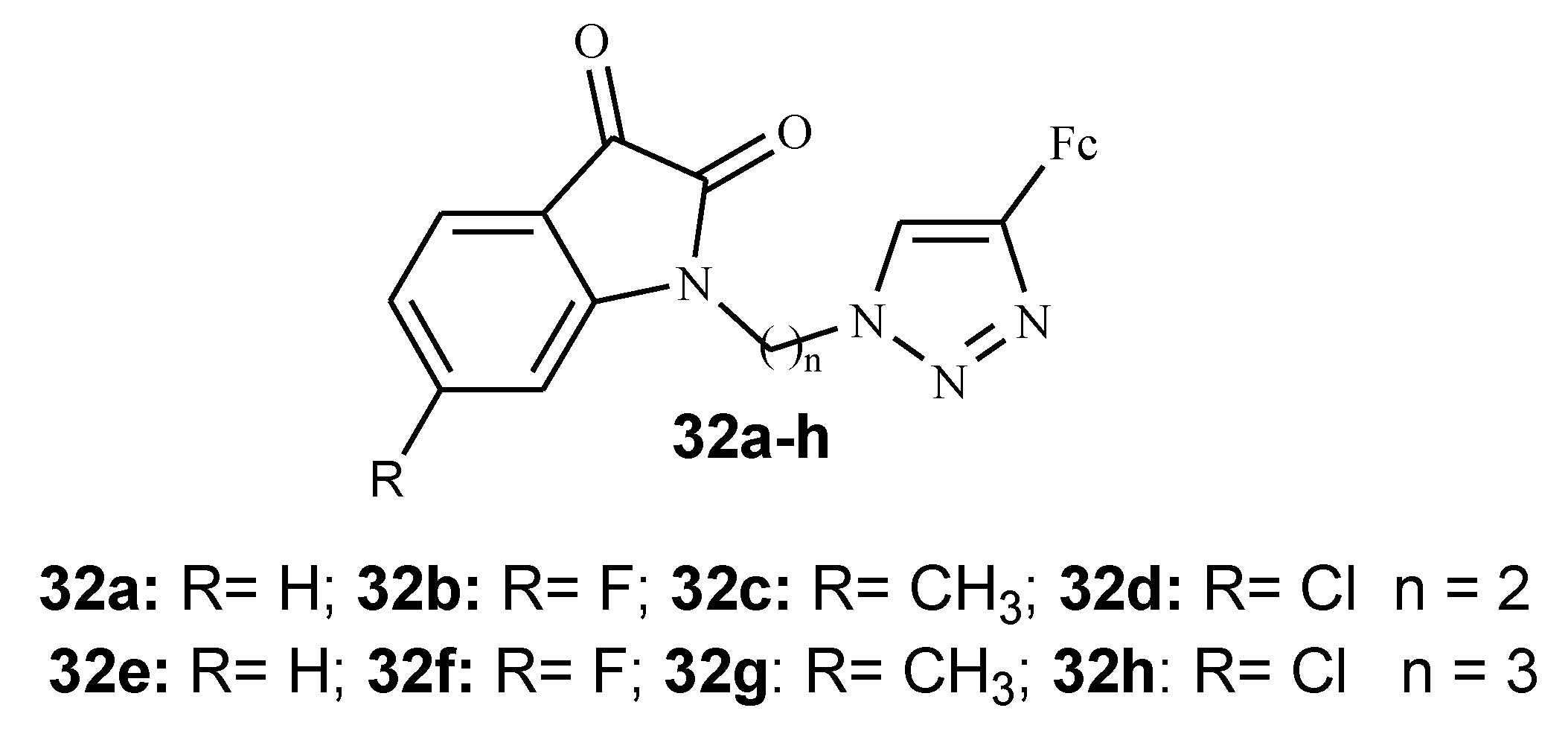
3.5. Isatin–Ferrocene Conjugates
3.6. Ferrocene–Pyrimidine Conjugates
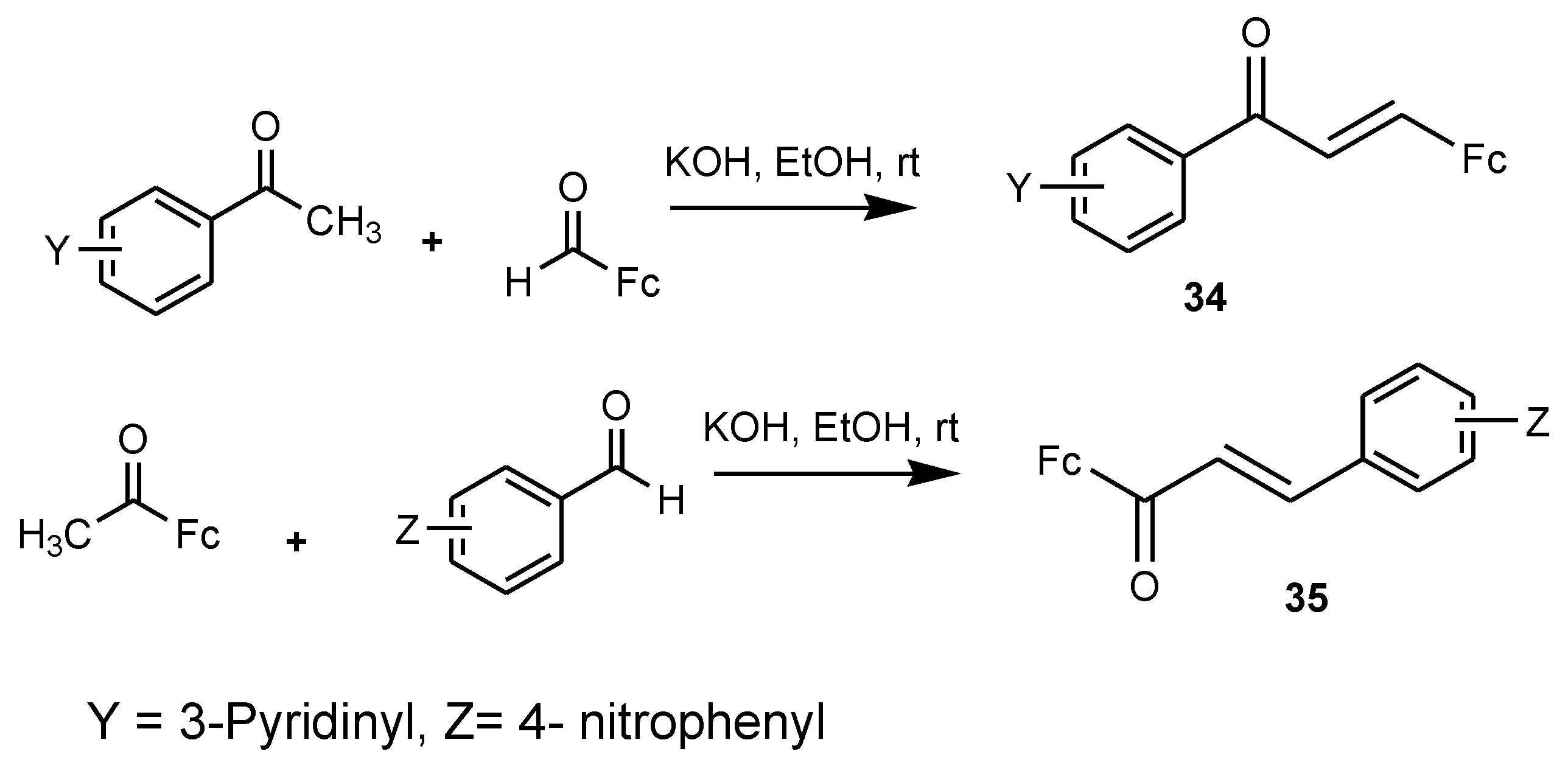
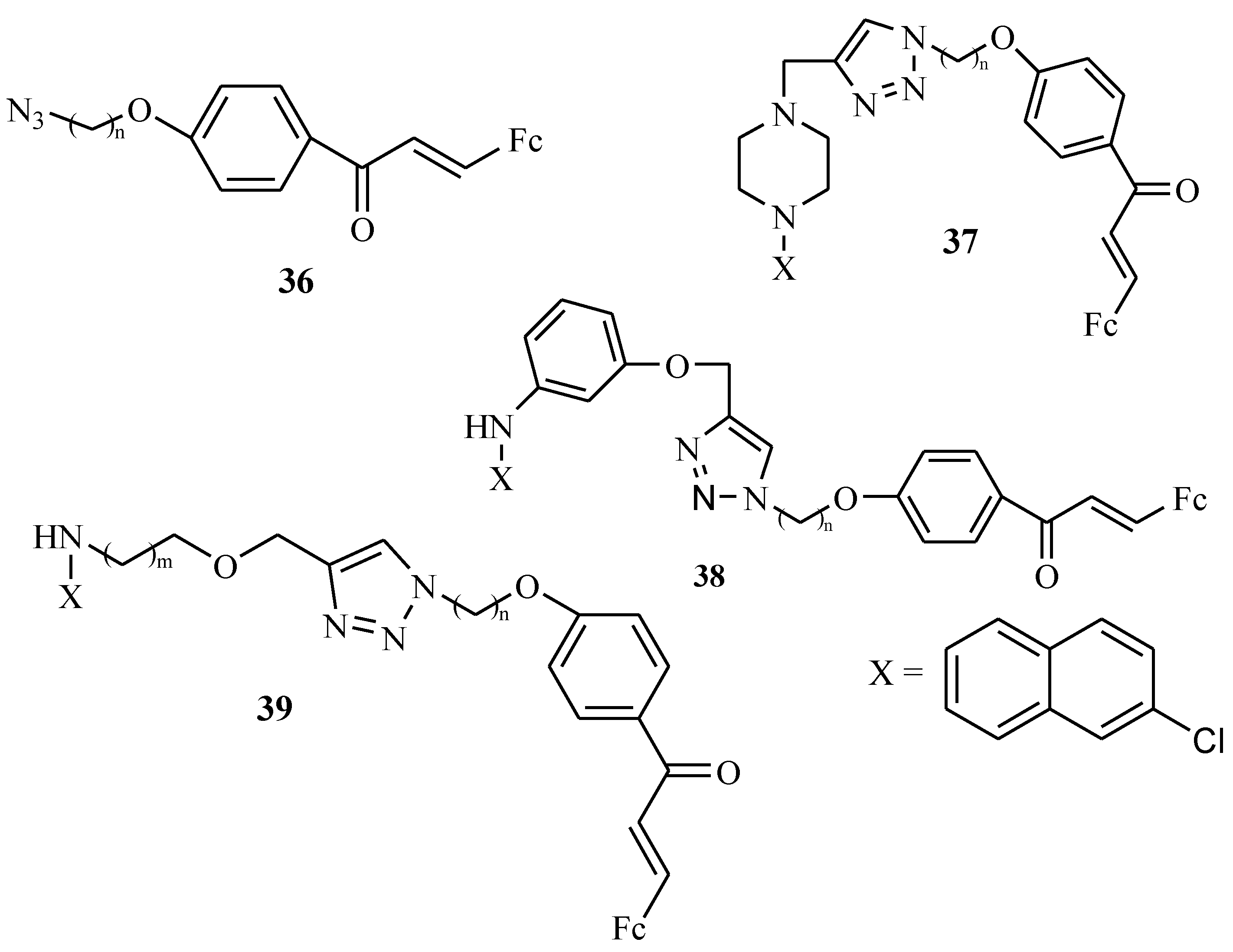
3.7. Ferrocenyl Chalcones
4. Ferrocene-Based Compounds with Anticancer Activity
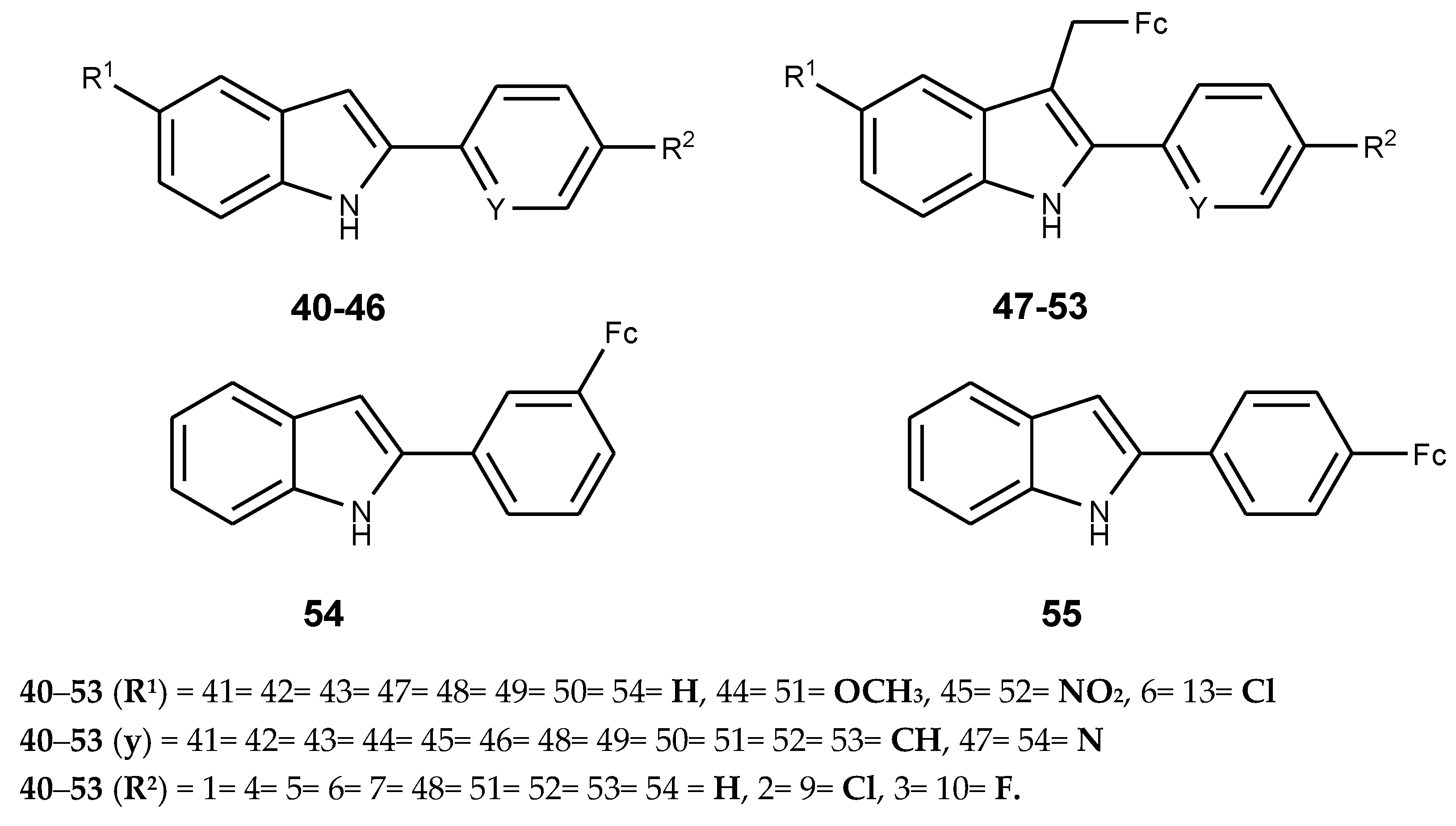
4.1. Ferrocene–Indole Hybrids
4.2. 1,2,4-Trioxane–Ferrocene Hybrids
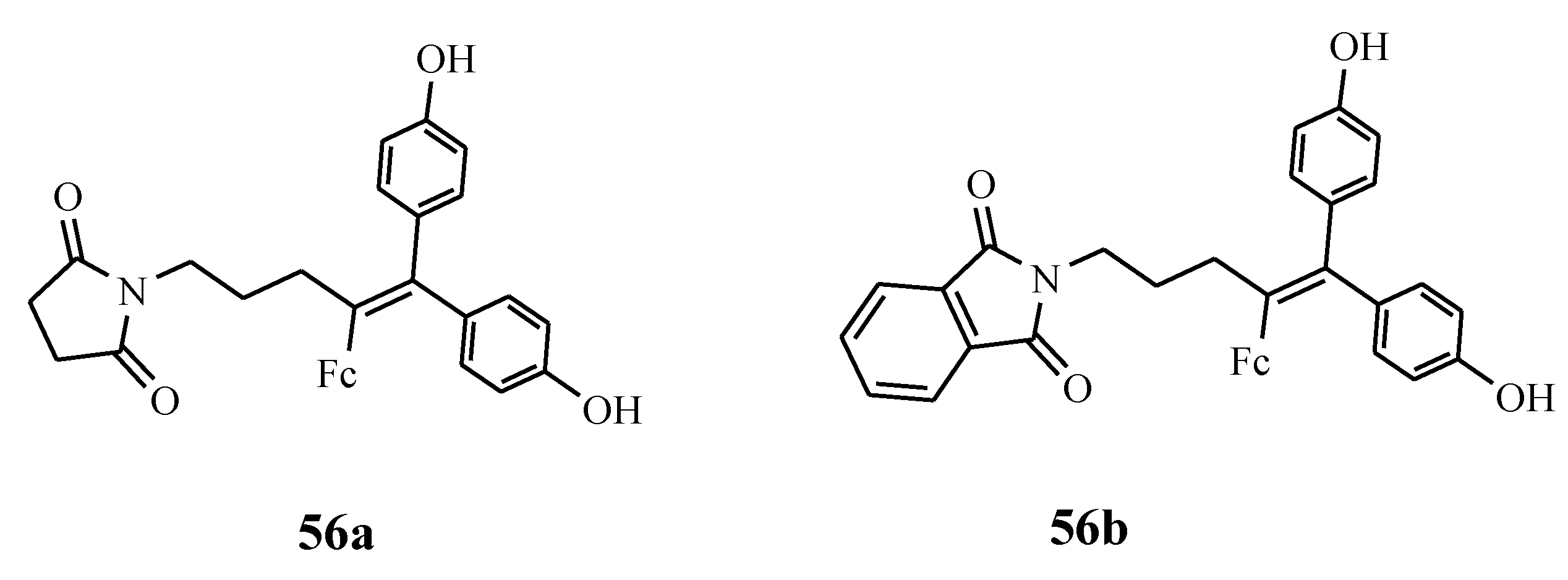
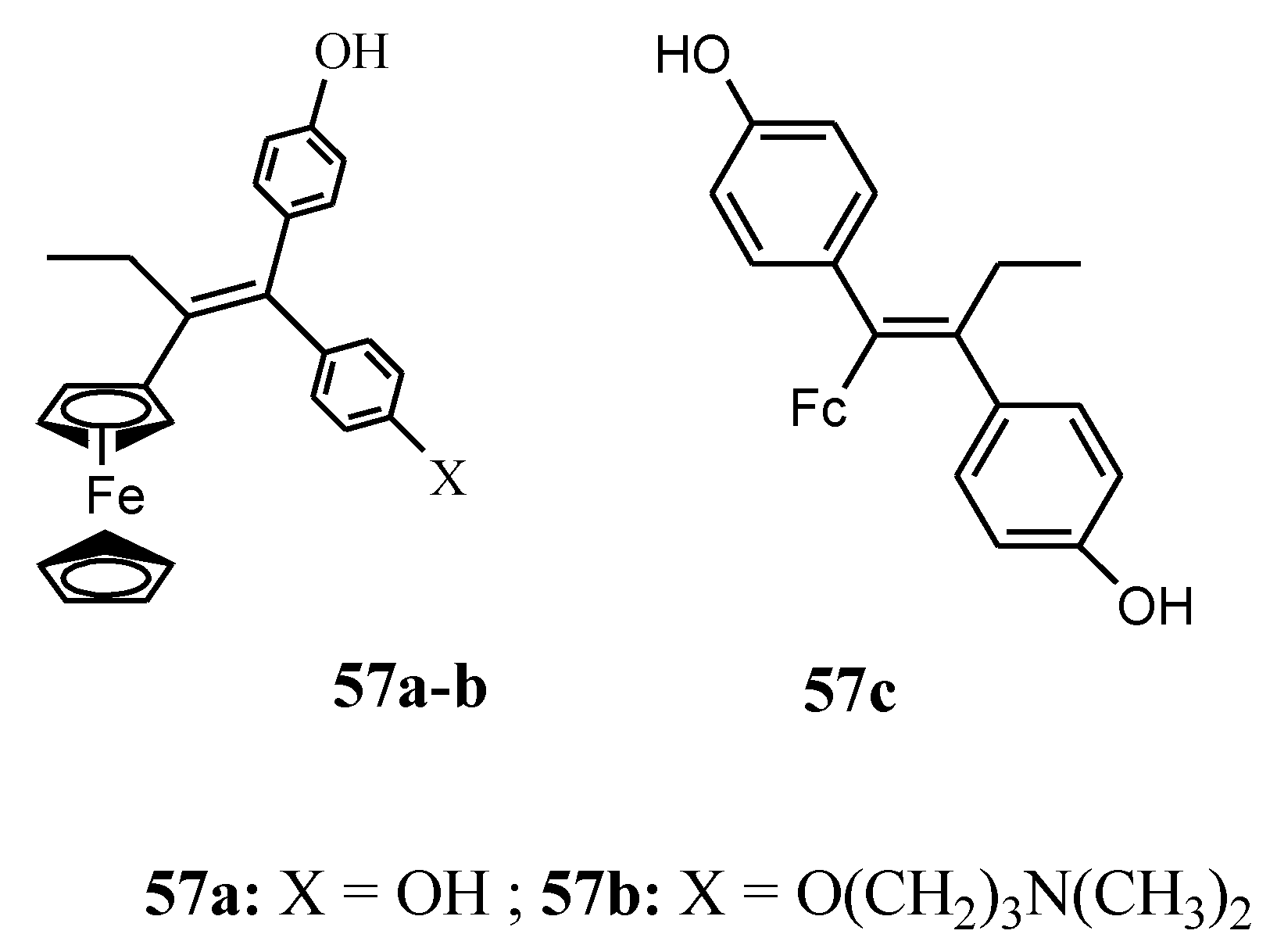
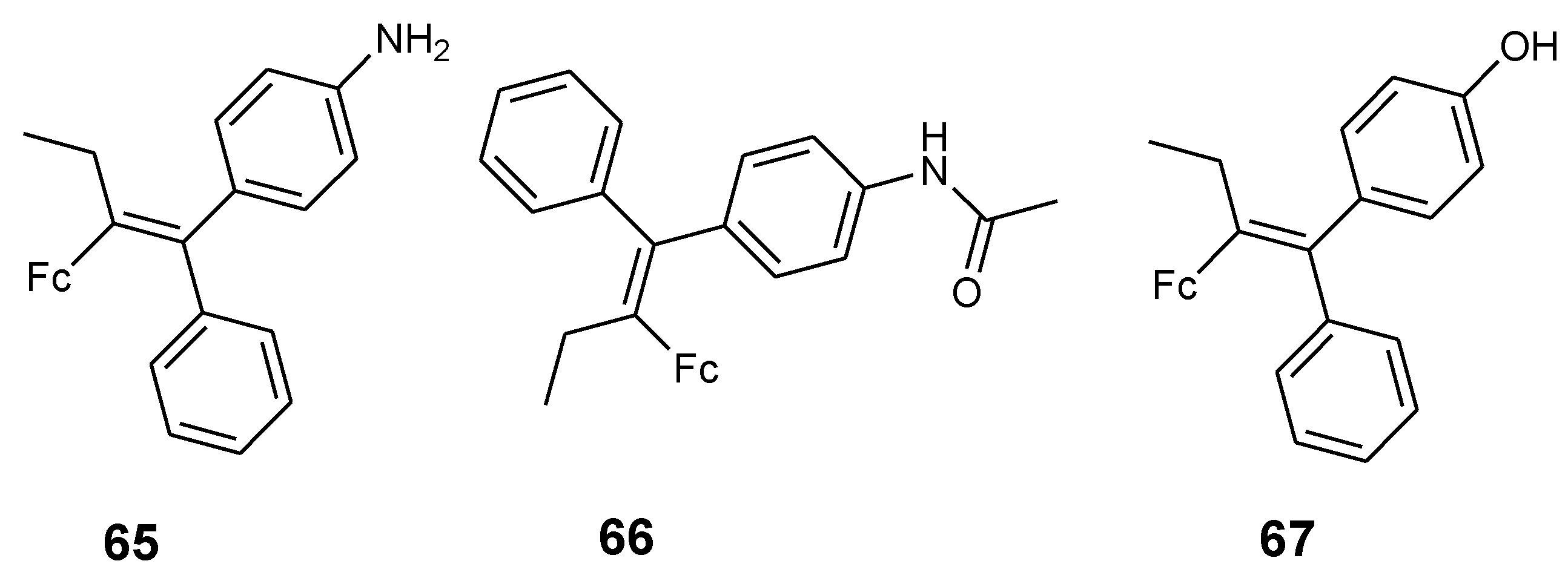
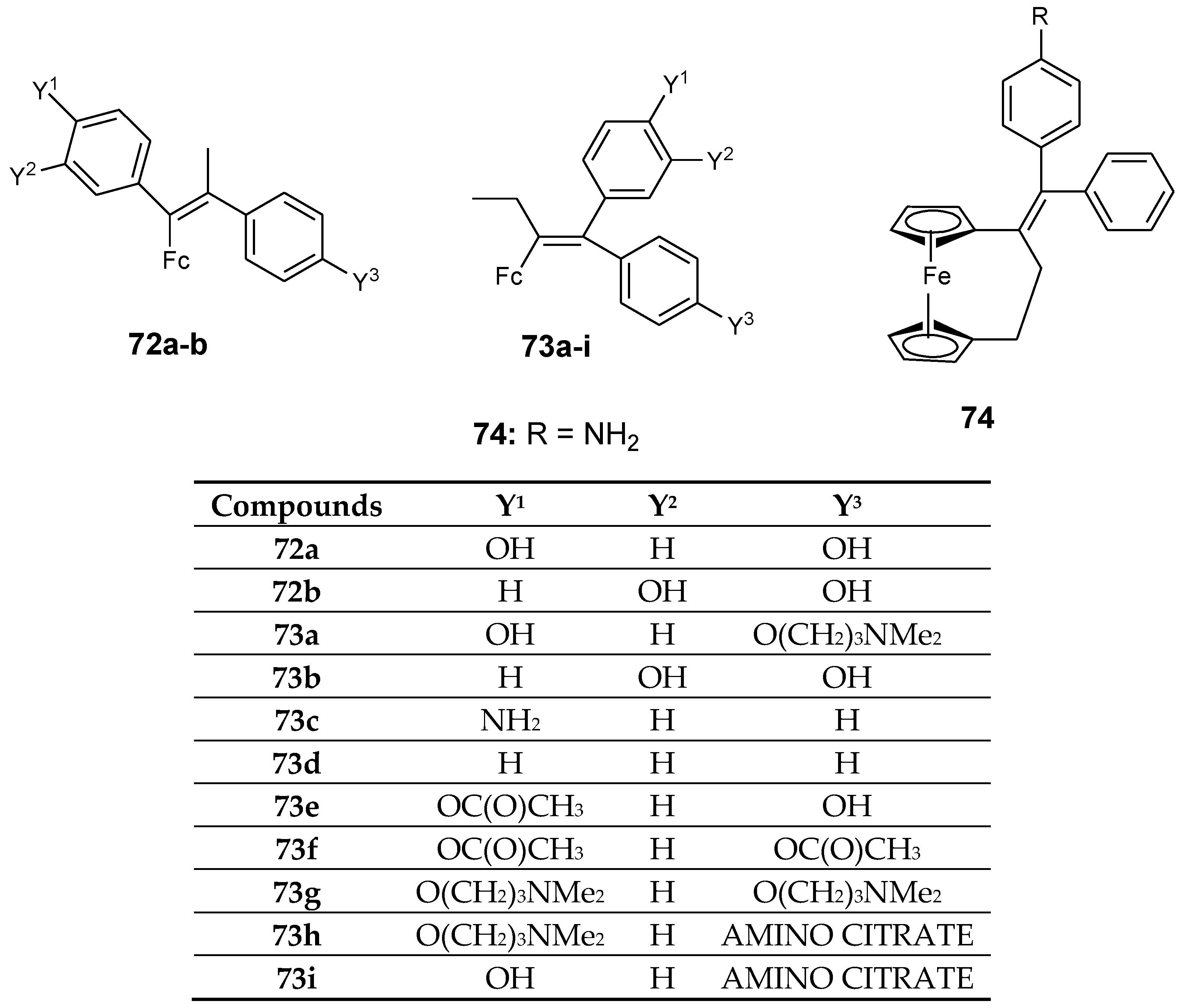
4.3. Ferrociphenols
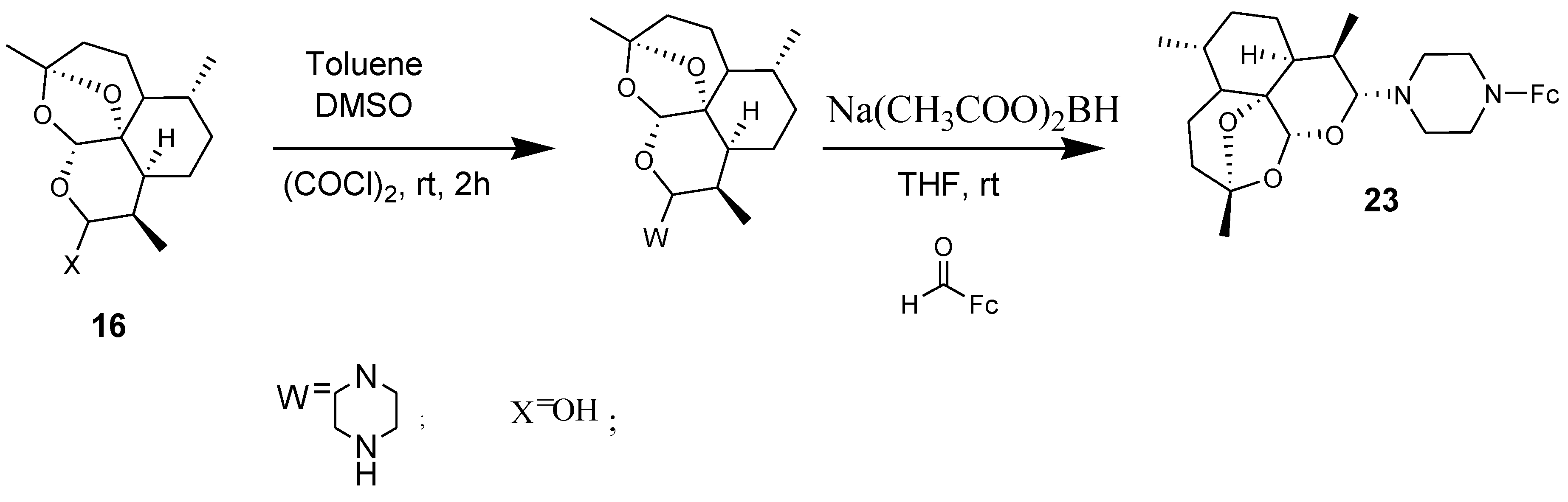
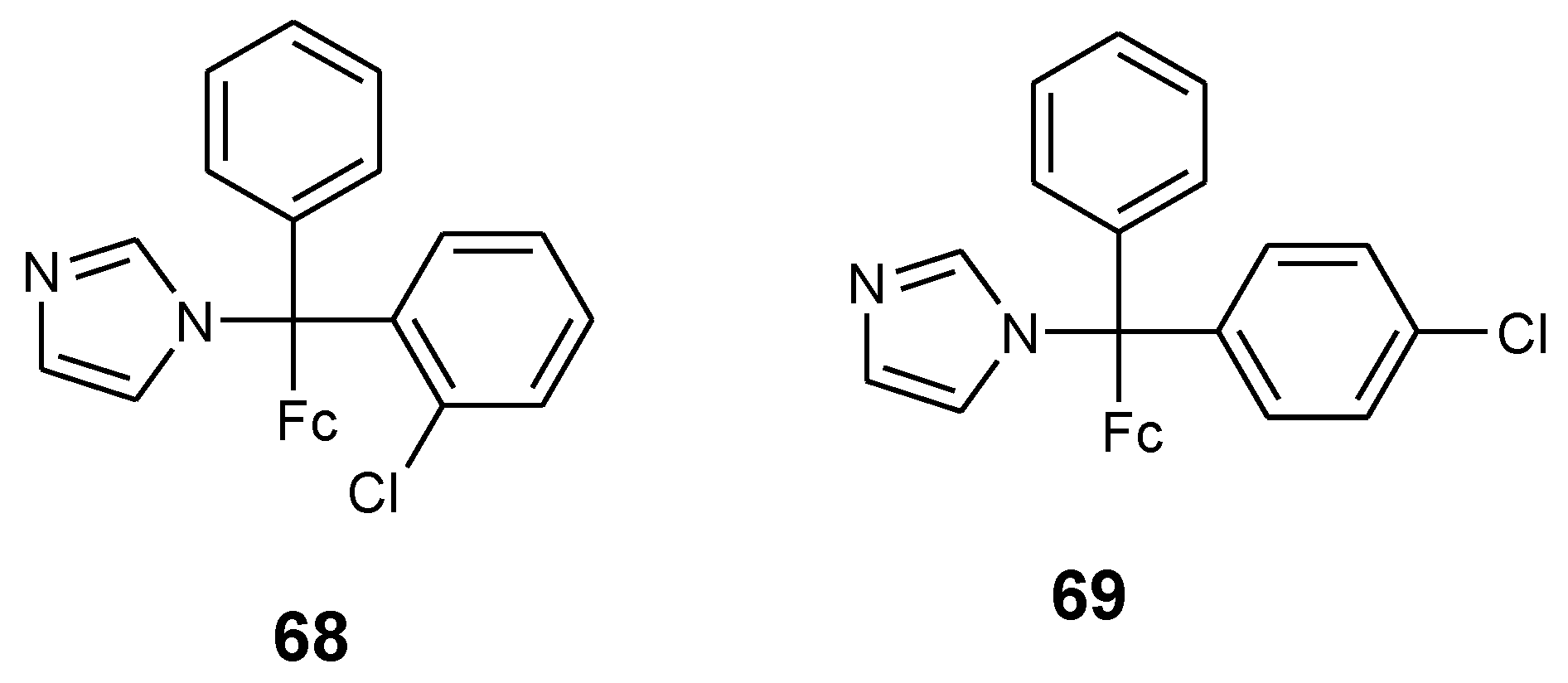
4.4. Ferrocenyl Derivatives of Clotrimazole Drug
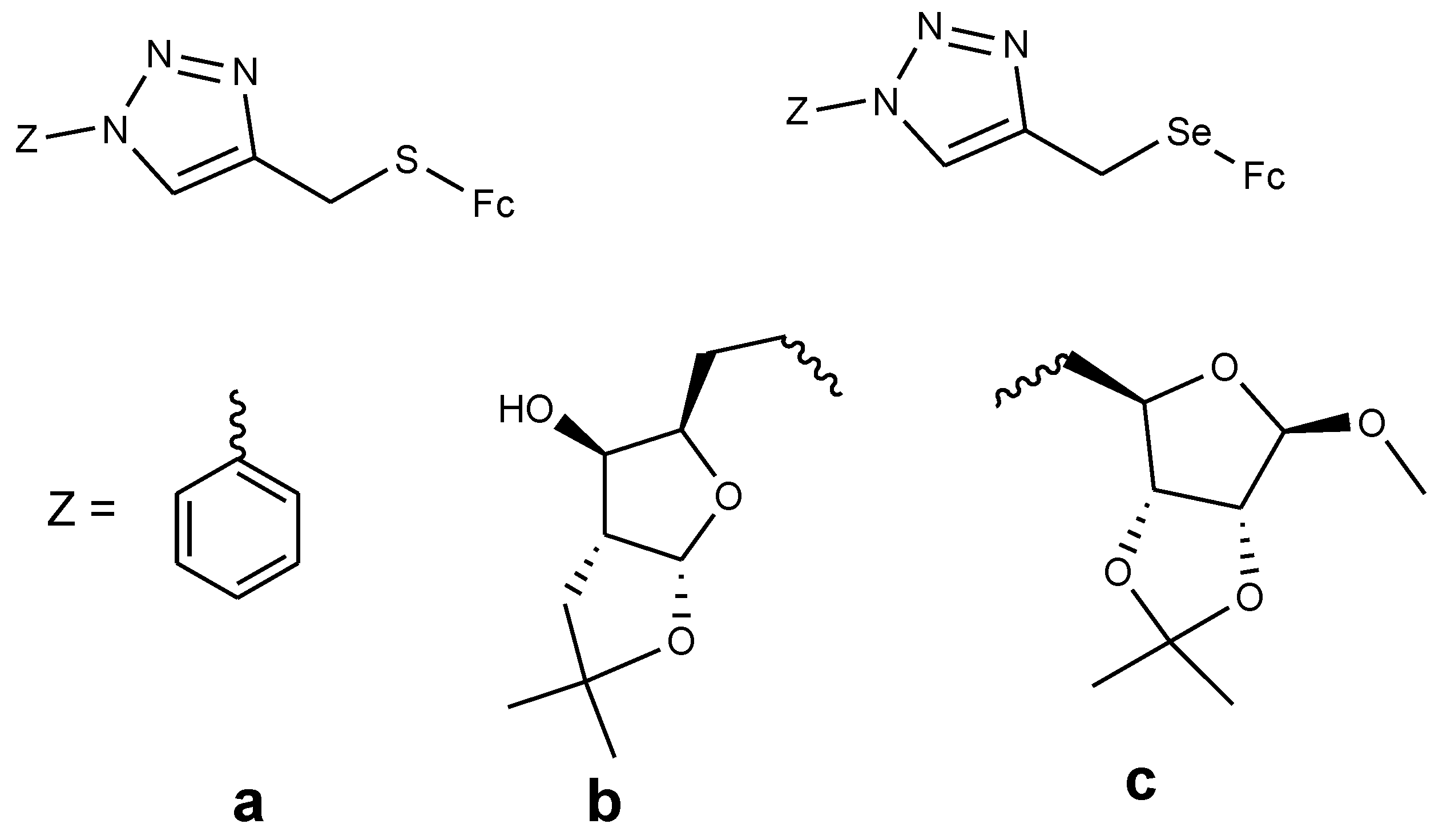
4.5. Ferrocenyl Chalcogeno Triazole Conjugates
4.6. Ferrocenyl–Olefin Derivatives
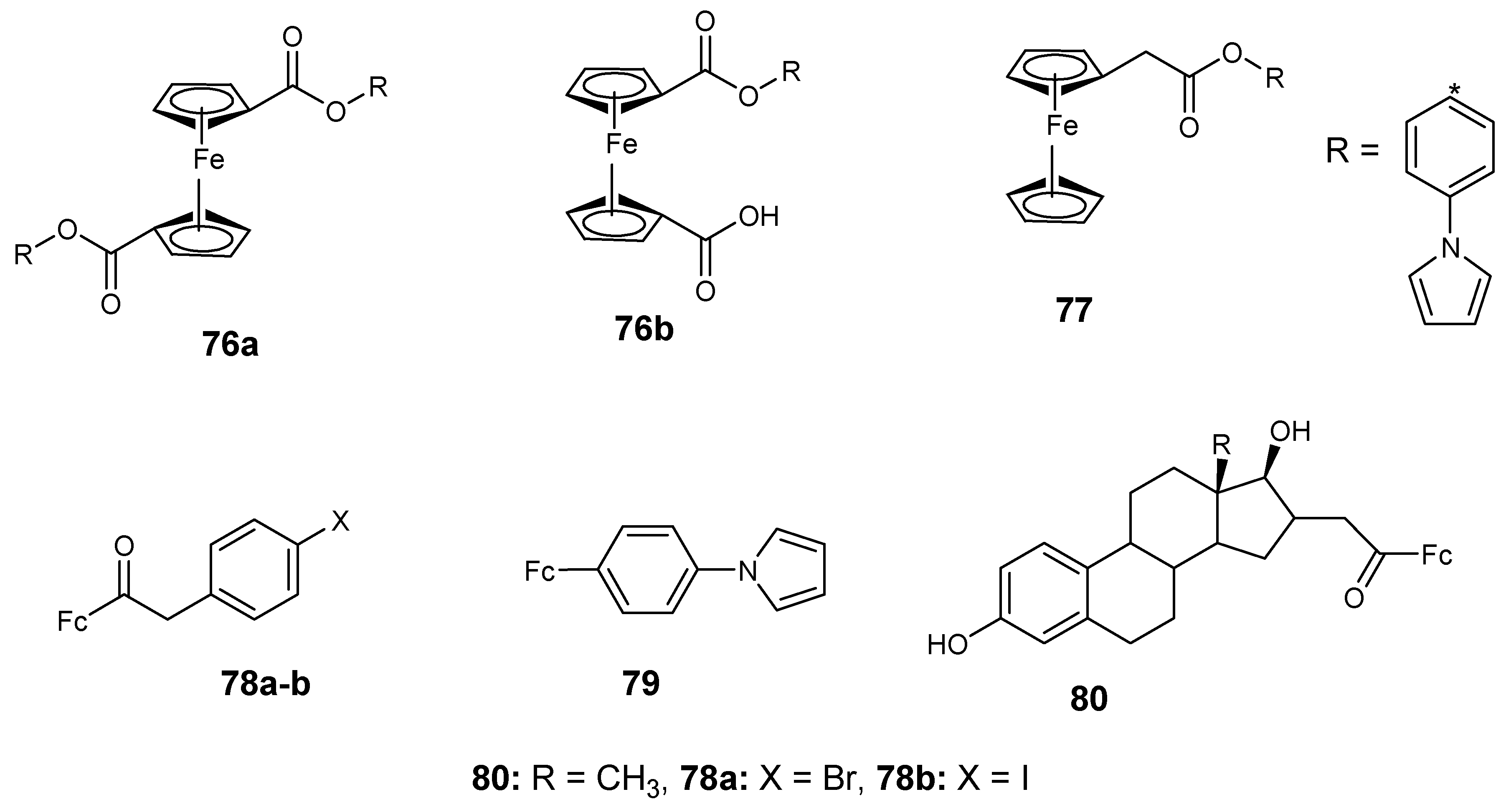
4.7. Ferrocene–Carboxylate Derivatives
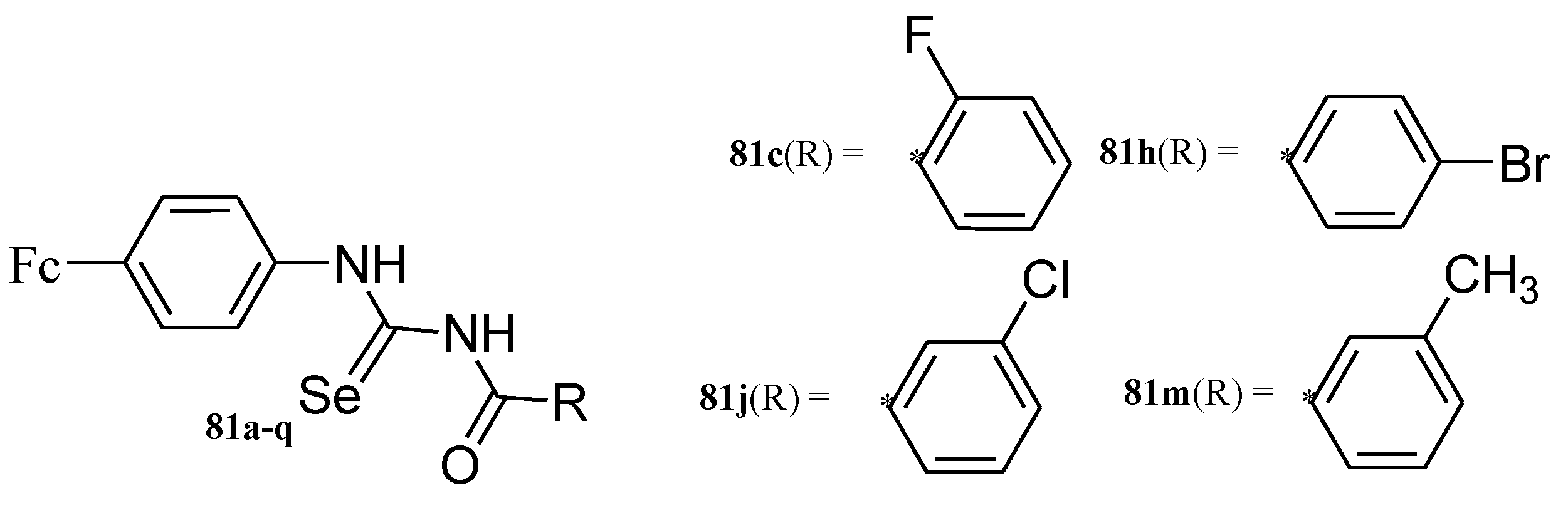
4.8. Ferrocene Incorporated Selenourea Derivatives
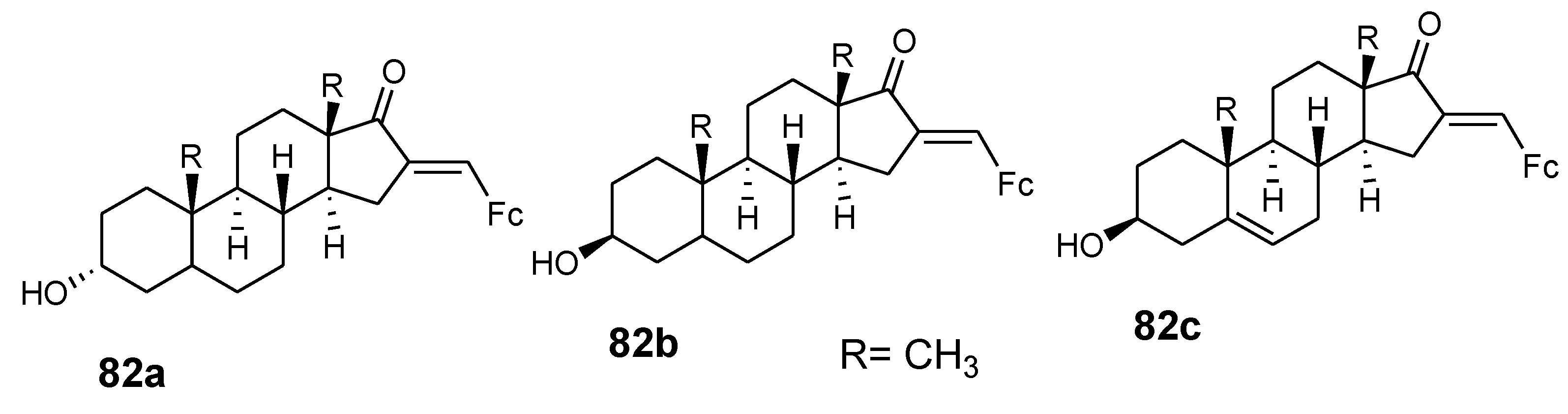
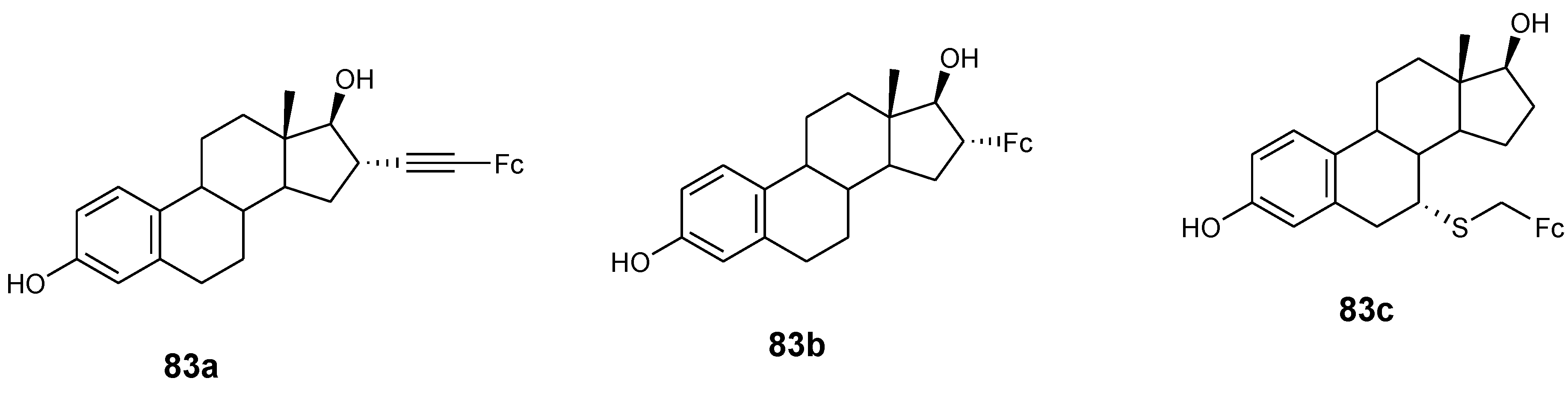
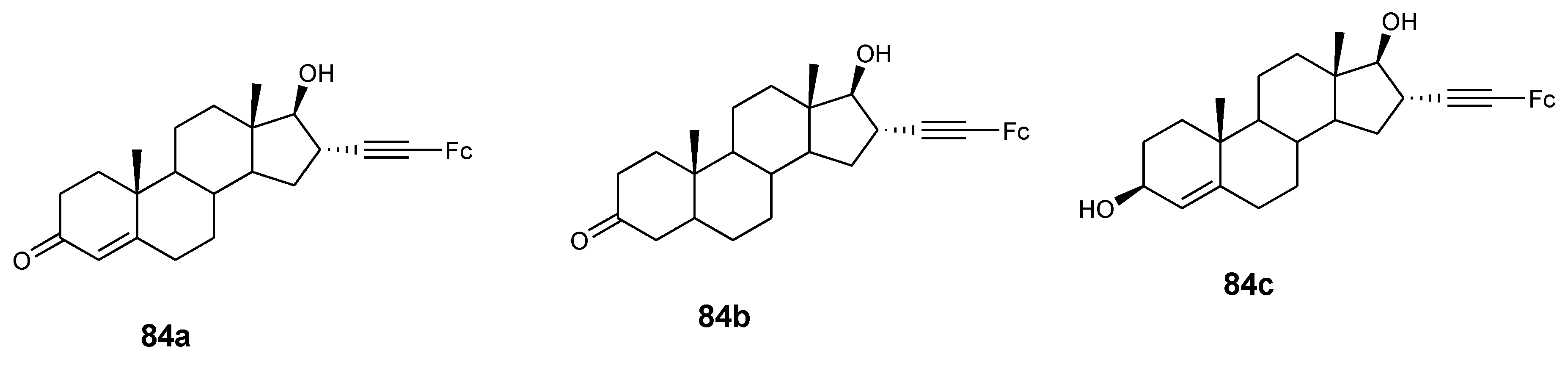
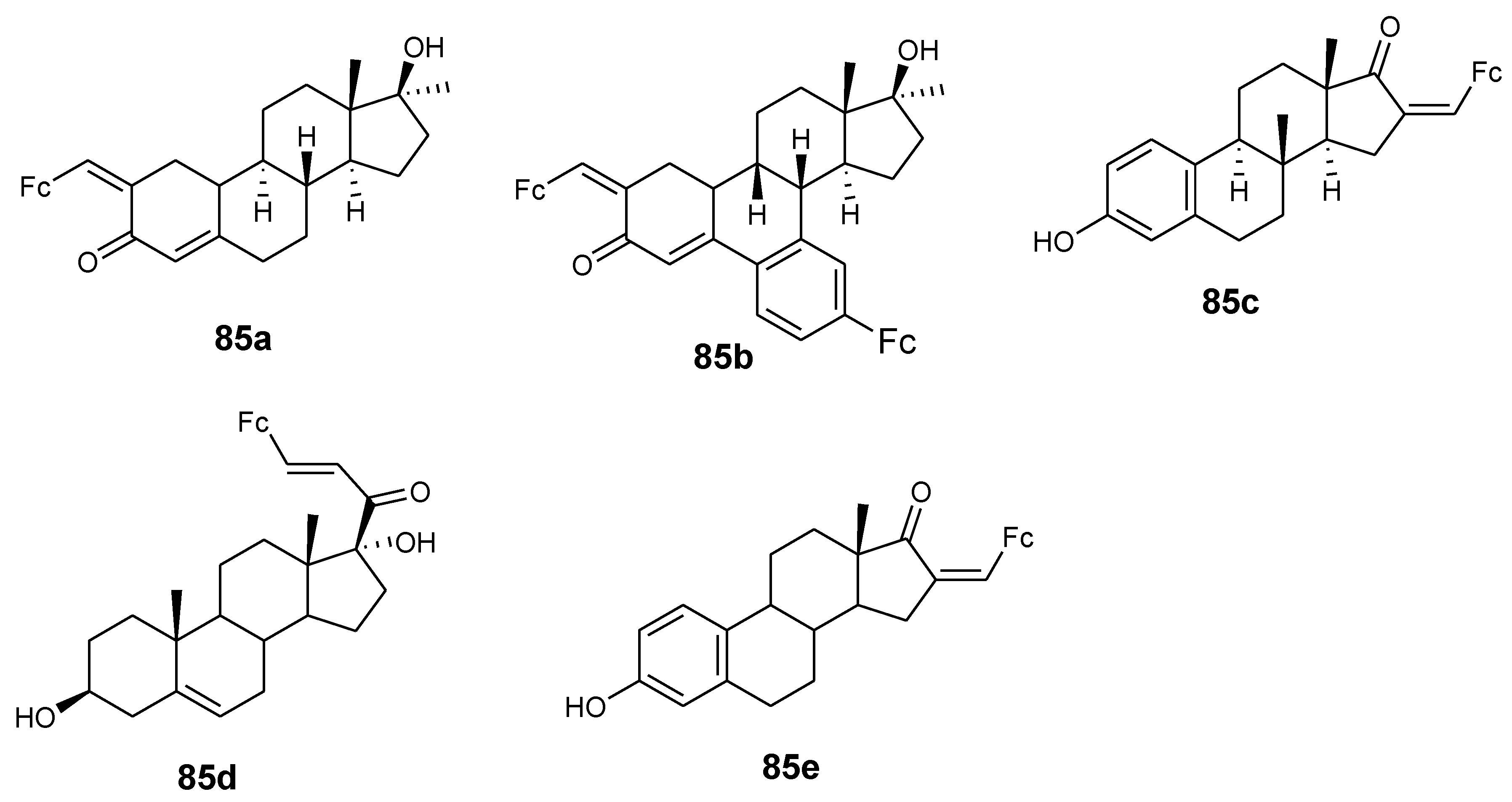
4.9. Ferrocene–Steroid Conjugates
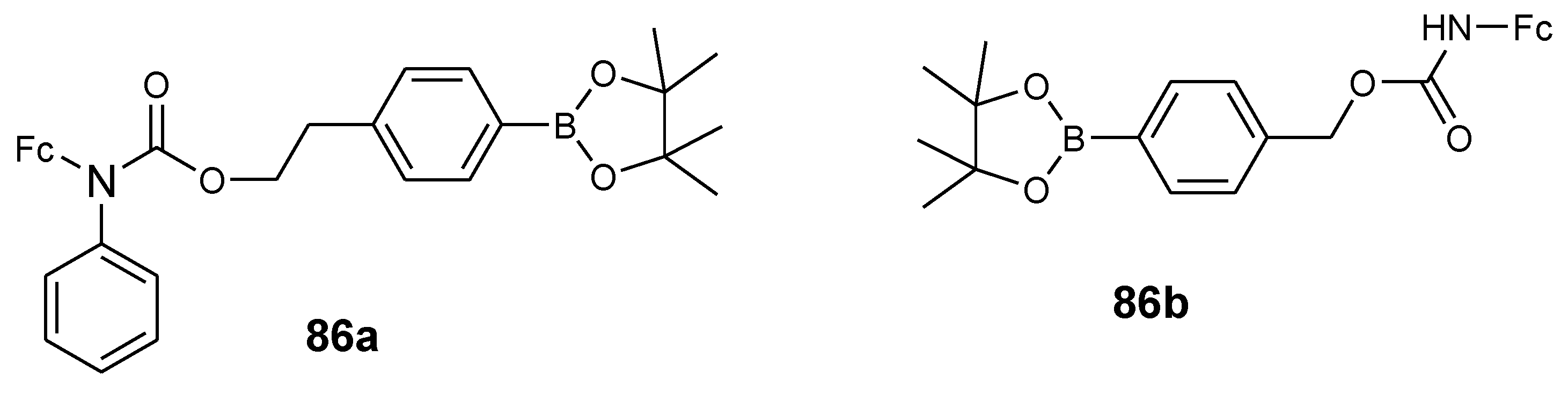
4.10. Aminoferrocene-Based Derivatives
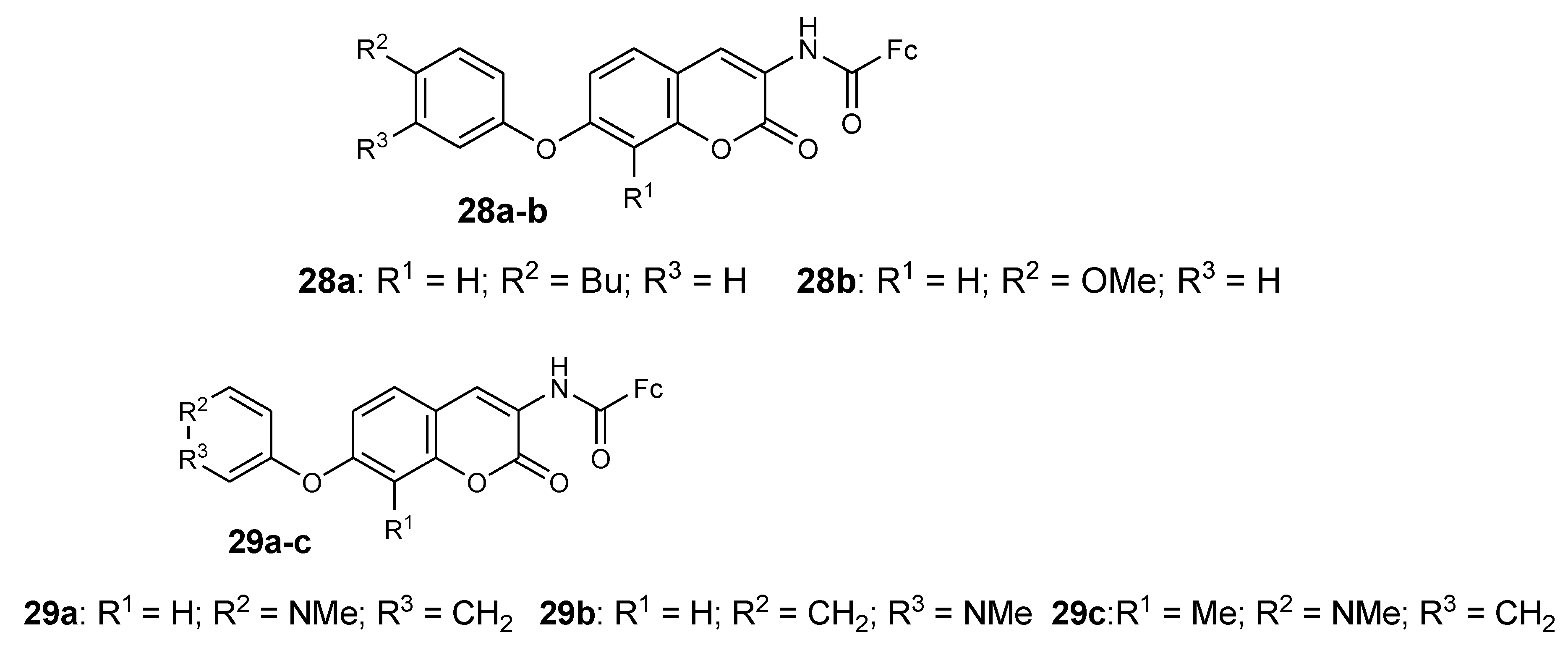
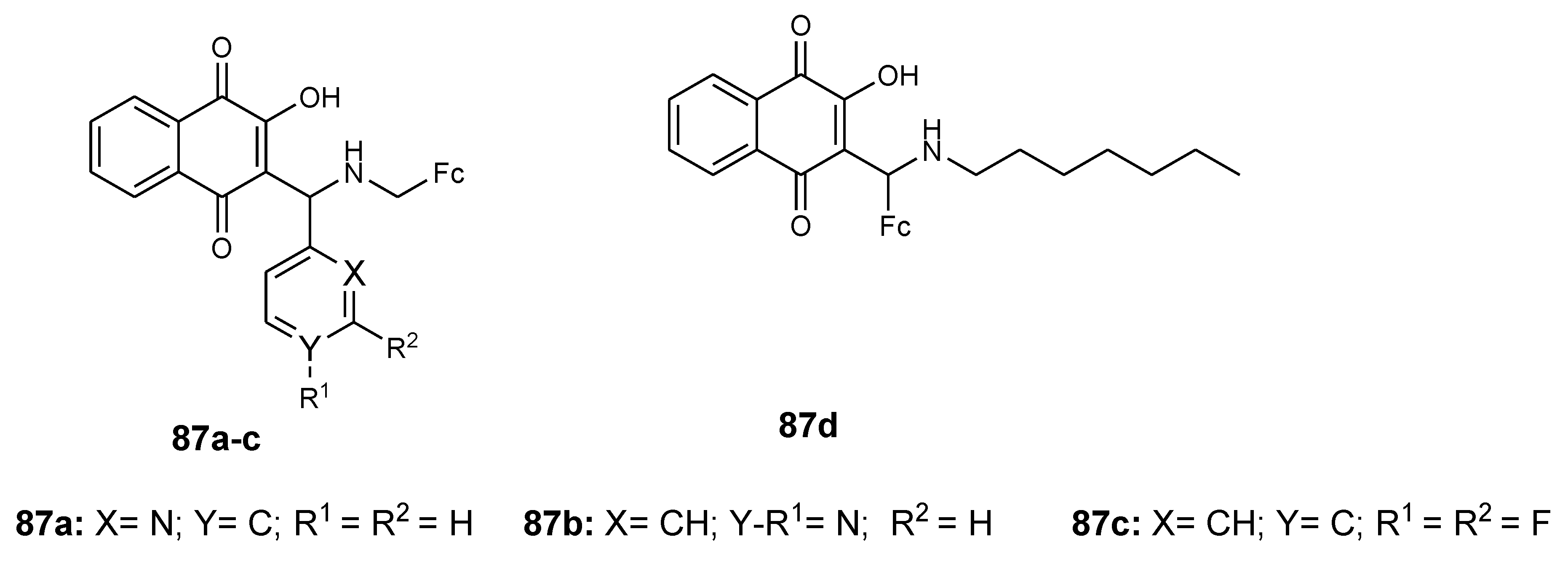
4.11. Ferrocene–Lawsone Mannich Derivatives
5. Conclusions
Funding
Conflicts of Interest
References
- Glennon, E.K.K.; Dankwa, S.; Smith, J.D.; Kaushasky, A. Opportunities for host-targeted therapies for malaria. Trends Parasitol. 2018, 34, 843–860. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Q.; Gao, C.; Zhang, S.; Xu, L.; Xu, S.; Feng, L.S.; Wu, X.; Zhao, F. Quinoline hybrids and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2017, 139, 22–47. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 8 July 2019).
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018; International Agency for Research on Cancer: Lyon, France, 2018. [Google Scholar]
- Okuhara, T.; Ishikawa, H.; Urakubo, A.; Hayakama, M.; Yamaki, T.; Tarayama, T.; Kuchi, T. Cancer information needs according to cancer type: A content analysis of data from Japan’s largest cancer information website. Prev. Med. Rep. 2018, 12, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bhardwaj, T.R.; Prasad, D.N.; Singh, R.K. Biomedicine & Pharmacotherapy Drug targets for resistant malaria: Historic to future perspectives. Biomed. Pharm. 2018, 104, 8–27. [Google Scholar]
- Zanetti, R.; Saccetto, L.; Coebergh, J.W.; Rosso, S. ScienceDirect to accelerate cancer prevention in Europe: Cancer Challenges for registries. Eur. J. Cancer 2018, 104, 151–159. [Google Scholar] [CrossRef]
- Wilkinson, G.; Rosenblum, M.; Whiting, M.C.; Woodward, R.B. Ferrocene. J. Am. Chem. Soc. 1952, 74, 1–10. [Google Scholar]
- Singh, A.; Lumb, I.; Mehra, V.; Kumar, V. Ferrocene-appended pharmacophores: An exciting approach for modulating the biological potential of organic scaffolds. Dalton Trans. 2019, 48, 2840–2860. [Google Scholar] [CrossRef]
- Narváez-Pita, X.; Rheingold, A.L.; Meléndez, E. Ferrocene-steroid conjugates: Synthesis, structure and biological activity. J. Organomet. Chem. 2017, 846, 113–120. [Google Scholar] [CrossRef]
- Biomol, O.; Hottin, A.; Dubar, F.; Steenackers, A.; Delannoy, P. Iminosugar–ferrocene conjugates as potential anticancer agents. Org. Biomol. Chem. 2012, 10, 5592–5597. [Google Scholar]
- Huang, X.F.; Tang, J.F.; Ji, J. L.; Wang, X.L.; Ruan, B.F. Synthesis, characterization and antitumor activity of novel amide derivatives containing ferrocenyl pyrazol-moiety. J. Organomet. Chem. 2012, 706, 113–123. [Google Scholar] [CrossRef]
- Pedotti, S.; Ussia, M.; Patti, A.; Musso, N.; Barresi, V.; Condorelli, D.F. Synthesis of the ferrocenyl analogue of clotrimazole drug. J. Organomet. Chem. 2017, 830, 56–61. [Google Scholar] [CrossRef]
- Krishna, A.D.S.; Panda, G.; Kondapi, A.K. Mechanism of action of ferrocene derivatives on the catalytic activity of topoisomerase IIα and β-Distinct mode of action of two derivatives. Arch. Biochem. Biophys. 2005, 438, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.A.; Mukaya, H.E. Polymer Therapeutics: Design, Application, and Pharmacokinetics. In Nano-and Microscale Drug Delivery Systems; Elsevier: Johannesburg, South Africa, 2017; pp. 33–48. [Google Scholar]
- Köpf-Maier, P.; Köpf, H.; Neuse, E.W. Ferricenium complexes: A new type of water-soluble antitumor agent. J. Cancer Res. Clin. Oncol. 1984, 108, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.A.; Jameel, E.; Baig, U.; Mumtazuddin, S.; Hun, L.T. Ferroquine and its derivatives: New generation of antimalarial agents. Eur. J. Med. Chem. 2015, 101, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Atteke, C.; Ndong, J.M.M.; Aubouy, A.; Maciejewski, L.; Brocard, J.; Lebibi, J.; Deloron, P. In vitro susceptibility to a new antimalarial organometallic analogue, ferroquine, of Plasmodium falciparum from the Haut-Ogooué region of Gabon. J. Antimicrob. Chemother. 2003, 51, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Nqoro, X.; Tobeka, N.; Aderibigbe, B.A. Quinoline-based hybrid compounds with antimalarial activity. Molecules 2017, 22, 2268. [Google Scholar] [CrossRef]
- White, N.J. The treatment of malaria. N. Engl. J. Med. 1996, 335, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, S.; D’Hooghe, M. Quinoline-based antimalarial hybrid compounds. Bioorg. Med. Chem. 2015, 23, 5098–5119. [Google Scholar] [CrossRef]
- Martinelli, A.; Moreira, R.; Cravo, P.V.L. Malaria combination therapies: Advantages and shortcomings. Mini Rev. Med. Chem. 2008, 8, 201–212. [Google Scholar] [CrossRef]
- Long, T.T.; Nakazawa, S.; Onizuka, S.; Huaman, M.S.; Kanbara, H. Influence of CD4+CD25+ T cells on Plasmodium berghei NK65 infection in BALB/c mice. Int. J. Parasitol. 2003, 33, 175–183. [Google Scholar] [CrossRef]
- Dubar, F.; Khalife, J.; Brocard, J.; Dive, D.; Biot, C. Ferroquine, an ingenious antimalarial drug: Thoughts on the mechanism of action. Molecules 2008, 13, 2900–2907. [Google Scholar] [CrossRef] [PubMed]
- Biot, C.; Daher, W.; Chavain, N.; Fandeur, T.; Khalife, J.; Dive, D.; De Clercq, E. Design and synthesis of hydroxyferroquine derivatives with antimalarial and antiviral activities. J. Med. Chem. 2006, 49, 2845–2849. [Google Scholar] [CrossRef] [PubMed]
- Bellot, F.; Cosledan, F.; Vendier, L.; Brocard, J.; Meunier, B.; Robert, A. Trioxaferroquines as new hybrid antimalarial drugs. J. Med. Chem. 2010, 53, 4103–4109. [Google Scholar] [CrossRef] [PubMed]
- Salas, P.F.; Herrmann, C.; Cawthray, J.F.; Nimphius, C.; Kenkel, A.; Chen, J.; De Kock, C.; Smith, P.J.; Patrick, B.O.; Adam, M.J.; et al. Structural characteristics of chloroquine-bridged ferrocenophane analogues of ferroquine may obviate malaria drug resistance mechanisms. J. Med. Chem. 2013, 56, 1596–1613. [Google Scholar] [CrossRef] [PubMed]
- Biot, C.; Dessolin, J.; Ricard, I.; Dive, D. Easily synthesized antimalarial ferrocene triazacyclononane quinoline conjugates. J. Organomet. Chem. 2004, 689, 4678–4682. [Google Scholar] [CrossRef]
- Domarle, O.; Blampain, G.; Agnaniet, H.; Nzadiyabi, T.; Lebibi, J.; Brocard, J.; Maciejewski, L.; Biot, C.; Georges, A.J.; Millet, P. In vitro antimalarial activity of a new organometallic analog, ferrocene-chloroquine. Antimicrob. Agents Chem. 1998, 42, 540–544. [Google Scholar] [CrossRef]
- N’Da, D.D.; Smith, P.J. Synthesis, in vitro antiplasmodial and antiproliferative activities of a series of quinoline–ferrocene hybrids. Med. Chem. Res. 2014, 23, 1214–1224. [Google Scholar] [CrossRef]
- Biot, C.; Pradines, B.; Sergeant, M.H.; Gut, J.; Rosenthal, P.J.; Chibale, K. Design, synthesis, and antimalarial activity of structural chimeras of thiosemicarbazone and ferroquine analogues. Bioorg. Med. Chem. Lett. 2007, 17, 6434–6438. [Google Scholar] [CrossRef]
- David, D.N.; Breytenbach, J.C.; Smith, P.J.; Lategan, C. Synthesis and in vitro antiplasmodial activity of quinoline-ferrocene esters. Arzneimittelforschung 2011, 61, 358–365. [Google Scholar]
- Herrmann, C.; Salas, P.F.; Cawthray, J.F.; de Kock, C.; Patrick, B.O.; Smith, P.J.; Adam, M.J.; Orvig, C. 1,1’-Disubstituted ferrocenyl carbohydrate chloroquine conjugates as potential antimalarials. Organometallics 2012, 31, 5736–5747. [Google Scholar] [CrossRef]
- Herrmann, C.; Salas, P.F.; Patrick, B.O.; Kock, C.D.; Smith, P.J.; Adam, M.J.; Orvig, C. Modular synthesis of 1,2- and 1,10-disubstituted ferrocenyl carbohydrate chloroquine and mefloquine conjugates as potential antimalarial agents. Organometallics 2012, 31, 5748–5759. [Google Scholar] [CrossRef]
- Chavain, N.; Davioud-Charvet, E.; Trivelli, X.; Mbeki, L.; Rottmann, M.; Brun, R.; Biot, C. Antimalarial activities of ferroquine conjugates with either glutathione reductase inhibitors or glutathione depletors via a hydrolyzable amide linker. Bioorg. Med. Chem. 2009, 17, 8048–8059. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.; Fröhlich, T.; Zeino, M.; Zeino, M.; Marschall, M.; Bahsi, H.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Hampel, F.; et al. New efficient artemisinin derived agents against human leukemia cells, human cytomegalovirus and Plasmodium falciparum: 2nd generation 1,2,4-trioxane-ferrocene hybrids. Eur. J. Med. Chem. 2015, 97, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. Artemisinin anti-malarial drugs in China. Acta Pharm. Sin. B 2016, 6, 115–124. [Google Scholar] [CrossRef]
- Reiter, C.; Çapcı Karagöz, A.; Fröhlich, T.; Klein, V.; Zeino, M.; Viertel, K.; Held, J.; Mordmüller, B.; Emirdağ Öztürk, S.; Anıl, H.; et al. Synthesis and study of cytotoxic activity of 1,2,4-trioxane- and egonol-derived hybrid molecules against Plasmodium falciparum and multidrug-resistant human leukemia cells. Eur. J. Med. Chem. 2014, 75, 403–412. [Google Scholar] [CrossRef]
- de Lange, C.; Coertzen, D.; Smit, F.J.; Wentzel, J.F.; Wonga, H.N.; Birkholtz, L.M.; Haynes, R.K.; N’Da, D.D. Synthesis, in vitro antimalarial activities and cytotoxicities of amino-artemisinin-ferrocene derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 289–292. [Google Scholar] [CrossRef]
- Delhaes, L.; Biot, C.; Berry, L.; Maciejewski, L.A.; Camus, D.; Brocard, J.S.; Dive, D. Novel ferrocenic artemisinin derivatives: Synthesis, in vitro antimalarial activity and affinity of binding with ferroprotoporphyrin IX. Bioorg. Med. Chem. 2000, 8, 2739–2745. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef]
- Mbaba, M.; Mabhula, A.N.; Boel, N.; Edkins, A.L.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Ferrocenyl and organic novobiocin derivatives: Synthesis and their in vitro biological activity. J. Inorg. Biochem. 2017, 172, 88–93. [Google Scholar] [CrossRef]
- Mbaba, M.; de la Mare, J.A.; Sterrenberg, J.N.; Kajewole, D.; Maharaj, S.; Edkins, A.L.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Novobiocin–ferrocene conjugates possessing anticancer and antiplasmodial activity independent of HSP90 inhibition. J. Biol. Inorg. Chem. 2019, 24, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Guillon, J.; Mouray, E.; Moreau, S.; Sinou, V.; Forfar, I.; Fabre, S.B.; Desplat, V.; Millet, P.; Parzy, D.; Jarry, C.; et al. New ferrocenic pyrrolo[1,2-a]quinoxaline derivatives: Synthesis, and in vitro antimalarial activity-Part II. Eur. J. Med. Chem. 2011, 46, 2310–2326. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Fong, G.; Liu, J.; Wu, Y.H.; Chang, K.; Park, W.; Kim, J.; Tam, C.; Cheng, L.W.; Land, K.M.; et al. Synthesis and Preliminary Antimicrobial Analysis of Isatin-Ferrocene and Isatin-Ferrocenyl Chalcone Conjugates. ACS Omega 2018, 3, 5808–5813. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Pradines, B.; Madamet, M.; Amalvict, R.; Benoit, N.; Kumar, V. 1H-1,2,3-triazole tethered isatin-ferrocene conjugates: Synthesis and in vitro antimalarial evaluation. Eur. J. Med. Chem. 2014, 87, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Maalik, A.; Noor, T.; Zaidi, A.; Farooq, U.; Bukhari, S.M. Advances in pharmacology of isatin and its derivatives: A review. Trop. J. Pharm. Res. 2015, 14, 1937–1942. [Google Scholar] [CrossRef]
- Chopra, R.; de Kock, C.; Smith, P.; Chibale, K.; Singh, K. Ferrocene-pyrimidine conjugates: Synthesis, electrochemistry, physicochemical properties and antiplasmodial activities. Eur. J. Med. Chem. 2015, 100, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wilairat, P.; Go, M.L. Antimalarial activity of ferrocenyl chalcones. Bioorg. Med. Chem. Lett. 2002, 12, 2299–2302. [Google Scholar] [CrossRef]
- Syahri, J.; Yuanita, E.; Nurohmah, B.A.; Armunanto, R.; Purwono, B. Chalcone analogue as potent anti-malarial compounds against Plasmodium falciparum: Synthesis, biological evaluation, and docking simulation study. Asian Pac. J. Trop. Biomed. 2017, 7, 675–679. [Google Scholar] [CrossRef]
- Kumar, S.; Saini, A.; Gut, J.; Rosenthal, P.J.; Raj, R.; Kumar, V. 4-Aminoquinoline-chalcone/-N-acetylpyrazoline conjugates: Synthesis and antiplasmodial evaluation. Eur. J. Med. Chem. 2017, 138, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, A.; Ocayo, F.; Artigas, V.; Santos, J.C.; Jara-Ulloa, P.; Kahlal, S.; Saillard, J.Y.; Fuentealba, M.; Escobar, C.A. New ferrocenyl-chalcones and bichalcones: Synthesis and characterization. Tetrahedron Lett. 2017, 58, 437–441. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Delavan, B.; Roberts, R.; Tong, W. Lessons learned from two decades of anticancer drugs. Trends Pharm. Sci. 2017, 38, 852–872. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dansette, P.M.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Mansuy, D.; Jaouen, G. A new generation of ferrociphenols leads to a great diversity of reactive metabolites, and exhibits remarkable antiproliferative properties. Chem. Sci. 2017, 9, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Quirante, J.; Dubar, F.; González, A.; Lopez, C.; Cascante, M.; Cortés, R.; Forfar, I.; Pradines, B.; Biot, C. Ferrocene-indole hybrids for cancer and malaria therapy. J. Organomet. Chem. 2011, 696, 1011–1017. [Google Scholar] [CrossRef]
- Radulović, N.S.; Zlatkovic, D.B.; Mitic´, K.V.; Randjelovic, P.J.; Stojanovic, N.M. Synthesis, spectral characterization, cytotoxicity and enzyme-inhibiting activity of new ferrocene-indole hybrids. Polyhedron 2014, 80, 134–141. [Google Scholar] [CrossRef]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Wang, Y.; Pigeon, P.; Mcglinchey, M.J.; Top, S.; Jaouen, G. Synthesis and antiproliferative evaluation of novel hydroxypropyl-ferrociphenol derivatives, resulting from the modification of hydroxyl groups. J. Organomet. Chem. 2017, 829, 108–115. [Google Scholar] [CrossRef]
- Pigeon, P.; Wang, Y.; Top, S.; Najlaoui, F.; Alvarez, M.C.G.; Mcglinchey, M.; Jaouen, G. A new series of succinimido-ferrociphenols and related heterocyclic species induce strong antiproliferative effects, especially against ovarian cancer cells resistant to cisplatin. J. Med. Chem. 2017, 60, 8358–8368. [Google Scholar] [CrossRef]
- Lu, L. Novel Ferrocenyl Peptide Bioconjugates as Anti-Cancer Agents. Doctoral Dissertation, Dublin City University, Dublin, Ireland, 2018. [Google Scholar]
- Zanellato, I.; Heldt, J.M.; Vessières, A.; Jaouen, G.; Osella, D. Antiproliferative effect of ferrocifen drug candidates on malignant pleural mesothelioma cell lines. Inorg. Chim. Acta 2009, 362, 4037–4042. [Google Scholar] [CrossRef]
- Vessieres, A.; Top, S.; Pigeon, P.; Hillard, E.; Boubeker, L.; Spera, D.; Jaouen, G. Modification of the estrogenic properties of diphenols by the incorporation of ferrocene. Generation of antiproliferative effects in vitro. J. Med. Chem. 2005, 48, 3937–3940. [Google Scholar]
- Plazuk, D.; Rychlik, B.; B1auz, A.; Domaga1a, S. Synthesis, electrochemistry and anticancer activity of novel ferrocenyl phenols prepared via azide-alkyne 1,3-cycloaddition reaction. J. Organomet. Chem. 2012, 715, 102–112. [Google Scholar] [CrossRef]
- Pigeon, P.; Top, S.; Zekri, O.; Hillard, E.A.; Vessières, A.; Plamont, M.A.; Buriez, O.; Labbé, E.; Huché, M.; Boutamine, S.; et al. The replacement of a phenol group by an aniline or acetanilide group enhances the cytotoxicity of 2-ferrocenyl-1, 1-diphenyl-but-l-ene compounds against breast cancer cells. J. Organomet. Chem. 2009, 694, 895–901. [Google Scholar] [CrossRef]
- Panaka, S.; Trivedi, R.; Jaipal, K.; Giribabu, L.; Sujitha, P.; Kumar, C.G.; Sridhar, B. Ferrocenyl chalcogeno (sugar) triazole conjugates: Synthesis, characterization and anticancer properties. J. Organomet. Chem. 2016, 813, 125–130. [Google Scholar] [CrossRef]
- De Oliveira, A.C.; Hillard, E.A.; Pigeon, P.; Rocha, D.D.; Rodrigues, F.A.R.; Montenegro, R.C.; Costa-Lotufo, L.V.; Goulart, M.O.F.; Jaouen, G. Biological evaluation of twenty-eight ferrocenyl tetrasubstituted olefins: Cancer cell growth inhibition, ROS production and hemolytic activity. Eur. J. Med. Chem. 2011, 46, 3778–3787. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, J.; Juvekar, A.; Kurane, R.; Khanapure, S.; Salunkhe, R.; Rashinkar, G. Remarkable anti-breast cancer activity of ferrocene tagged multi-functionalized 1,4-dihydropyrimidines. Eur. J. Med. Chem. 2013, 65, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Pérez, W.I.; Soto, Y.; Ortíz, C.; Matta, J.; Meléndez, E. Ferrocenes as potential chemotherapeutic drugs: Synthesis, cytotoxic activity, reactive oxygen species production and micronucleus assay. Bioorg. Med. Chem. 2015, 23, 471–479. [Google Scholar] [CrossRef][Green Version]
- Vera, J.L.; Rullán, J.; Santos, N.; Jiménez, J.; Rivera, J.; Santana, A.; Briggs, J.; Rhenigold, A.L.; Matta, J.; Meléndez, E. Functionalized ferrocenes: The role of the para substituent on the phenoxy pendant group. J. Organomet. Chem. 2014, 749, 204. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.A.; Badshah, A.; Pezzuto, J.M.; Ahmed, N.; Kondratyuk, T.P.; Park, E.J. Ferrocene incorporated selenoureas as anticancer agents. J. Photochem. Photobiol. B Biol. 2015, 148, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Manosroi, J.; Rueanto, K.; Boonpisuttinant, K.; Manosroi, W.; Biot, C.; Akazawa, H.; Akihisa, T.; Issarangporn, W.; Manosroi, A. Novel ferrocenic steroidal drug de- rivatives and their bioactivities. J. Med. Chem. 2010, 53, 3937–3943. [Google Scholar] [CrossRef] [PubMed]
- Schikora, M.; Reznikov, A.; Chaykovskaya, L.; Sachinska, O.; Polyakova, L.; Mokhir, A. Activity of aminoferrocene-based prodrugs against prostate cancer. Bioorg. Med. Chem. Lett. 2015, 25, 3447–3450. [Google Scholar] [CrossRef]
- Ahmad, A.; Mahal, K.; Padhye, S.; Sarkar, F.H.; Schobert, R.; Biersack, B. New ferrocene modified lawsone Mannich bases with anti-proliferative activity against tumor cells. J. Saudi Chem. Soc. 2017, 21, 105–110. [Google Scholar] [CrossRef]



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
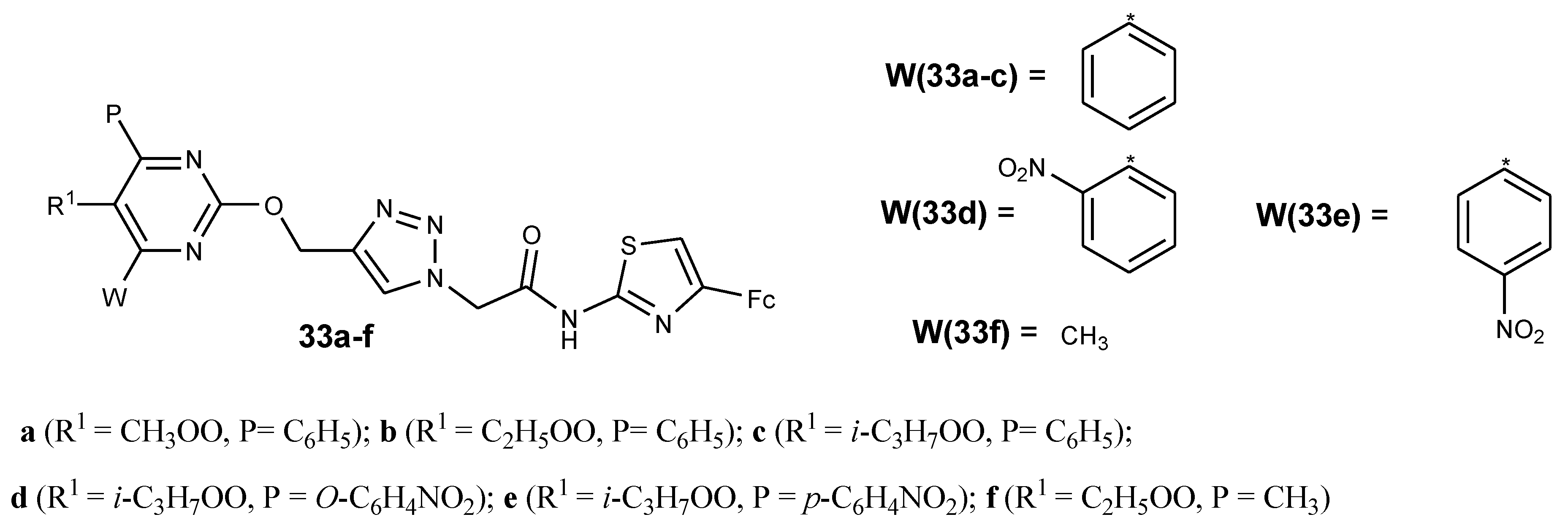{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Values (nM) | ||
|---|---|---|
| Compound | 3D7 | W2 |
| 1a | 15.4 | 133.2 |
| 1b | 21.5 | 30 |
| 1c | 11.7 | 20.4 |
| Ferroquine | 7.8 | 9.7 |
| Chloroquine | 10.6 | 138.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. https://doi.org/10.3390/molecules24193604
Peter S, Aderibigbe BA. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules. 2019; 24(19):3604. https://doi.org/10.3390/molecules24193604
Chicago/Turabian StylePeter, Sijongesonke, and Blessing Atim Aderibigbe. 2019. "Ferrocene-Based Compounds with Antimalaria/Anticancer Activity" Molecules 24, no. 19: 3604. https://doi.org/10.3390/molecules24193604
APA StylePeter, S., & Aderibigbe, B. A. (2019). Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules, 24(19), 3604. https://doi.org/10.3390/molecules24193604