Inside the Black Box: What Makes SELEX Better?


{kind=link}
Abstract
1. Introduction
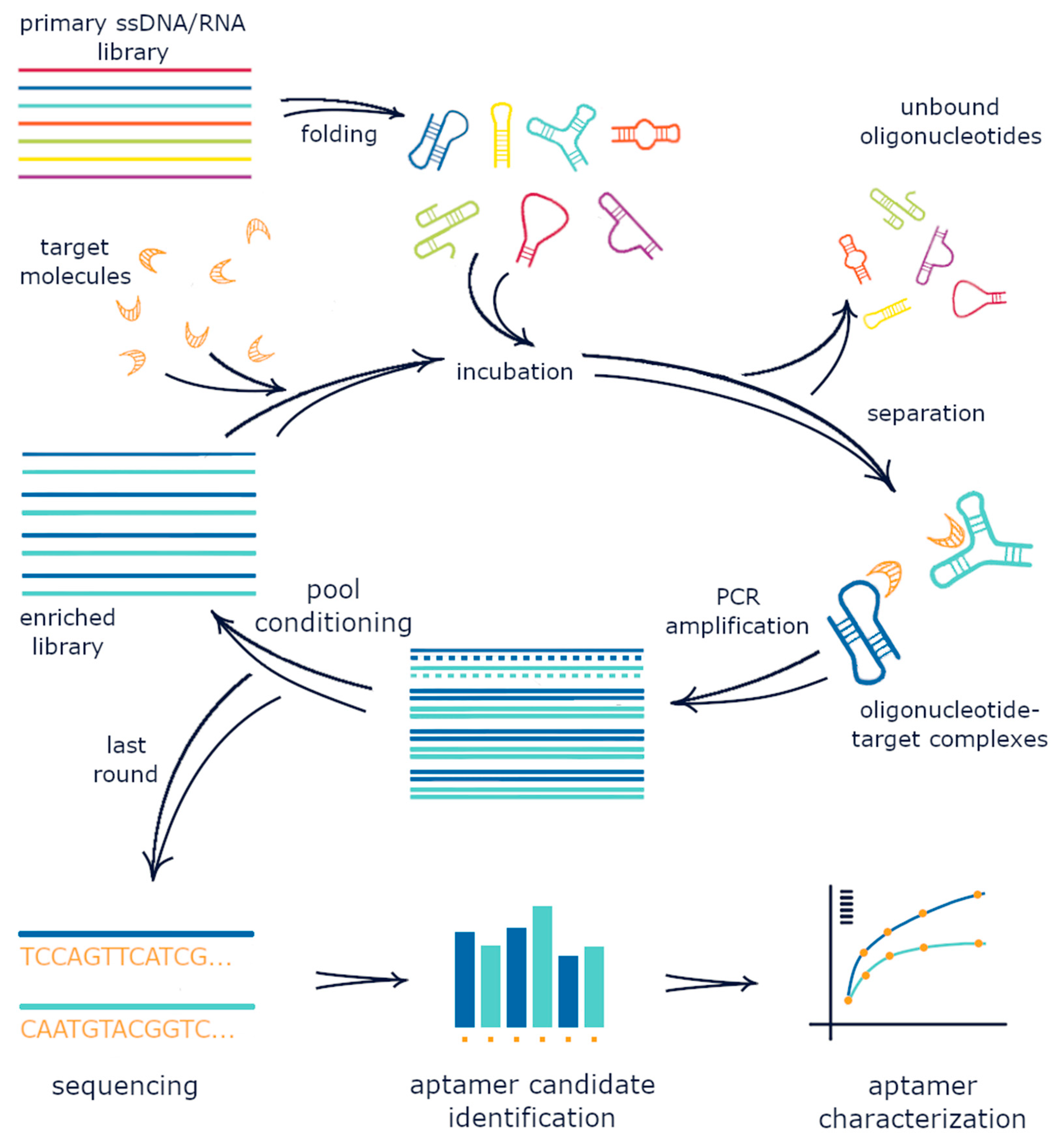
2. The Conventional SELEX Protocol
3. Library Design
3.1. The Length of the Random Region
3.2. Primer-Binding Site Sequences
3.3. Structural Features of the Library
3.4. Chemical Modifications of Nucleic Acids
4. Selection Conditions
4.1. Target (Amount and Concentration)
4.2. Incubation Conditions and the Separation of Target-Bound and Unbound Oligonucleotides
5. Amplification
6. Pool Conditioning
6.1. Transcription to RNA
6.2. ssDNA Regeneration
7. Monitoring of Selection Progress and Selection Cycle Number
8. Sequencing and Candidate Aptamer Identification (Sanger vs. Next-Generation Sequencing (NGS))
9. Aptamer Characterization
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaur, H.; Bruno, J.G.; Kumar, A.; Sharma, T.K. Aptamers in the Therapeutics and Diagnostics Pipelines. Theranostics 2018, 8, 4016–4032. [Google Scholar] [CrossRef] [PubMed]
- Ilgu, M.; Nilsen-Hamilton, M. Aptamers in analytics. Analyst 2016, 141, 1551–1568. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; Derosa, M.C. Challenges and opportunities for small molecule aptamer development. J. Nucleic Acids 2012, 2012, 748913. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Bae, H.; Ren, S.; Kang, J.; Kim, M.; Jiang, Y.; Jin, M.M.; Min, I.M.; Kim, S. Sol-Gel SELEX Circumventing Chemical Conjugation of Low Molecular Weight Metabolites Discovers Aptamers Selective to Xanthine. Nucleic Acid Ther. 2013, 23, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, E.; Foley, J.H.; Conway, E.M.; Haynes, C. Hi-Fi SELEX: A High-Fidelity Digital-PCR Based Therapeutic Aptamer Discovery Platform. Biotechnol. Bioeng. 2015, 112, 1506–1522. [Google Scholar] [CrossRef]
- Pestourie, C.; Cerchia, L.; Gombert, K.; Aissouni, Y.; Boulay, J.; De Franciscis, V.; Libri, D.; Tavitian, B.; Ducongé, F. Comparison of Different Strategies to Select Aptamers Against a Transmembrane Protein Target. Oligonucleotides 2006, 16, 323–335. [Google Scholar] [CrossRef]
- Hamula, C.L.A.; Peng, H.; Wang, Z.; Newbigging, A.M.; Tyrrell, G.J.; Li, X.F.; Le, X.C. The Effects of SELEX Conditions on the Resultant Aptamer Pools in the Selection of Aptamers Binding to Bacterial Cells. J. Mol. Evol. 2015, 81, 194–209. [Google Scholar] [CrossRef]
- Hall, B.; Micheletti, J.M.; Satya, P.; Ogle, K.; Pollard, J.; Ellington, A.D. Design, Synthesis, and Amplification of DNA Pools for In Vitro Selection. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Takahashi, M.; Wu, X.; Ho, M.; Chomchan, P.; Rossi, J.J.; Burnett, J.C.; Zhou, J. High throughput sequencing analysis of RNA libraries reveals the influences of initial library and PCR methods on SELEX efficiency. Sci. Rep. 2016, 6, 33697. [Google Scholar] [CrossRef]
- Wilson, R.; Bourne, C.; Chaudhuri, R.R.; Gregory, R.; Kenny, J.; Cossins, A. Single-Step Selection of Bivalent Aptamers Validated by Comparison with SELEX Using High-Throughput Sequencing. PLoS ONE 2014, 9, 100572. [Google Scholar] [CrossRef] [PubMed]
- Vanbrabant, J.; Leirs, K.; Vanschoenbeek, K.; Lammertyn, J.; Michiels, L. reMelting curve analysis as a tool for enrichment monitoring in the SELEX process. Analyst 2014, 139, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.; Smith, D. Estimation of SELEX pool size by measurement of DNA renaturation rates. RNA 1999, 5, 1326–1332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schütze, T.; Arndt, P.F.; Menger, M.; Wochner, A.; Vingron, M.; Erdmann, V.A.; Lehrach, H.; Kaps, C.; Glökler, J. A calibrated diversity assay for nucleic acid libraries using DiStRO—a Diversity Standard of Random Oligonucleotides. Nucleic Acids Res. 2010, 38, 23. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Vorobyeva, M.; Davydova, A.; Vorobjev, P.; Pyshnyi, D.; Venyaminova, A. Key Aspects of Nucleic Acid Library Design for in Vitro Selection. Int. J. Mol. Sci. 2018, 19, 470. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T. Aptamers and SELEX: The technology. World Pat. Inf. 2003, 25, 123–129. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef]
- Salehi-Ashtiani, K.; Szostak, J.W. In vitro evolution suggests multiple origins for the hammerhead ribozyme. Nature 2001, 414, 82–84. [Google Scholar] [CrossRef]
- Lozupone, C. Selection of the simplest RNA that binds isoleucine. RNA 2003, 9, 1315–1322. [Google Scholar] [CrossRef]
- Legiewicz, M. Size, constant sequences, and optimal selection. RNA 2005, 11, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Coleman, T.M.; Huang, F. RNA-Catalyzed Thioester Synthesis. Chem. Biol. 2002, 9, 1227–1236. [Google Scholar] [CrossRef]
- Coleman, T.M.; Huang, F. Optimal Random Libraries for the Isolation of Catalytic RNA. RNA Biol. 2005, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Conaty, J.; Hendry, P.; Lockett, T. Selected classes of minimised hammerhead ribozyme have very high cleavage rates at low Mg2+ concentration. Nucleic Acids Res. 1999, 27, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Cowperthwaite, M.C.; Ellington, A.D. Bioinformatic Analysis of the Contribution of Primer Sequences to Aptamer Structures. J. Mol. Evol. 2008, 67, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Musheev, M.U.; Krylov, S.N. Selection of aptamers by systematic evolution of ligands by exponential enrichment: Addressing the polymerase chain reaction issue. Anal. Chim. Acta 2006, 564, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Hesselberth, J.R.; Miller, D.; Robertus, J.; Ellington, A.D. In Vitro Selection of RNA Molecules That Inhibit the Activity of Ricin A-chain. J. Biol. Chem. 2000, 275, 4937–4942. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Stewart, C.A.; Mcnamara, J.O., II; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and Optimization of Murine IL-10 Receptor Blocking Oligonucleotide Aptamers Using High-throughput Sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef]
- Hicke, B.J.; Marion, C.; Chang, Y.F.; Gould, T.; Lynott, C.K.; Parma, D.; Schmidt, P.G.; Warren, S. Tenascin-C Aptamers Are Generated Using Tumor Cells and Purified Protein. J. Biol. Chem. 2001, 276, 48644–48654. [Google Scholar] [CrossRef]
- Pan, W.; Xin, P.; Clawson, G.A. Minimal primer and primer-free SELEX protocols for selection of aptamers from random DNA libraries. Biotechniques 2008, 44, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.D. Selection of genomic sequences that bind tightly to Ff gene 5 protein: Primer-free genomic SELEX. Nucleic Acids Res. 2004, 32, 182. [Google Scholar] [CrossRef] [PubMed]
- Legiewicz, M.; Yarus, M. A More Complex Isoleucine Aptamer with a Cognate Triplet. J. Biol. Chem. 2005, 280, 19815–19822. [Google Scholar] [CrossRef] [PubMed]
- Connell, G.J.; Illangesekare, M.; Yarus, M. Three small ribooligonucleotides with specific arginine sites. Biochemistry 1993, 32, 5497–5502. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Khrapov, M.; Shaw, C.A. The scene of a frozen accident. RNA 2000, 6, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Clawson, G. The Shorter the Better: Reducing Fixed Primer Regions of Oligonucleotide Libraries for Aptamer Selection. Molecules 2009, 14, 1353–1369. [Google Scholar] [CrossRef] [PubMed]
- Tolle, F.; Wilke, J.; Wengel, J.; Mayer, G. By-Product Formation in Repetitive PCR Amplification of DNA Libraries during SELEX. PLoS ONE 2014, 9, 114693. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.K.; Chou, C.F.; Chen, L.C. Selection of aptamers for AMACR detection from DNA libraries with different primers. RSC Adv. 2018, 8, 19067–19074. [Google Scholar] [CrossRef]
- Boiziau, C.; Toulmé, J.J. A Method to Select Chemically Modified Aptamers Directly. Antisense Nucleic Acid Drug Dev. 2001, 11, 379–385. [Google Scholar] [CrossRef]
- Vater, A. Short bioactive Spiegelmers to migraine-associated calcitonin gene-related peptide rapidly identified by a novel approach: Tailored-SELEX. Nucleic Acids Res. 2003, 31, 130. [Google Scholar] [CrossRef]
- Eulberg, D. Development of an automated in vitro selection protocol to obtain RNA-based aptamers: Identification of a biostable substance P antagonist. Nucleic Acids Res. 2005, 33, 45. [Google Scholar] [CrossRef] [PubMed]
- Jarosch, F. In vitro selection using a dual RNA library that allows primerless selection. Nucleic Acids Res. 2006, 34, 86. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.T.; DeStefano, J.J. A primer-free method that selects high-affinity single-stranded DNA aptamers using thermostable RNA ligase. Anal. Biochem. 2011, 414, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.M.; Lai, J.C.; Horng, H.E.; Liu, T.C.; Hong, C.Y. Generation of Aptamers from A Primer-Free Randomized ssDNA Library Using Magnetic-Assisted Rapid Aptamer Selection. Sci. Rep. 2017, 7, 45478. [Google Scholar] [CrossRef] [PubMed]
- Shtatland, T. Interactions of Escherichia coli RNA with bacteriophage MS2 coat protein: Genomic SELEX. Nucleic Acids Res. 2000, 28, 93. [Google Scholar] [CrossRef] [PubMed]
- Hamm, J. Characterisation of Antibody-Binding RNAs Selected from Structurally Constrained Libraries. Nucleic Acids Res. 1996, 24, 2220–2227. [Google Scholar] [CrossRef]
- Ouellet, E.; Lagally, E.T.; Cheung, K.C.; Haynes, C.A. A simple method for eliminating fixed-region interference of aptamer binding during SELEX. Biotechnol. Bioeng. 2014, 111, 2265–2279. [Google Scholar] [CrossRef]
- Oh, S.S.; Plakos, K.; Lou, X.; Xiao, Y.; Soh, H.T. In vitro selection of structure-switching, self-reporting aptamers. Proc. Natl. Acad. Sci. USA 2010, 107, 14053–14058. [Google Scholar] [CrossRef]
- Gevertz, J. In vitro RNA random pools are not structurally diverse: A computational analysis. RNA 2005, 11, 853–863. [Google Scholar] [CrossRef]
- Davis, J.H.; Szostak, J.W. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc. Natl. Acad. Sci. USA 2002, 99, 11616–11621. [Google Scholar] [CrossRef]
- Zhu, L.; Li, C.; Zhu, Z.; Liu, D.; Zou, Y.; Wang, C.; Fu, H.; Yang, C.J. In Vitro Selection of Highly Efficient G-Quadruplex-Based DNAzymes. Anal. Chem. 2012, 84, 8383–8390. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; McKeague, M.; Pitre, S.; Dumontier, M.; Green, J.; Golshani, A.; Derosa, M.C.; Dehne, F. Computational approaches toward the design of pools for the in vitro selection of complex aptamers. RNA 2010, 16, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Shin, J.S.; Elmetwaly, S.; Gan, H.H.; Schlick, T. RAGPOOLS: RNA-As-Graph-Pools a web server for assisting the design of structured RNA pools for in vitro selection. Bioinformatics 2007, 23, 2959–2960. [Google Scholar] [CrossRef] [PubMed]
- Carothers, J.M.; Oestreich, S.C.; Davis, J.H.; Szostak, J.W. Informational Complexity and Functional Activity of RNA Structures. J. Am. Chem. Soc. 2004, 126, 5130–5137. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Gan, H.H.; Schlick, T. A computational proposal for designing structured RNA pools for in vitro selection of RNAs. RNA 2007, 13, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Snyder, T.M.; Liu, D.R. Enhanced Functional Potential of Nucleic Acid Aptamer Libraries Patterned to Increase Secondary Structure. J. Am. Chem. Soc. 2010, 132, 9453–9464. [Google Scholar] [CrossRef] [PubMed]
- Carothers, J.M. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA 2006, 12, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.A.; Pei, R.; Stefanovic, D.; Stojanovic, M.N. Optimizing Cross-reactivity with Evolutionary Search for Sensors. J. Am. Chem. Soc. 2012, 134, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Trevino, S.G.; Levy, M. High-Throughput Bead-Based Identification of Structure-Switching Aptamer Beacons. ChemBioChem 2014, 15, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Lipi, F.; Chen, S.; Chakravarthy, M.; Rakesh, S.; Veedu, R.N. In vitro evolution of chemically-modified nucleic acid aptamers: Pros and cons, and comprehensive selection strategies. RNA Biol. 2016, 13, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Lapa, S.A.; Chudinov, A.V.; Timofeev, E.N. The Toolbox for Modified Aptamers. Mol. Biotechnol. 2016, 58, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Diafa, S.; Hollenstein, M. Generation of Aptamers with an Expanded Chemical Repertoire. Molecules 2015, 20, 16643–16671. [Google Scholar] [CrossRef] [PubMed]
- Stovall, G.M.; Bedenbaugh, R.S.; Singh, S.; Meyer, A.J.; Hatala, P.J.; Ellington, A.D.; Hall, B. In Vitro Selection Using Modified or Unnatural Nucleotides. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Li, M.; Lin, N.; Huang, Z.; Du, L.; Altier, C.; Fang, H.; Wang, B. Selecting Aptamers for a Glycoprotein through the Incorporation of the Boronic Acid Moiety. J. Am. Chem. Soc. 2008, 130, 12636–12638. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y.; Kasahara, Y.; Fujita, H.; Kitadume, S.; Ozaki, H.; Endoh, T.; Kuwahara, M.; Sugimoto, N. Efficacy of Base-Modification on Target Binding of Small Molecule DNA Aptamers. J. Am. Chem. Soc. 2013, 135, 9412–9419. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J. Aptamer-Based Multiplexed Proteomic Technology for Biomarker Discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [PubMed]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar] [CrossRef]
- Irvine, D.; Tuerk, C.; Gold, L. Selexion: Systematic evolution of ligands by exponential enrichment with integrated optimization by non-linear analysis. J. Mol. Biol. 1991, 222, 739–761. [Google Scholar] [CrossRef]
- Levine, H.A.; Nilsen-Hamilton, M. A mathematical analysis of SELEX. Comput. Biol. Chem. 2007, 31, 11–35. [Google Scholar] [CrossRef]
- Levine, H.A.; Seo, Y.J. Discrete Dynamical Systems in Multiple Target and Alternate SELEX. SIAM J. Appl. Dyn. Syst. 2015, 14, 1048–1101. [Google Scholar] [CrossRef]
- Sun, F.; Galas, D.; Waterman, M.S. A Mathematical Analysis ofin VitroMolecular Selection – Amplification. J. Mol. Biol. 1996, 258, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rudzinski, J.F.; Gong, Q.; Soh, H.T.; Atzberger, P.J. Influence of Target Concentration and Background Binding on In Vitro Selection of Affinity Reagents. PLoS ONE 2012, 7, e43940. [Google Scholar] [CrossRef] [PubMed]
- Aita, T.; Nishigaki, K.; Husimi, Y. Theoretical consideration of selective enrichment in in vitro selection: Optimal concentration of target molecules. Math. Biosci. 2012, 240, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K. Complex SELEX against target mixture: Stochastic computer model, simulation, and analysis. Comput. Methods Programs Biomed. 2007, 87, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Vant-Hull, B.; Payano-Baez, A.; Davis, R.H.; Gold, L. The mathematics of SELEX against complex targets. J. Mol. Biol. 1998, 278, 579–597. [Google Scholar] [CrossRef]
- Chen, C.K.; Kuo, T.L.; Chan, P.C.; Lin, L.Y. Subtractive SELEX against two heterogeneous target samples: Numerical simulations and analysis. Comput. Biol. Med. 2007, 37, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; White, B.S.; Lis, J.T.; Shalloway, D. Density-dependent cooperative non-specific binding in solid-phase SELEX affinity selection. Nucleic Acids Res. 2013, 41, 7167–7175. [Google Scholar] [CrossRef]
- Gopinath, S.C.B. Methods developed for SELEX. Anal. Bioanal. Chem. 2006, 387, 171–182. [Google Scholar] [CrossRef]
- Zhuo, Z.; Yu, Y.; Wang, M.; Li, J.; Zhang, Z.; Liu, J.; Wu, X.; Lu, A.; Zhang, G.; Zhang, B. Recent Advances in SELEX Technology and Aptamer Applications in Biomedicine. Int. J. Mol. Sci. 2017, 18, 2142. [Google Scholar] [CrossRef]
- Darmostuk, M.; Rimpelova, S.; Gbelcova, H.; Ruml, T. Current approaches in SELEX: An update to aptamer selection technology. Biotechnol. Adv. 2015, 33, 1141–1161. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, Z.; Wang, S.; Wu, Y.; Ma, Y.; Liu, J. Introduction of SELEX and Important SELEX Variants. In Aptamers for Analytical Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp. 1–25. [Google Scholar]
- Zhang, Y.; Lai, B.; Juhas, M. Recent Advances in Aptamer Discovery and Applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Pagano, J.M.; Lis, J.T. New Technologies Provide Quantum Changes in the Scale, Speed, and Success of SELEX Methods and Aptamer Characterization. Mol. Ther. Nucleic Acids 2014, 3, e183. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H. Recent developments in cell-SELEX technology for aptamer selection. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Nikolaus, N.; Strehlitz, B. Capture-SELEX: Selection of DNA Aptamers for Aminoglycoside Antibiotics. J. Anal. Methods Chem. 2012, 2012, 415697. [Google Scholar] [CrossRef]
- Mondal, B.; Ramlal, S.; Lavu, P.S.R.; Murali, H.S.; Batra, H.V. A combinatorial systematic evolution of ligands by exponential enrichment method for selection of aptamer against protein targets. Appl. Microbiol. Biotechnol. 2015, 99, 9791–9803. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cload, S.T.; Hamaguchi, N.; Kurz, J.; Keene, S.; Kurz, M.; Boomer, R.M.; Blanchard, J.; Epstein, D.; Wilson, C.; et al. ADP-Specific Sensors Enable Universal Assay of Protein Kinase Activity. Chem. Biol. 2004, 11, 499–508. [Google Scholar] [CrossRef]
- Shao, K.; Ding, W.; Wang, F.; Li, H.; Ma, D.; Wang, H. Emulsion PCR: A High Efficient Way of PCR Amplification of Random DNA Libraries in Aptamer Selection. PLoS ONE 2011, 6, e24910. [Google Scholar] [CrossRef]
- Williams, R.; Peisajovich, S.G.; Miller, O.J.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Amplification of complex gene libraries by emulsion PCR. Nat. Methods 2006, 3, 545–550. [Google Scholar] [CrossRef]
- Day, D. Identification of non-amplifying CYP21 genes when using PCR-based diagnosis of 21-hydroxylase deficiency in congenital adrenal hyperplasia (CAH) affected pedigrees. Hum. Mol. Genet. 1996, 5, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX Process with Next-Generation Sequencing. PLoS ONE 2011, 6, e29604. [Google Scholar] [CrossRef] [PubMed]
- Czerny, T. High Primer Concentration Improves PCR Amplification from Random Pools. Nucleic Acids Res. 1996, 24, 985–986. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Schütze, T.; Rubelt, F.; Repkow, J.; Greiner, N.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Glökler, J. A streamlined protocol for emulsion polymerase chain reaction and subsequent purification. Anal. Biochem. 2011, 410, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Levay, A.; Brenneman, R.; Hoinka, J.; Sant, D.; Cardone, M.; Trinchieri, G.; Przytycka, T.M.; Berezhnoy, A. Identifying high-affinity aptamer ligands with defined cross-reactivity using high-throughput guided systematic evolution of ligands by exponential enrichment. Nucleic Acids Res. 2015, 43, 82. [Google Scholar] [CrossRef]
- Meyerhans, A.; Vartanian, J.P.; Wain-Hobson, S. DNA recombination during PCR. Nucleic Acids Res. 1990, 18, 1687–1691. [Google Scholar] [CrossRef]
- Zhu, Z.; Jenkins, G.; Zhang, W.; Zhang, M.; Guan, Z.; Yang, C.J. Single-molecule emulsion PCR in microfluidic droplets. Anal. Bioanal. Chem. 2012, 403, 2127–2143. [Google Scholar] [CrossRef]
- Witt, M.; Phung, N.L.; Stalke, A.; Walter, J.G.; Stahl, F.; von Neuhoff, N.; Scheper, T. Comparing two conventional methods of emulsion PCR and optimizing of Tegosoft-based emulsion PCR. Eng. Life Sci. 2017, 17, 953–958. [Google Scholar] [CrossRef]
- Yufa, R.; Krylova, S.M.; Bruce, C.; Bagg, E.A.; Schofield, C.J.; Krylov, S.N. Emulsion PCR Significantly Improves Nonequilibrium Capillary Electrophoresis of Equilibrium Mixtures-Based Aptamer Selection: Allowing for Efficient and Rapid Selection of Aptamer to Unmodified ABH2 Protein. Anal. Chem. 2015, 87, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Hirabayashi, N.; Kato, S.; Akitomi, J.; Egashira, H.; Tanaka, T.; Waga, I.; Ohtsu, T. Effective isolation of RNA aptamer through suppression of PCR bias. Biochem. Biophys. Res. Commun. 2009, 386, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Wyatt Thiel, K.; Dassie, J.P.; Rockey, W.M.; Howell, C.A.; Liu, X.Y.; Dupuy, A.J.; Huang, L.; Owczarzy, R.; et al. Nucleotide Bias Observed with a Short SELEX RNA Aptamer Library. Nucleic Acid Ther. 2011, 21, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, C.; Tang, T.H.; Tominaga, J.; Tan, S.C.; Gopinath, S.C.B. Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst 2012, 137, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Svobodová, M.; Pinto, A.; Nadal, P.; O’ Sullivan, C.K. Comparison of different methods for generation of single-stranded DNA for SELEX processes. Anal. Bioanal. Chem. 2012, 404, 835–842. [Google Scholar] [CrossRef]
- Paul, A.; Avci-Adali, M.; Ziemer, G.; Wendel, H.P. Streptavidin-Coated Magnetic Beads for DNA Strand Separation Implicate a Multitude of Problems During Cell-SELEX. Oligonucleotides 2009, 19, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R. Preparation of Single-Stranded DNA from PCR Products with Streptavidin Magnetic Beads. Nucleic Acid Ther. 2011, 21, 437–440. [Google Scholar] [CrossRef]
- Hamedani, N.S.; Blümke, F.; Tolle, F.; Rohrbach, F.; Rühl, H.; Oldenburg, J.; Mayer, G.; Pötzsch, B.; Müller, J. Capture and Release (CaR): A simplified procedure for one-tube isolation and concentration of single-stranded DNA during SELEX. Chem. Commun. 2015, 51, 1135–1138. [Google Scholar] [CrossRef]
- Sheng, Y.; Bowser, M.T. Isolating single stranded DNA using a microfluidic dialysis device. Analyst 2014, 139, 215–224. [Google Scholar] [CrossRef]
- Liang, C.; Li, D.; Zhang, G.; Li, H.; Shao, N.; Liang, Z.; Zhang, L.; Lu, A.; Zhang, G. Comparison of the methods for generating single-stranded DNA in SELEX. Analyst 2015, 140, 3439–3444. [Google Scholar] [CrossRef]
- Damase, T.R.; Ellington, A.D.; Allen, P.B. Purification of single-stranded DNA by co-polymerization with acrylamide and electrophoresis. Biotechniques 2017, 62, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K. The enzymatic basis of processivity in lambda exonuclease. Nucleic Acids Res. 2003, 31, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Komarova, N.V.; Glukhov, S.I.; Andrianova, M.S.; Kuznetsov, A.E. Use of the Cy3 and Cy5 Fluorescent Labels to Protect a DNA Strand from Degradation under λ Exonuclease Treatment. Moscow Univ. Chem. Bull. 2018, 73, 19–26. [Google Scholar] [CrossRef]
- Citartan, M.; Tang, T.H.; Tan, S.C.; Gopinath, S.C.B. Conditions optimized for the preparation of single-stranded DNA (ssDNA) employing lambda exonuclease digestion in generating DNA aptamer. World J. Microbiol. Biotechnol. 2010, 27, 1167–1173. [Google Scholar] [CrossRef]
- Citartan, M.; Tang, T.H.; Tan, S.C.; Hoe, C.H.; Saini, R.; Tominaga, J.; Gopinath, S.C.B. Asymmetric PCR for good quality ssDNA generation towards DNA aptamer production. Songklanakarin J. Sci. Technol. 2012, 34, 125–132. [Google Scholar]
- He, C.Z.; Zhang, K.H.; Wang, T.; Wan, Q.S.; Hu, P.P.; Hu, M.D.; Huang, D.Q.; Lv, N.H. Single-primer-limited amplification: A method to generate random single-stranded DNA sub-library for aptamer selection. Anal. Biochem. 2013, 440, 63–70. [Google Scholar] [CrossRef]
- Heiat, M.; Ranjbar, R.; Latifi, A.M.; Rasaee, M.J.; Farnoosh, G. Essential strategies to optimize asymmetric PCR conditions as a reliable method to generate large amount of ssDNA aptamers. Biotechnol. Appl. Biochem. 2017, 64, 541–548. [Google Scholar] [CrossRef]
- Tolnai, Z.; Harkai, Á.; Szeitner, Z.; Scholz, É.N.; Percze, K.; Gyurkovics, A.; Mészáros, T. A simple modification increases specificity and efficiency of asymmetric PCR. Anal. Chim. Acta 2019, 1047, 225–230. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Paul, A.; Wilhelm, N.; Ziemer, G.; Wendel, H.P. Upgrading SELEX technology by using lambda exonuclease digestion for single-stranded DNA generation. Molecules 2009, 15, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H.; Zhou, H.; Wu, F.; Su, Y.; Liang, Y.; Zhou, D. Indirect purification method provides high yield and quality ssDNA sublibrary for potential aptamer selection. Anal. Biochem. 2015, 476, 84–90. [Google Scholar] [CrossRef]
- Civit, L.; Fragoso, A.; O’Sullivan, C.K. Evaluation of techniques for generation of single-stranded DNA for quantitative detection. Anal. Biochem. 2012, 431, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Wang, C.H.; Hsu, K.F.; Chou, C.Y.; Lee, G.B. An on-chip Cell-SELEX process for automatic selection of high-affinity aptamers specific to different histologically classified ovarian cancer cells. Lab Chip 2014, 14, 4017–4028. [Google Scholar] [CrossRef] [PubMed]
- Spiga, F.M.; Maietta, P.; Guiducci, C. More DNA-Aptamers for Small Drugs: A Capture-SELEX Coupled with Surface Plasmon Resonance and High-Throughput Sequencing. ACS Comb. Sci. 2015, 17, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Li, H.; Wilkop, T.; Liu, X.; Yu, X.; Cheng, Q.; Xu, D.; Chen, H.Y. Silver decahedral nanoparticles empowered SPR imaging-SELEX for high throughput screening of aptamers with real-time assessment. Biosens. Bioelectron. 2018, 109, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Wochner, A.; Glökler, J. Nonradioactive fluorescence microtiter plate assay monitoring aptamer selections. Biotechniques 2007, 42, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. FluMag-SELEX as an advantageous method for DNA aptamer selection. Anal. Bioanal. Chem. 2005, 383, 83–91. [Google Scholar] [CrossRef]
- Yang, D.K.; Chen, L.C.; Lee, M.Y.; Hsu, C.H.; Chen, C.S. Selection of aptamers for fluorescent detection of alpha-methylacyl-CoA racemase by single-bead SELEX. Biosens. Bioelectron. 2014, 62, 106–112. [Google Scholar] [CrossRef]
- Shangguan, D.; Bing, T.; Zhang, N. Cell-SELEX: Aptamer Selection Against Whole Cells. In Aptamers Selected by Cell-SELEX for Theranostics; Springer: Berlin, Germany, 2015; pp. 13–33. [Google Scholar]
- Kunii, T.; Ogura, S.; Mie, M.; Kobatake, E. Selection of DNA aptamers recognizing small cell lung cancer using living cell-SELEX. Analyst 2011, 136, 1310. [Google Scholar] [CrossRef]
- Nabavinia, M.S.; Charbgoo, F.; Alibolandi, M.; Mosaffa, F.; Gholoobi, A.; Ramezani, M.; Abnous, K. Comparison of Flow Cytometry and ELASA for Screening of Proper Candidate Aptamer in Cell-SELEX Pool. Appl. Biochem. Biotechnol. 2018, 184, 444–452. [Google Scholar] [CrossRef]
- Dastjerdi, K.; Tabar, G.H.; Dehghani, H.; Haghparast, A. Generation of an enriched pool of DNA aptamers for an HER2-overexpressing cell line selected by Cell SELEX. Biotechnol. Appl. Biochem. 2011, 58, 226–230. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Wilhelm, N.; Perle, N.; Stoll, H.; Schlensak, C.; Wendel, H.P. Absolute Quantification of Cell-Bound DNA Aptamers During SELEX. Nucleic Acid Ther. 2013, 23, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G.; Ahmed, M.-S.L.; Dolf, A.; Endl, E.; Knolle, P.A.; Famulok, M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat. Protoc. 2010, 5, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; He, L.; Wang, J.; Fang, X.; Zhang, L. Developing a combined strategy for monitoring the progress of aptamer selection. Analyst 2017, 142, 3136–3139. [Google Scholar] [CrossRef] [PubMed]
- Amano, R.; Aoki, K.; Miyakawa, S.; Nakamura, Y.; Kozu, T.; Kawai, G.; Sakamoto, T. NMR monitoring of the SELEX process to confirm enrichment of structured RNA. Sci. Rep. 2017, 7, 283. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; El-Maarri, O.; Oldenburg, J.; Pötzsch, B.; Mayer, G. Monitoring the progression of the in vitro selection of nucleic acid aptamers by denaturing high-performance liquid chromatography. Anal. Bioanal. Chem. 2008, 390, 1033–1037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mencin, N.; Šmuc, T.; Vraničar, M.; Mavri, J.; Hren, M.; Galeša, K.; Krkoč, P.; Ulrich, H.; Šolar, B. Optimization of SELEX: Comparison of different methods for monitoring the progress of in vitro selection of aptamers. J. Pharm. Biomed. Anal. 2014, 91, 151–159. [Google Scholar] [CrossRef]
- Gu, G.; Wang, T.; Yang, Y.; Xu, X.; Wang, J. An Improved SELEX-Seq Strategy for Characterizing DNA-Binding Specificity of Transcription Factor: NF-κB as an Example. PLoS ONE 2013, 8, e76109. [Google Scholar] [CrossRef]
- Amano, R.; Takada, K.; Tanaka, Y.; Nakamura, Y.; Kawai, G.; Kozu, T.; Sakamoto, T. Kinetic and Thermodynamic Analyses of Interaction between a High-Affinity RNA Aptamer and Its Target Protein. Biochemistry 2016, 55, 6221–6229. [Google Scholar] [CrossRef]
- Civit, L.; Taghdisi, S.M.; Jonczyk, A.; Haßel, S.K.; Gröber, C.; Blank, M.; Stunden, H.J.; Beyer, M.; Schultze, J.; Latz, E.; et al. Systematic evaluation of cell-SELEX enriched aptamers binding to breast cancer cells. Biochimie 2018, 145, 53–62. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Strehlitz, B. Refining the Results of a Classical SELEX Experiment by Expanding the Sequence Data Set of an Aptamer Pool Selected for Protein A. Int. J. Mol. Sci. 2018, 19, 642. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Schubert, T.; Strehlitz, B. In vitro Selection and Interaction Studies of a DNA Aptamer Targeting Protein A. PLoS ONE 2015, 10, e0134403. [Google Scholar] [CrossRef] [PubMed]
- Blank, M. Next-Generation Analysis of Deep Sequencing Data: Bringing Light into the Black Box of SELEX Experiments. In Nucleic Acid Aptamers. Selection, Characterization, and Application; Humana Press: New York, NY, USA, 2016; pp. 85–95. [Google Scholar]
- McKeague, M.; De Girolamo, A.; Valenzano, S.; Pascale, M.; Ruscito, A.; Velu, R.; Frost, N.R.; Hill, K.; Smith, M.; McConnell, E.M.; et al. Comprehensive Analytical Comparison of Strategies Used for Small Molecule Aptamer Evaluation. Anal. Chem. 2015, 87, 8608–8612. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598. https://doi.org/10.3390/molecules24193598
Komarova N, Kuznetsov A. Inside the Black Box: What Makes SELEX Better? Molecules. 2019; 24(19):3598. https://doi.org/10.3390/molecules24193598
Chicago/Turabian StyleKomarova, Natalia, and Alexander Kuznetsov. 2019. "Inside the Black Box: What Makes SELEX Better?" Molecules 24, no. 19: 3598. https://doi.org/10.3390/molecules24193598
APA StyleKomarova, N., & Kuznetsov, A. (2019). Inside the Black Box: What Makes SELEX Better? Molecules, 24(19), 3598. https://doi.org/10.3390/molecules24193598