
Investigation of 5’-Norcarbocyclic Nucleoside Analogues as Antiprotozoal and Antibacterial Agents

 , , , and
, , , and 
Abstract
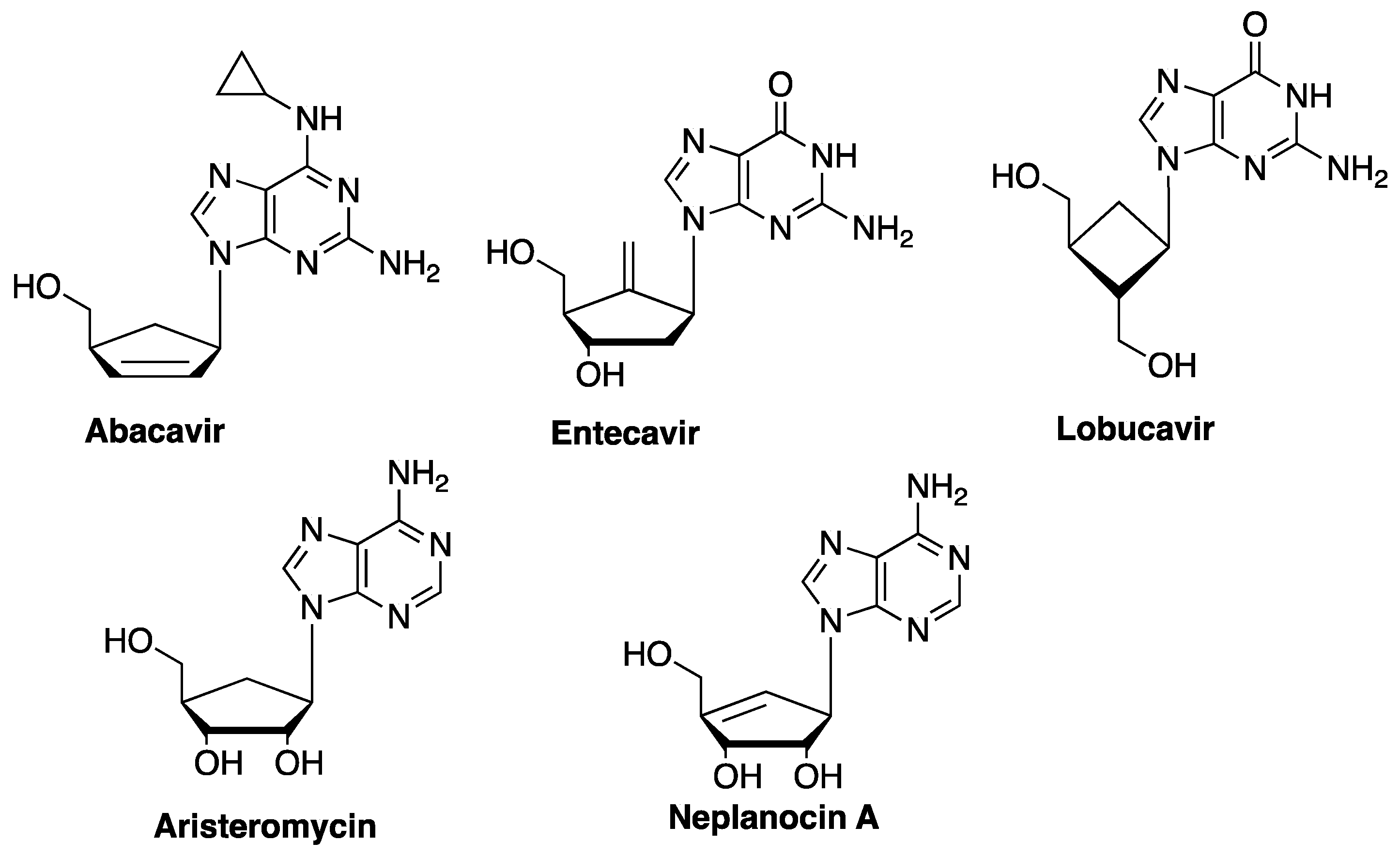
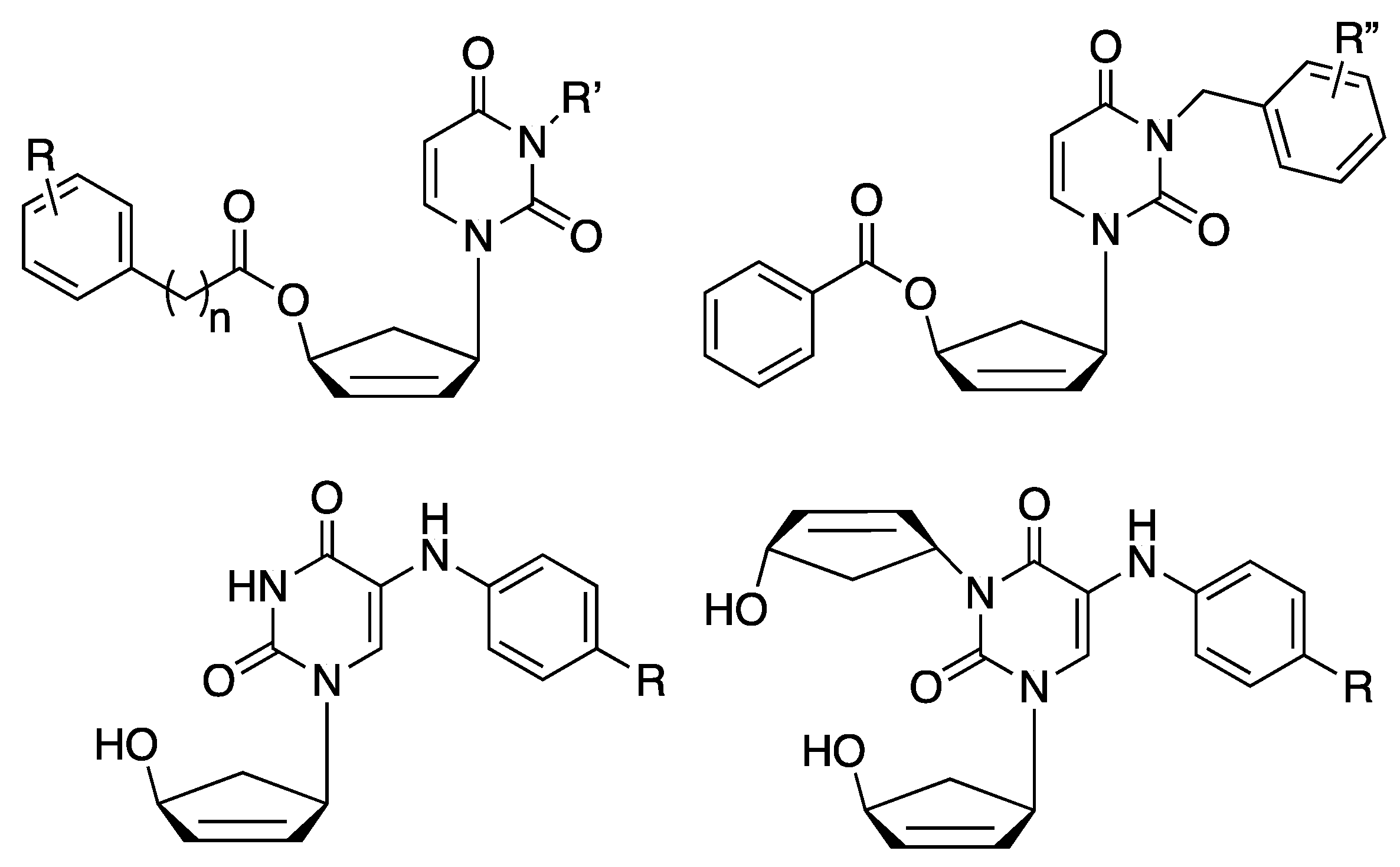
1. Introduction
2. Results
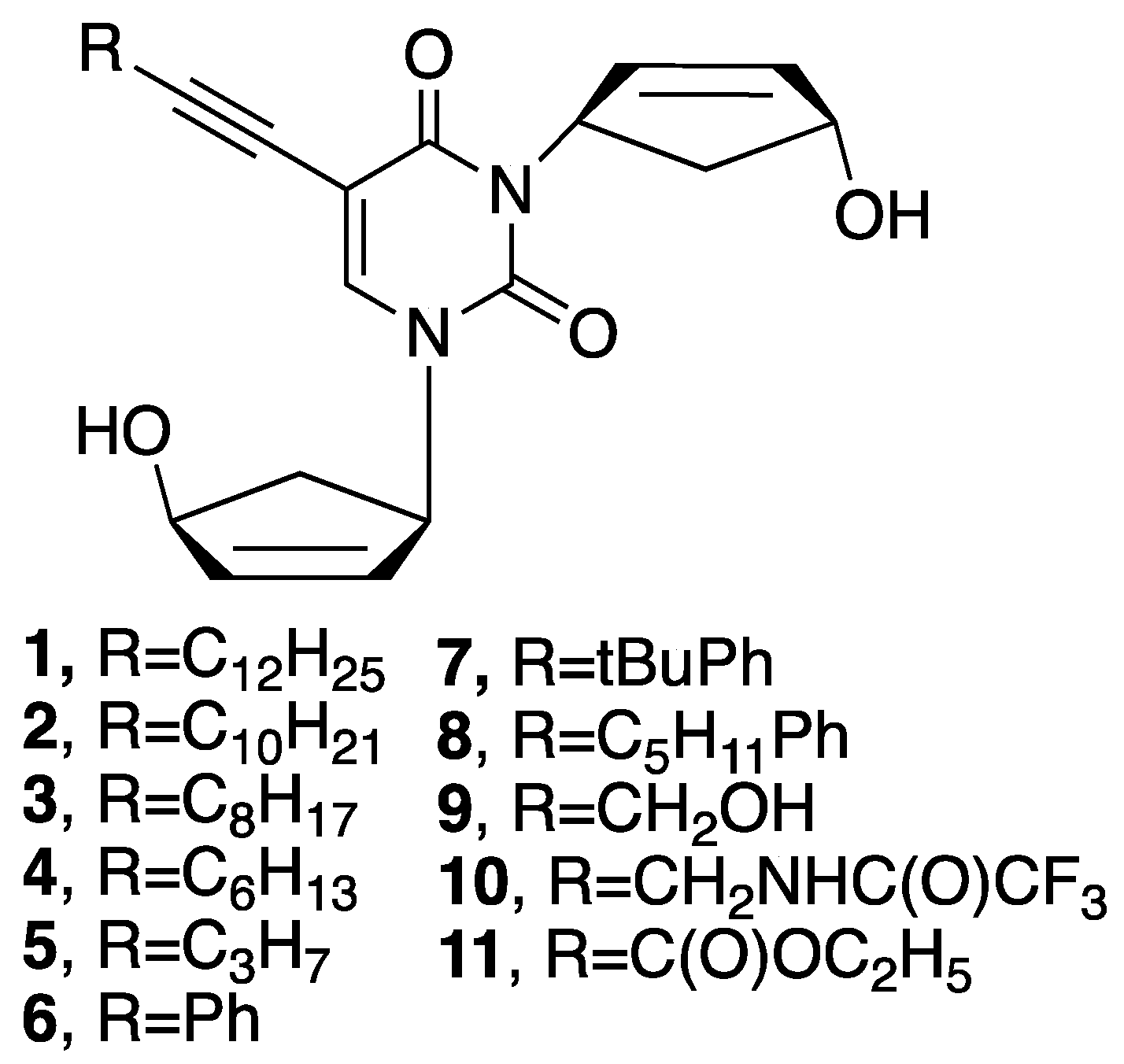
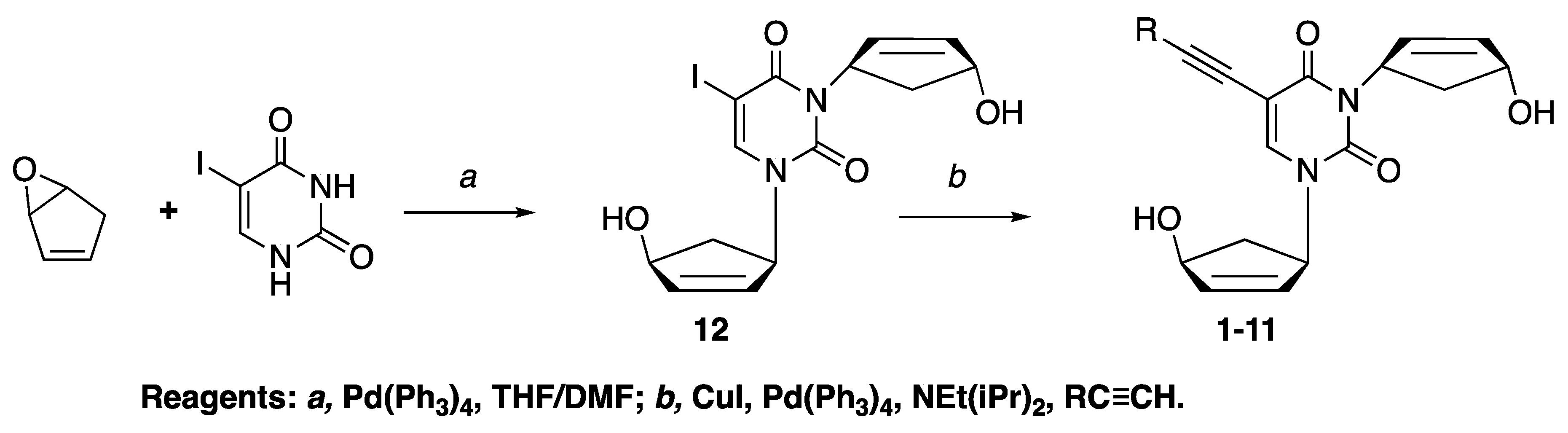
2.1. Chemistry
2.2. Biological Evaluation
3. Discussion
4. Materials and Methods
4.1. Chemistry Experimental
4.1.1. General
4.1.2. Compound Synthesis and Characterization
4.2. Antiparasite Experiments
4.2.1. Parasite Strains and Cultures
Trypanosoma brucei brucei
Leishmania mexicana
Trichomonas vaginalis
4.2.2. Determination of EC50 Values for Protozoan Parasites
Trypanosoma brucei
Leishmania mexicana
Trichomonas vaginalis
4.3. Antibacterial Experiments
4.3.1. Bacterial Strains
4.3.2. Determination of Minimal Inhibitory Concentration Against M. tuberculosis ATCC 25,177
4.3.3. Determination of Minimal Inhibitory Concentration Against M. bovis ATCC 35,737
4.3.4. Antituberculosis Tests with Virulent Strains of M. tuberculosis
4.4. Evaluation of Cytotoxicity
4.4.1. U-937 Cells
4.4.2. Evaluation of U-937 Cytotoxicity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.S.; Khandazhinskaya, A.L.; Kochetkov, S.N. Carbocyclic nucleoside analogues: Classification, target enzymes, mechanisms of action and synthesis. Russ. Chem. Rev. 2012, 81, 729–746. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Valuev-Elliston, V.T.; Babkov, D.A.; Novikov, M.S.; Ivanov, A.V.; Kochetkov, S.N.; Balzarini, J.; Seley-Radtke, K.L.; Khandazhinskaya, A.L. 5′-Nor carbocyclic nucleosides: Unusual nonnucleoside inhibitors of HIV-1 reverse transcriptase. Medchemcomm 2013, 4, 741–748. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Valuev-Elliston, V.T.; Geisman, A.N.; Novikov, M.S.; Chizhov, A.O.; Kochetkov, S.N.; Seley-Radtke, K.L.; Khandazhinskaya, A.L. Structure-activity evaluation of new uracil-based non-nucleoside inhibitors of HIV reverse transcriptase. Medchemcomm 2013, 4, 1443–1451. [Google Scholar] [CrossRef]
- Matyugina, E.; Novikov, M.; Babkov, D.; Ozerov, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Karpenko, I.; Chizhov, A.; Murthu, P.; et al. 5-Arylaminouracil Derivatives: New Inhibitors of Mycobacterium tuberculosis. Chem. Biol. Drug Des. 2015, 86, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.; Khandazhinskaya, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Chizhov, A.; Karpenko, I.; Kochetkov, S.; Alexandrova, L. The synthesis and antituberculosis activity of 5′-nor carbocyclic uracil derivatives. Bioorg. Med. Chem. 2012, 20, 6680–6686. [Google Scholar] [CrossRef]
- Alzahrani, K.J.; Matyugina, E.S.; Khandazhinskaya, A.L.; Kochetkov, S.N.; Seley-Radtke, K.L.; Koning, H.P. Evaluation of the antiprotozoan properties of 5′-norcarbocyclic pyrimidine nucleosides. Bioorg. Med. Chem. Lett. 2017, 27, 3081–3086. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Logashenko, E.B.; Zenkova, M.A.; Kochetkov, S.N.; Khandazhinskaya, A.L. 5′-Norcarbocyclic analogues of furano [2,3-d]pyrimidine nucleosides. Heterocycl. Commun. 2015, 21, 259–262. [Google Scholar] [CrossRef]
- Agrofoglio, L.; Suhas, E.; Farese, A.; Condom, R.; Challand, S.R.; Earl, R.A.; Guedj, R. Synthesis of carbocyclic nucleosides. Tetrahedron 1994, 50, 10611–10670. [Google Scholar] [CrossRef]
- Agrofoglio, L.A.; Gillaizeau, I.; Saito, Y. Palladium-Assisted Routes to Nucleosides. Chem. Rev. 2003, 103, 1875–1916. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.A.M.; Tagoe, D.N.A.; Munday, J.C.; Donachie, A.; Morrison, L.J.; de Koning, H.P. Pyrimidine Biosynthesis Is Not an Essential Function for Trypanosoma brucei Bloodstream Forms. PLOS ONE 2013, 8, e58034. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.A.M.; Creek, D.J.; Burgess, K.; Allison, H.C.; Field, M.C.; Maser, P.; De Koning, H.P. Pyrimidine Salvage in Trypanosoma brucei Bloodstream Forms and the Trypanocidal Action of Halogenated Pyrimidines. Mol. Pharmacol. 2013, 83, 439–453. [Google Scholar] [CrossRef]
- Hughes, D.; Andersson, D.I. Evolutionary Trajectories to Antibiotic Resistance. Annu. Rev. Microbiol. 2017, 71, 579–596. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P. Drug resistance in protozoan parasites. Ermg. Top. Life Sci. 2017, 1, 627–632. [Google Scholar] [CrossRef]
- Paine, M.F.; Wang, M.Z.; Generaux, C.N.; Boykin, D.W.; Wilson, W.D.; De Koning, H.P.; Olson, C.A.; Pohlig, G.; Burri, C.; Brun, R.; et al. Diamidines for human African trypanosomiasis. Curr. Opin. Investig. Drugs 2010, 11, 876–883. [Google Scholar]
- Millan, C.R.; Acosta-Reyes, F.J.; Lagartera, L.; Ebiloma, G.U.; Lemgruber, L.; Martinez, J.J.N.; Saperas, N.; Dardonville, C.; de Koning, H.P.; Campos, J.L. Functional and structural analysis of AT-specific minor groove binders that disrupt DNA-protein interactions and cause disintegration of the Trypanosoma brucei kinetoplast. Nucl. Acids Res. 2017, 45, 8378–8391. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Csh. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.H. Tetracycline Antibiotics and Resistance. Csh. Perspect. Med. 2016, 6, a025387. [Google Scholar] [CrossRef]
- Bridges, D.J.; Gould, M.K.; Nerima, B.; Maser, P.; Burchmore, R.J.S.; de Koning, H.P. Loss of the high-affinity pentamidine transporter is responsible for high levels of cross-resistance between arsenical and diamidine drugs in African trypanosomes. Mol. Pharmacol. 2007, 71, 1098–1108. [Google Scholar] [CrossRef]
- Teka, I.A.; Kazibwe, A.J.N.; El-Sabbagh, N.; Al-Salabi, M.I.; Ward, C.P.; Eze, A.A.; Munday, J.C.; Maser, P.; Matovu, E.; Barrett, M.P.; et al. The Diamidine Diminazene Aceturate Is a Substrate for the High-Affinity Pentamidine Transporter: Implications for the Development of High Resistance Levels in Trypanosomes. Mol. Pharmacol. 2011, 80, 110–116. [Google Scholar] [CrossRef]
- Ward, C.P.; Wong, P.E.; Burchmore, R.J.; de Koning, H.P.; Barrett, M.P. Trypanocidal Furamidine Analogues: Influence of Pyridine Nitrogens on Trypanocidal Activity, Transport Kinetics, and Resistance Patterns. Antimicrob. Agents Chemother. 2011, 55, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Matovu, E.; Stewart, M.L.; Geiser, F.; Brun, R.; Maser, P.; Wallace, L.J.M.; Burchmore, R.J.; Enyaru, J.C.K.; Barrett, M.P.; Kaminsky, R.; et al. Mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot. Cell 2003, 2, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Geiser, F.; Luscher, A.; de Koning, H.P.; Seebeck, T.; Maser, P. Molecular pharmacology of adenosine transport in Trypanosoma brucei: P1/P2 revisited. Mol. Pharmacol. 2005, 68, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Vodnala, S.K.; Lundback, T.; Yeheskieli, E.; Sjoberg, B.; Gustavsson, A.L.; Svensson, R.; Olivera, G.C.; Eze, A.A.; de Koning, H.P.; Hammarstrom, L.G.J.; et al. Structure-Activity Relationships of Synthetic Cordycepin Analogues as Experimental Therapeutics for African Trypanosomiasis. J. Med. Chem. 2013, 56, 9861–9873. [Google Scholar] [CrossRef]
- Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Andres, D.A.; Natto, M.J.; Teka, I.A.; McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [Google Scholar] [CrossRef]
- Baker, N.; de Koning, H.P.; Maser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef]
- Munday, J.C.; Tagoe, D.N.A.; Eze, A.A.; Krezdorn, J.A.M.; Lopez, K.E.R.; Alkhaldi, A.A.M.; McDonald, F.; Still, J.; Alzahrani, K.J.; Settimo, L.; et al. Functional analysis of drug resistance-associated mutations in the Trypanosoma brucei adenosine transporter 1 (TbAT1) and the proposal of a structural model for the protein. Mol. Microbiol. 2015, 96, 887–900. [Google Scholar] [CrossRef]
- Delespaux, V.; De Koning, H.P. Transporters in antiparasitic drug development and resistance. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery; Jäger, T., Koch, O., Flohe, L., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; pp. 335–349. [Google Scholar]
- Zoltner, M.; Horn, D.; de Koning, H.P.; Field, M.C. Exploiting the Achilles’ heel of membrane trafficking in trypanosomes. Curr. Opin. Microbiol. 2016, 34, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.S.; Barrett, M.P. A Molecular Mechanism for Eflornithine Resistance in African Trypanosomes. PLOS Pathog. 2010, 6, e1001204. [Google Scholar] [CrossRef]
- De Koning, H.P.; Jarvis, S.M. A highly Selective, High Affinity Transporter for Uracil in Trypanosoma brucei brucei; Evidence for Proton-Dependent Transport. Biochem. Cell Biol. 1998, 76, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, I.G.; Yakob, L.; Al Salabi, M.I.; Diallinas, G.; Soteriadou, K.P.; De Koning, H.P. Identification of the first pyrimidine nucleobase transporter in Leishmania: Similarities with the Trypanosoma brucei U1 transporter and antileishmanial activity of uracil analogues. Parasitology 2005, 130, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Ad. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Meco-Navas, A.; Ebiloma, G.U.; Martin-Dominguez, A.; Martinez-Benayas, I.; Cueto-Diaz, E.J.; Alhejely, A.S.; Balogun, E.O.; Saito, M.; Matsui, M.; Arai, N.; et al. SAR of 4-Alkoxybenzoic Acid Inhibitors of the Trypanosome Alternative Oxidase. ACS Med. Chem. Lett. 2018, 9, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Ebiloma, G.U.; Ayuga, T.D.; Balogun, E.O.; Gil, L.A.; Donachie, A.; Kaiser, M.; Herraiz, T.; Inaoka, D.K.; Shiba, T.; Harada, S.; et al. Inhibition of trypanosome alternative oxidase without its N-terminal mitochondrial targeting signal (Delta MTS-TAO) by cationic and non-cationic 4-hydroxybenzoate and 4-alkoxybenzaldehyde derivatives active against T-brucei and T-congolense. Eur. J. Med. Chem. 2018, 150, 385–402. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P.; MacLeod, A.; Barrett, M.P.; Cover, B.; Jarvis, S.M. Further evidence for a link between melarsoprol resistance and P2 transporter function in African trypanosomes. Mol. Biochem. Parasitol. 2000, 106, 181–185. [Google Scholar] [CrossRef]
- Cerone, M.; Uliassi, E.; Prati, F.; Ebiloma, G.U.; Lemgruber, L.; Bergamini, C.; Watson, D.G.; Ferreira, T.D.M.; Cardoso, G.S.H.R.; Romeiro, L.A.S.; et al. Discovery of Sustainable Drugs for Neglected Tropical Diseases: Cashew Nut Shell Liquid (CNSL)-Based Hybrids Target Mitochondrial Function and ATP Production in Trypanosoma brucei. Chemmedchem 2019, 14, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Al-Salabi, M.I.; Wallace, L.J.M.; De Koning, H.P. A Leishmania major nucleobase transporter responsible for allopurinol uptake is a functional homolog of the Trypanosoma brucei H2 transporter. Mol. Pharmacol. 2003, 63, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Natto, M.J.; Savioli, F.; Quashie, N.B.; Dardonville, C.; Rodenko, B.; de Koning, H.P. Validation of novel fluorescence assays for the routine screening of drug susceptibilities of Trichomonas vaginalis. J. Antimicrob. Chemother. 2012, 67, 933–943. [Google Scholar] [CrossRef]
- Siheri, W.; Zhang, T.; Ebiloma, G.U.; Biddau, M.; Woods, N.; Hussain, M.Y.; Clements, C.J.; Fearnley, J.; Ebel, R.E.; Paget, T.; et al. Chemical and Antimicrobial Profiling of Propolis from Different Regions within Libya. PLOS ONE 2016, 11, e0155355. [Google Scholar] [CrossRef]
- Gould, M.K.; Vu, X.L.; Seebeck, T.; de Koning, H.P. Propidium iodide-based methods for monitoring drug action in the kinetoplastidae: Comparison with the Alamar Blue assay. Anal. Biochem. 2008, 382, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Natto, M.J.; Eze, A.A.; de Koning, H.P. Protocols for the routine screening of drug sensitivity in the human parasite Trichomonas vaginalis. Methods Mol. Biol. 2015, 1263, 103–110. [Google Scholar] [PubMed]
- Siddiqui, S.H.; Rusch-Gerdes, S. MGIT Procedure Manual; Foundation for Innovative New Diagnostics: Geneva, Switzerland, 2006. [Google Scholar]
- Shakya, N.; Garg, G.; Agrawal, B.; Kumar, R. Chemotherapeutic interventions against tuberculosis. Pharmaceuticals 2012, 5, 690–718. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | T. Brucei, EC50, µM | U-937 Cells | |||
|---|---|---|---|---|---|
| S427-WT | B48 | 2T1 PYR6-5 −/− | Tbb-5FURes | TC50, µM | |
| 1 | 8.0 ± 0.1 | 9.0 ± 0.1 | 9.2 ± 0.4 | 7.5 ± 0.1 | 66.3 |
| 2 | 3.8 ± 0.1 | 4.0 ± 0.4 | 5.5 ± 0.5 * | 5.2 ± 0.6 | 22.1 |
| 3 | 13.2 ± 0.6 | 14.7 ± 0.2 | 17.1 ± 1.2 * | 19.9 ± 0.7 *** | 2.2 |
| 4 | 13.6 ± 0.1 | 17.7 ± 1.0 * | 17.5 ± 1.2 * | 24.9 ± 0.5 *** | 73.8 |
| 5 | NE 1 | NE | NE | NE | 88.5 |
| 6 | NE | NE | NE | NE | 166.2 |
| 7 | 8.7 ± 0.4 | 10.8 ± 0.6 * | 14.7 ± 0.4 *** | 12.6 ± 1.4 | 22.5 |
| 8 | 7.8 ± 0.3 | 11.0 ± 0.6 * | 15.4 ± 0.7 *** | 12.0 ± 1.2 * | 17.3 |
| 9 | 66.8 ± 6.6 | 76.8 ± 4.4 ** | 59.9 ± 1.0 | 74.9 ± 3.5 | >100 |
| 10 | NE | NE | NE | NE | 78.0 |
| 11 | NE | NE | NE | NE | 21.6 |
| 5-Fluorouracil | 87 ± 6.4 | 101 ± 7 | 2.33 ± 0.35 *** | 2497 ± 76 *** | ND 2 |
| Pentamidine | 0.0032 ± 0.0004 | 0.71 ± 0.03 *** | 0.0031 ± 0.0004 | 0.0034 ± 0.0004 | ND |
| N9 | ND | ND | ND | ND | 6.5 µg/mL |
| Compound | L. Mexicana WT, EC50, µM | L. Mexicana 5FUR, EC50, µM | Trichomonas Vaginalis, EC50, µM |
|---|---|---|---|
| 1 | 11.8 ± 0.3 | 11.4 ± 0.3 | ND |
| 2 | 28.3 ± 3.0 | 51.1 ± 0.6 ** | 48.9 ± 0.05 |
| 3 | 13.8 ± 1.7 | 25.8 ± 0.4 *** | 32.0 ± 0.001 |
| 4 | 45.6 ± 0.9 | 47.3 ± 0.4 | 54.1 ± 0.002 |
| 5 | >100 | >100 | 73.5 ± 0.08 |
| 6 | NE 1 | NE | 70.2 ± 0.06 |
| 7 | 21.1 ± 2.9 | 45.3 ± 2.2 *** | 52.8 ± 0.007 |
| 8 | 13.0 ± 1.5 | 27.5 ± 2.3 ** | 36.9 ± 0.18 |
| 9 | NE | NE | 84.8 ± 0.004 |
| 10 | NE | NE | 69.2 ± 0.05 |
| 11 | NE | NE | 53.8 ± 0.74 |
| 5-Fluorouracil | 8.7 ± 2.1 | 3516 ± 440 *** | ND |
| Pentamidine | 1.2 ± 0.8 | 1.63 ± 0.18 | ND |
| Metronidazole | ND | ND | 0.53 ± 0.10 |
| Compound | M. Tuberculosis 25,177 | |
|---|---|---|
| MIC, µM | MIC, µg/mL | |
| 1 | 250 | 117.3 |
| 2 | 31 | 13.6 |
| 3 | 31 | 12.8 |
| 4 | 31 | 11.9 |
| 5 | >1000 | >342.4 |
| 6 | 500 | 188.2 |
| 7 | 62.5 | 27.0 |
| 8 | 31 | 13.9 |
| 9 | 1000 | 330.3 |
| 10 | >1000 | >425.4 |
| 11 | 1000 | 372.4 |
| DMSO, % | >5 | >0.05 |
| Kanamycin | 6.5 | 3.13 |
| Rifampicin | 0.19 | 0.16 |
| Compound | M. Tuberculosis H37Rv MIC, µg/mL | M. Tuberculosis MS-115 MIC, µg/mL |
|---|---|---|
| 1 | 50 | >50 |
| 2 | >50 | ND |
| 3 | 50 | 50 |
| 4 | >50 | ND |
| 7 | >50 | ND |
| 8 | >50 | ND |
| Rifampicin | 1 | >50 |
| Isoniazid | 0.1 | >100 |
| Levofloxacin | 1.5 | 1.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khandazhinskaya, A.L.; Matyugina, E.S.; Solyev, P.N.; Wilkinson, M.; Buckheit, K.W.; Buckheit, R.W., Jr.; Chernousova, L.N.; Smirnova, T.G.; Andreevskaya, S.N.; Alzahrani, K.J.; et al. Investigation of 5’-Norcarbocyclic Nucleoside Analogues as Antiprotozoal and Antibacterial Agents. Molecules 2019, 24, 3433. https://doi.org/10.3390/molecules24193433
Khandazhinskaya AL, Matyugina ES, Solyev PN, Wilkinson M, Buckheit KW, Buckheit RW Jr., Chernousova LN, Smirnova TG, Andreevskaya SN, Alzahrani KJ, et al. Investigation of 5’-Norcarbocyclic Nucleoside Analogues as Antiprotozoal and Antibacterial Agents. Molecules. 2019; 24(19):3433. https://doi.org/10.3390/molecules24193433
Chicago/Turabian StyleKhandazhinskaya, Anastasia L., Elena S. Matyugina, Pavel N. Solyev, Maggie Wilkinson, Karen W. Buckheit, Robert W. Buckheit, Jr., Larisa N. Chernousova, Tatiana G. Smirnova, Sofya N. Andreevskaya, Khalid J. Alzahrani, and et al. 2019. "Investigation of 5’-Norcarbocyclic Nucleoside Analogues as Antiprotozoal and Antibacterial Agents" Molecules 24, no. 19: 3433. https://doi.org/10.3390/molecules24193433
APA StyleKhandazhinskaya, A. L., Matyugina, E. S., Solyev, P. N., Wilkinson, M., Buckheit, K. W., Buckheit, R. W., Jr., Chernousova, L. N., Smirnova, T. G., Andreevskaya, S. N., Alzahrani, K. J., Natto, M. J., Kochetkov, S. N., de Koning, H. P., & Seley-Radtke, K. L. (2019). Investigation of 5’-Norcarbocyclic Nucleoside Analogues as Antiprotozoal and Antibacterial Agents. Molecules, 24(19), 3433. https://doi.org/10.3390/molecules24193433