
Development of a Fragment-Based Screening Assay for the Focal Adhesion Targeting Domain Using SPR and NMR

 ,
,
Abstract
1. Introduction
2. Results
2.1. Development of a Surface Plasmon Resonance Screening Assay for the FAT Fomain
2.2. Optimization of SPR Assay Buffer Conditions
2.3. Comparison of OneStep vs. Fixed Concentration Injection Kinetics
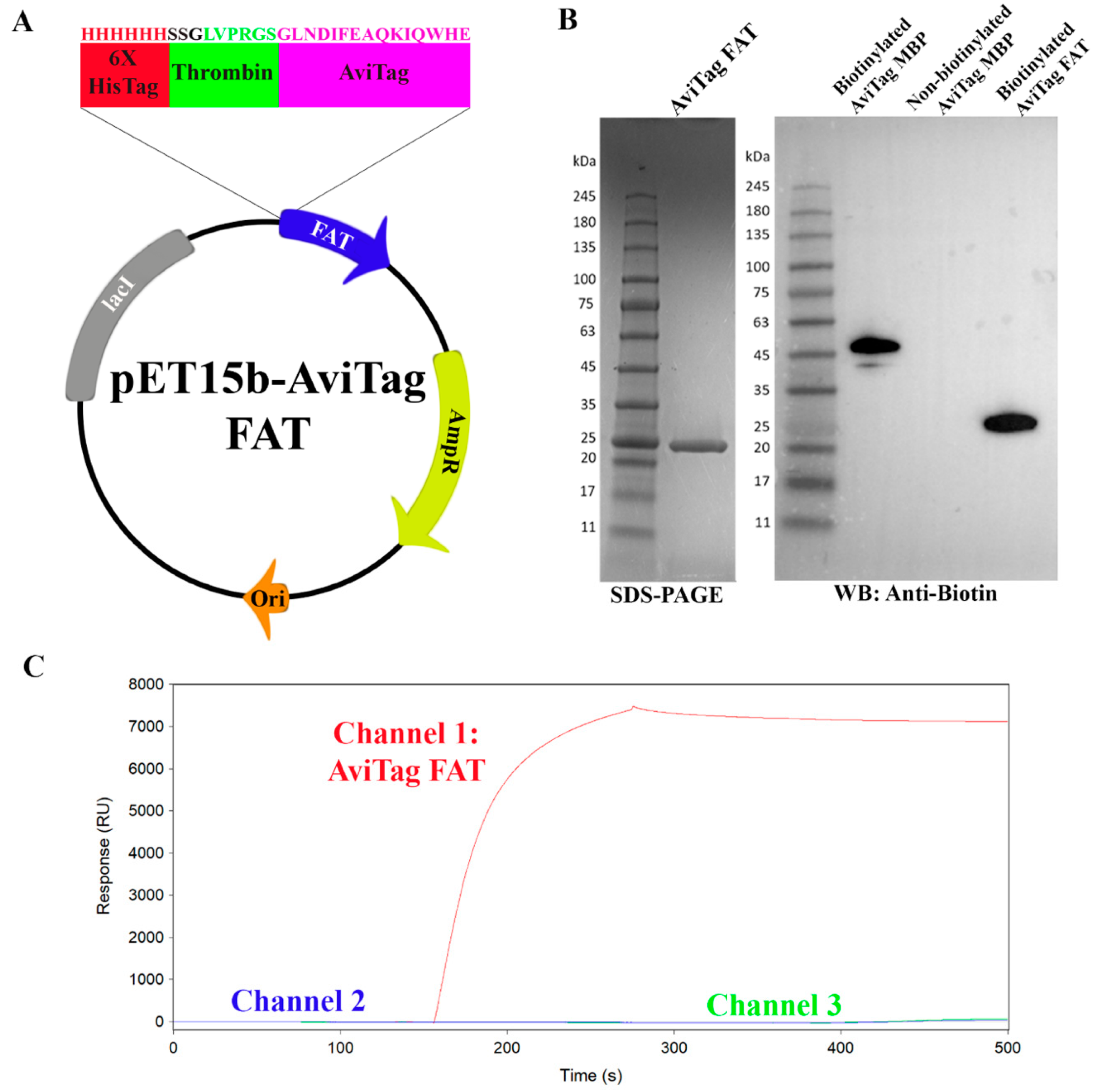
2.4. Mutant AviTag FAT Constructs for SPR Assay Controls
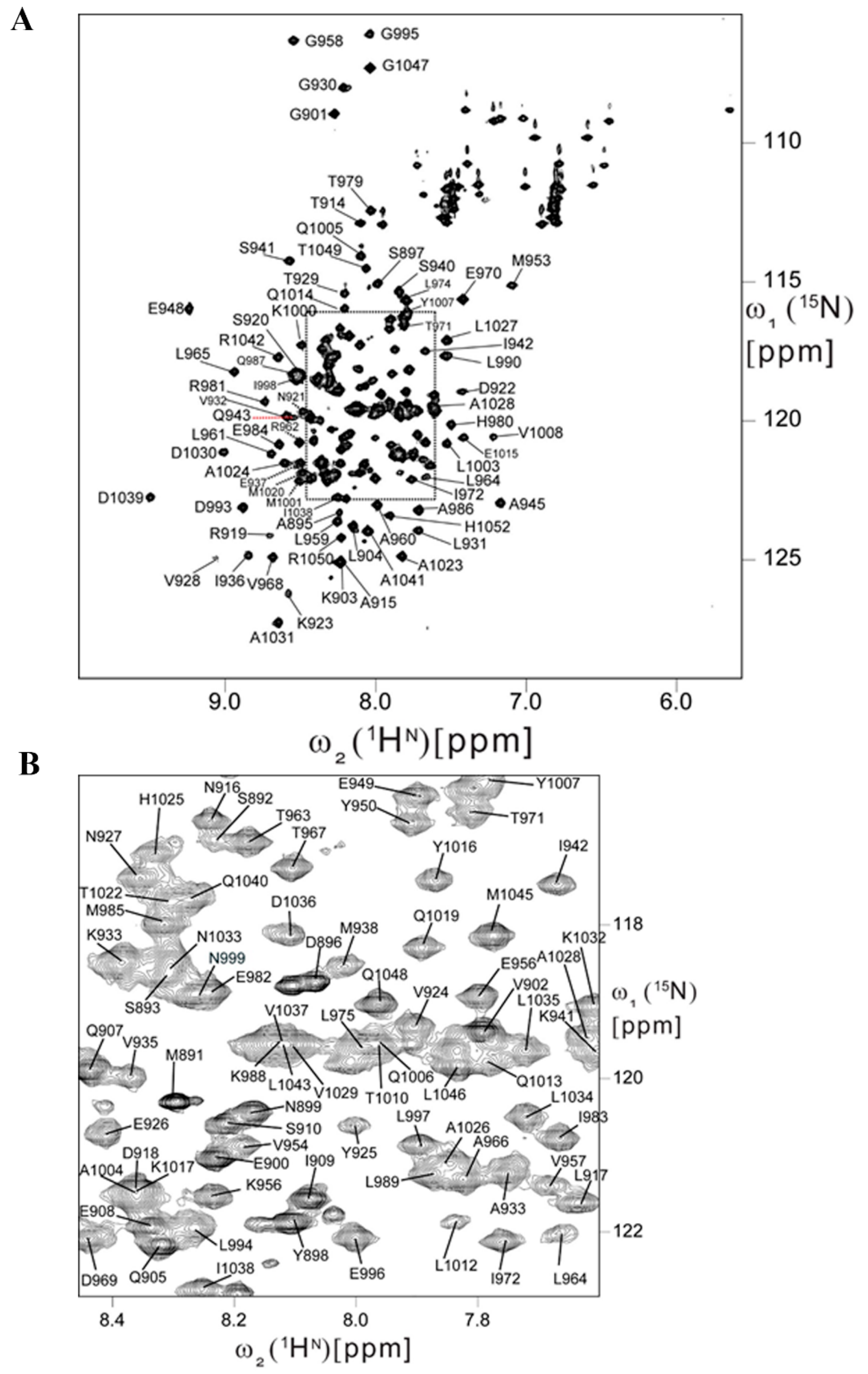
2.5. Backbone Resonance Assignments for Human FAT Domain
2.6. Feasibility of HSQC-NMR Assay
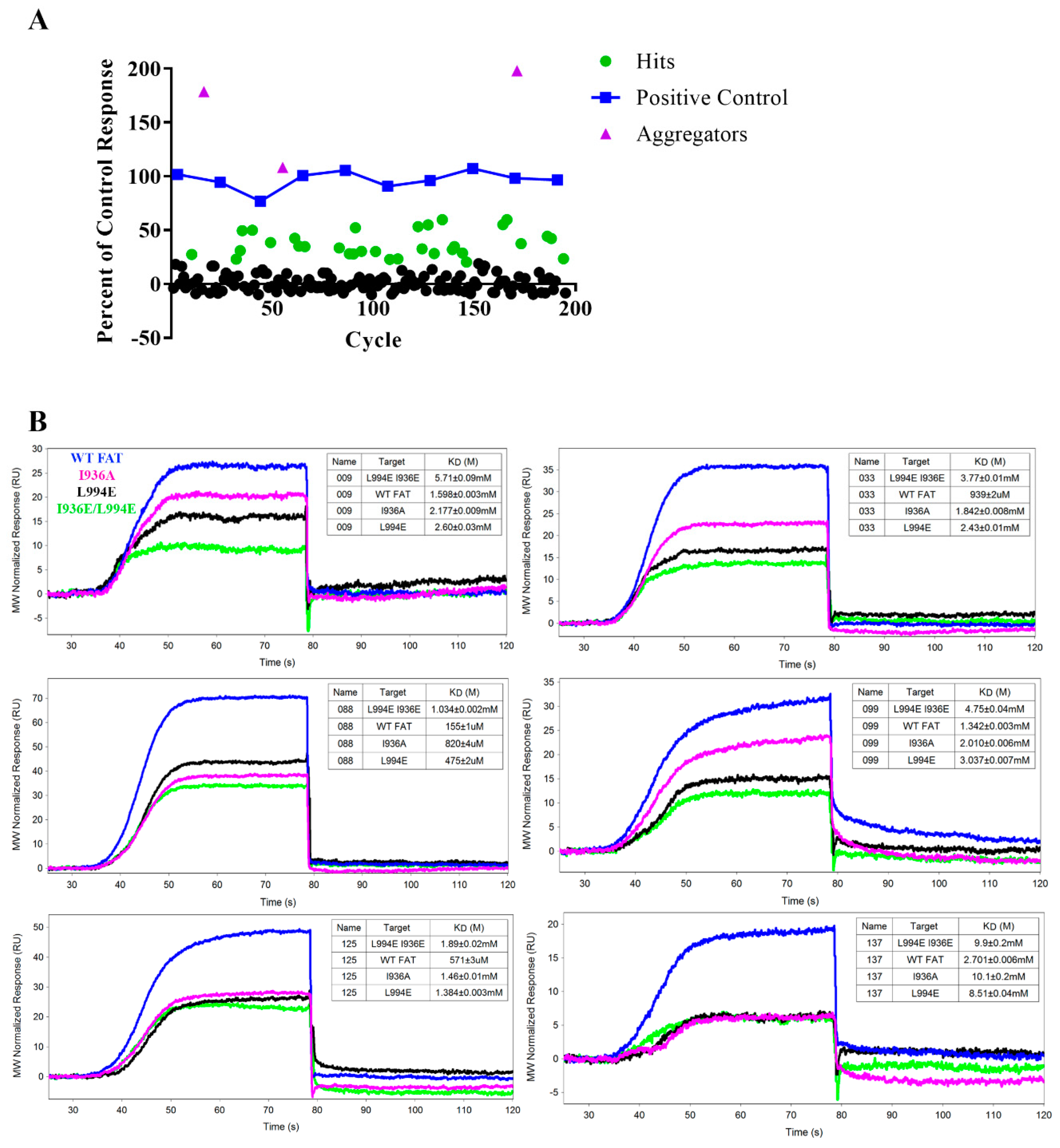
2.7. Pilot SPR Screening and Hit Validation for A Small Fragment Library
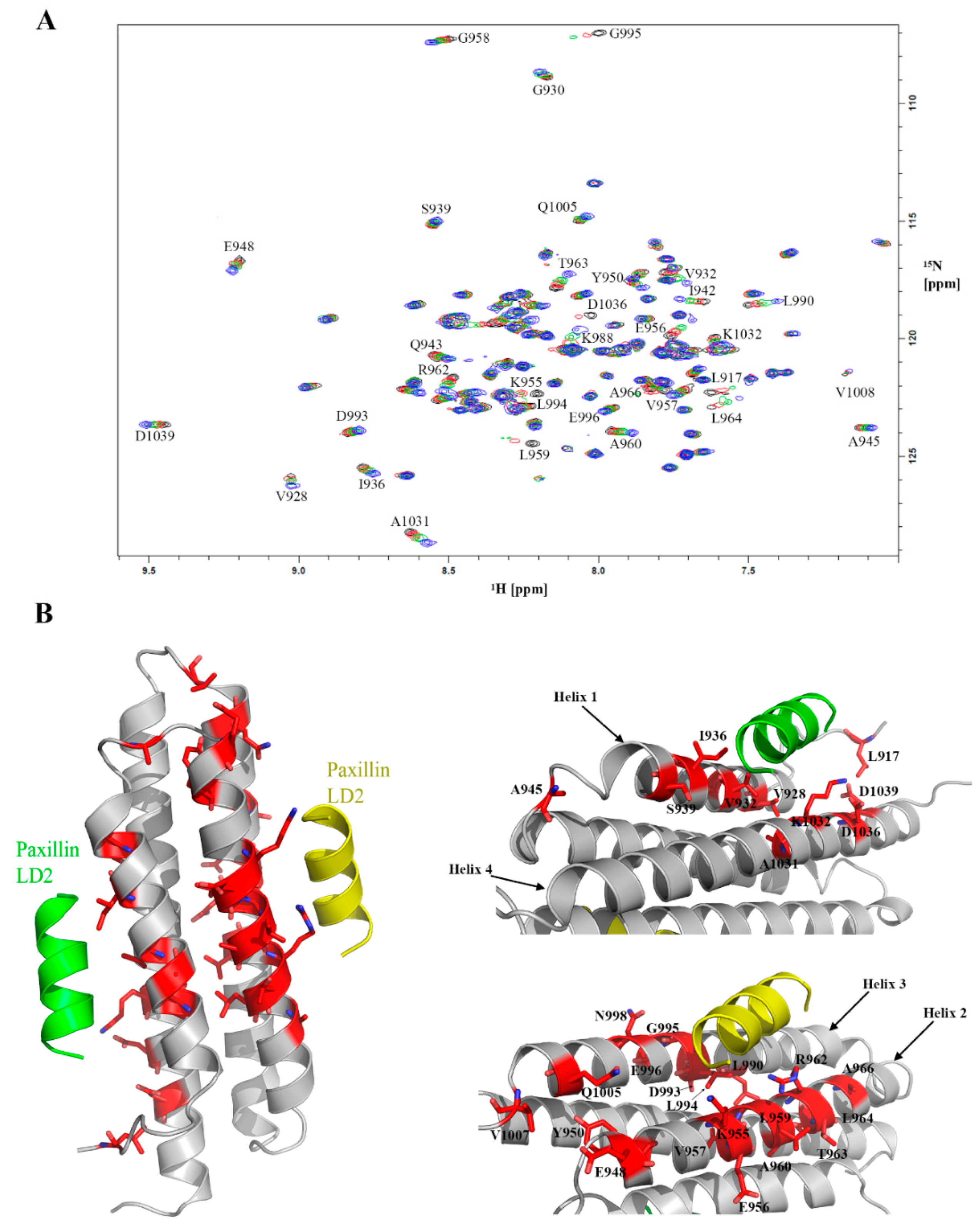
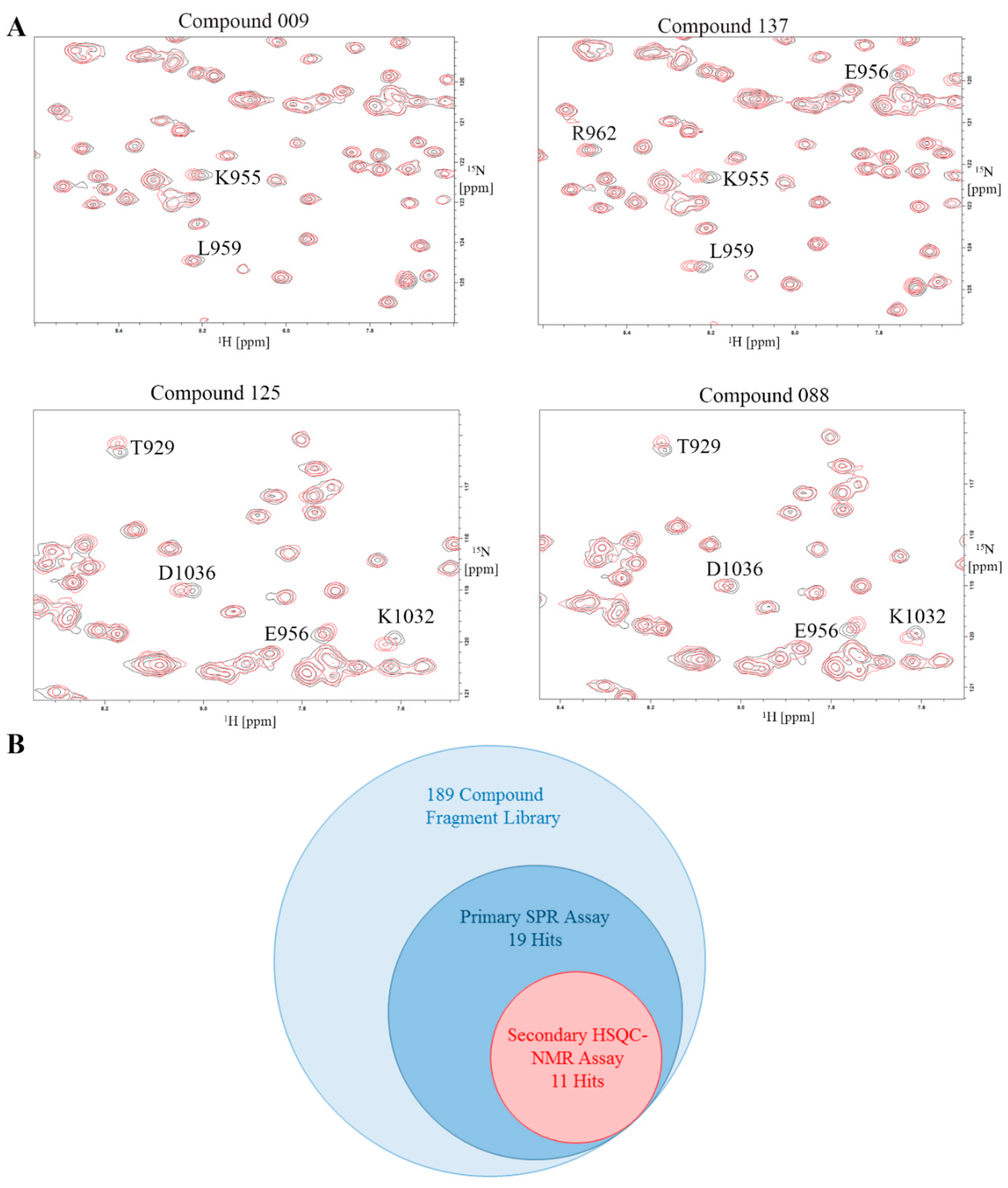
2.8. Validation of Hits Using HSQC-NMR
3. Discussion
4. Materials and Methods
4.1. Construct Design, Cloning, Expression, and Purification of Biotinylated FAT
4.2. Surface Plasmon Resonance (SPR)
4.3. Purification of Isotopically Labeled Protein
4.4. FAT Backbone Resonance Assignments
4.5. D HSQC-NMR
4.6. Site-Directed Mutagenesis of AviTag FAT
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weiner, T.M.; Liu, E.T.; Craven, R.J.; Cance, W.G. Expression of focal adhesion kinase gene and invasive cancer. Lancet 1993, 342, 1024–1025. [Google Scholar] [CrossRef]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125fak) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar] [PubMed]
- Lark, A.L.; Livasy, C.A.; Calvo, B.; Caskey, L.; Moore, D.T.; Yang, X.; Cance, W.G. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: Immunohistochemistry and real-time pcr analyses. Clin. Cancer Res. 2003, 9, 215–222. [Google Scholar] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. Pp125fak a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Ryzhova, L.; Shin, N.Y.; Brabek, J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front. Biosci. 2003, 8, d982–d996. [Google Scholar] [CrossRef] [PubMed]
- Siesser, P.M.F.; Hanks, S.K. The signaling and biological implications of fak overexpression in cancer. Clin. Cancer Res. 2006, 12, 3233–3237. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Komiyama, N.H.; Serrels, B.; Asano, H.; Reynolds, L.; Conti, F.; Hodivala-Dilke, K.; Metzger, D.; Chambon, P.; Grant, S.G.; et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004, 18, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, H.; Sanguin-Gendreau, V.; Zuo, D.; Cardiff, R.D.; McLean, G.W.; Frame, M.C.; Muller, W.J. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc. Natl. Acad. Sci. USA 2007, 104, 20302–20307. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Beggs, H.E.; Keely, P.J. Mammary epithelial-specific disruption of focal adhesion kinase retards tumor formation and metastasis in a transgenic mouse model of human breast cancer. Am. J. Pathol. 2008, 173, 1551–1565. [Google Scholar] [CrossRef]
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and pi3k-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Investig. 2009, 119, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Yang, X.; Craven, R.J.; Cance, W.G. The cooh-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 1998, 9, 999–1005. [Google Scholar] [PubMed]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal. 2013, 6, pe10. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin a2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, T.A.; Lenzo, F.L.; Figel, S.A.; Grapes, A.T.; Cance, W.G. Oncogenic receptor tyrosine kinases directly phosphorylate focal adhesion kinase (fak) as a resistance mechanism to fak-kinase inhibitors. Mol. Cancer 2016, 15, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase i study of vs-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.J.A.; Steeghs, N.; Lolkema, M.P.; Hotte, S.J.; Hirte, H.W.; van der Biessen, D.A.J.; Abdul Razak, A.R.; De Vos, F.; Verheijen, R.B.; Schnell, D.; et al. Phase i study of bi 853520, an inhibitor of focal adhesion kinase, in patients with advanced or metastatic nonhematologic malignancies. Target. Oncol. 2019, 14, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kanteti, R.; Batra, S.K.; Lennon, F.E.; Salgia, R. Fak and paxillin, two potential targets in pancreatic cancer. Oncotarget 2016, 7, 31586–31601. [Google Scholar] [CrossRef]
- Sieg, D.J.; Hauck, C.R.; Ilic, D.; Klingbeil, C.K.; Schaefer, E.; Damsky, C.H.; Schlaepfer, D.D. Fak integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000, 2, 249–256. [Google Scholar] [CrossRef]
- Deramaudt, T.B.; Dujardin, D.; Noulet, F.; Martin, S.; Vauchelles, R.; Takeda, K.; Ronde, P. Altering fak-paxillin interactions reduces adhesion, migration and invasion processes. PLoS ONE 2014, 9, e92059. [Google Scholar] [CrossRef]
- Kaneda, T.; Sonoda, Y.; Ando, K.; Suzuki, T.; Sasaki, Y.; Oshio, T.; Tago, M.; Kasahara, T. Mutation of y925f in focal adhesion kinase (fak) suppresses melanoma cell proliferation and metastasis. Cancer Lett. 2008, 270, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Scheswohl, D.M.; Harrell, J.R.; Rajfur, Z.; Gao, G.; Campbell, S.L.; Schaller, M.D. Multiple paxillin binding sites regulate fak function. J. Mol. Signal. 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Hoellerer, M.K.; Noble, M.E.; Labesse, G.; Campbell, I.D.; Werner, J.M.; Arold, S.T. Molecular recognition of paxillin ld motifs by the focal adhesion targeting domain. Structure 2003, 11, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Prutzman, K.C.; King, M.L.; Scheswohl, D.M.; DeRose, E.F.; London, R.E.; Schaller, M.D.; Campbell, S.L. Nmr solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a paxillin ld peptide: Evidence for a two-site binding model. J. Biol. Chem. 2004, 279, 8441–8451. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I.; Vuori, K.; Liddington, R.C. The focal adhesion targeting (fat) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 2002, 9, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Bertolucci, C.M.; Guibao, C.D.; Zheng, J. Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly. Protein Sci. 2005, 14, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (fbld). Essays Biochem. 2017, 61, 453–464. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The ‘rule of three’ for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644–645. [Google Scholar] [CrossRef]
- Bembenek, S.D.; Tounge, B.A.; Reynolds, C.H. Ligand efficiency and fragment-based drug discovery. Drug Discov. Today 2009, 14, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.J. Amine coupling through edc/nhs: A practical approach. Methods Mol. Biol 2010, 627, 55–73. [Google Scholar] [PubMed]
- Cull, M.G.; Schatz, P.J. Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzym. 2000, 326, 430–440. [Google Scholar]
- Vachali, P.P.; Li, B.; Besch, B.M.; Bernstein, P.S. Protein-flavonoid interaction studies by a taylor dispersion surface plasmon resonance (spr) technique: A novel method to assess biomolecular interactions. Biosensors 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, T.; Alvarado, C.; Rivera, A.; Lenzo, F.; Nott, R.; Bondugji, D.; Montoya, J.; Hurley, A.; Kaplan, M.; Capaldi, A.; et al. Development of a high-throughput fluorescence polarization assay to detect inhibitors of the fak-paxillin interaction. SLAS 2019. In Press. [Google Scholar] [CrossRef] [PubMed]
- Arold, S.T.; Hoellerer, M.K.; Noble, M.E. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 2002, 10, 319–327. [Google Scholar] [CrossRef]
- Romasanta, A.K.S.; van der Sijde, P.; Hellsten, I.; Hubbard, R.E.; Keseru, G.M.; van Muijlwijk-Koezen, J.; de Esch, I.J.P. When fragments link: A bibliometric perspective on the development of fragment-based drug discovery. Drug Discov. Today 2018, 23, 1596–1609. [Google Scholar] [CrossRef]
- Jahnke, W.; Florsheimer, A.; Blommers, M.J.; Paris, C.G.; Heim, J.; Nalin, C.M.; Perez, L.B. Second-site nmr screening and linker design. Curr. Top. Med. Chem. 2003, 3, 69–80. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed src homology 2 domain studied by 15n nmr relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Güntert, P.; Dötsch, V.; Wider, G.; Wüthrich, K. Processing of multi-dimensional nmr data with the new software prosa. J. Biomol. NMR 1992, 2, 619–629. [Google Scholar] [CrossRef]
- Bartels, C.; Xia, T.H.; Billeter, M.; Guntert, P.; Wuthrich, K. The program xeasy for computer-supported nmr spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rochus, K. The Computer Aided Resonance Assignment Tutorial; CANTINA Verlag: Goldau, Switzerland, 2004. [Google Scholar]
- Wishart, D.S.; Sykes, B.D. The 13c chemical-shift index: A simple method for the identification of protein secondary structure using 13c chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Czisch, M.; Boelens, R. Sensitivity enhancement in the trosy experiment. J. Magn. Reson. 1998, 134, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.V.; Wider, G.; Wuthrich, K. Single transition-to-single transition polarization transfer (st2-pt) in [15n,1h]-trosy. J. Biomol. NMR 1998, 12, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Briand, J.; Sphirensen, O.W. Editing of multidimensional nmr spectra of partially deuterated proteins. Measurement of amide deuterium isotope effects on the chemical shifts of protein backbone nuclei. J. Biomol. NMR 1998, 12, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Loria, J.P.; Rance, M.; Palmer, A.G., 3rd. Transverse-relaxation-optimized (trosy) gradient-enhanced triple-resonance nmr spectroscopy. J. Magn. Reson. 1999, 141, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.M.; Sze, K.H.; Zhu, G. Gradient and sensitivity enhanced multiple-quantum coherence in heteronuclear multidimensional nmr experiments. J. Biomol. NMR 1999, 14, 133–140. [Google Scholar] [CrossRef]
Sample Availability: All DNA constructs available for sharing. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Wild Type KD ± SE (µM) | I936A Single Mutant KD ± SE (µM) | L994E Single Mutant KD ± SE (µM) | I936E/L994E Double Mutant KD ± SE (µM) | I936A Selectivity Ratio | L994E Selectivity Ratio | I936E/L994E Selectivity Ratio | Ligand Efficiency (WT) | Validated by HSQC NMR |
|---|---|---|---|---|---|---|---|---|---|
| 009 | 1598 ± 3 | 2177 ± 9 | 2600 ± 30 | 5710 ± 90 | 1.27 | 1.64 | 2.95 | 0.34 | X |
| 030 | ND | ND | ND | ND | 2.09 | 2.27 | 1.35 | ND | |
| 032 | ND | ND | ND | ND | 0.92 | 1.55 | 1.37 | ND | |
| 033 | 939 ± 2 | 1842 ± 8 | 2430 ± 10 | 3770 ± 10 | 1.57 | 2.11 | 2.62 | 0.31 | |
| 038 | ND | ND | ND | ND | 1.01 | 0.94 | 0.83 | ND | |
| 047 | 1780 ± 30 | 2400 ± 70 | 3000 ± 200 | 10,000 ± 2000 | 1.34 | 1.97 | 5.18 | 0.26 | |
| 059 | 2900 ± 10 | 2215 ± 9 | 857 ± 2 | 1286 ± 4 | 0.86 | 0.76 | 1.56 | 0.26 | X |
| 061 | ND | ND | ND | ND | 1.65 | 1.35 | 0.92 | ND | X |
| 063 | 884 ± 3 | 671 ± 3 | 418 ± 1 | 2088 ± 5 | 0.90 | 0.94 | 1.95 | 0.32 | X |
| 081 | 1815 ± 9 | 4310 ± 40 | 483 ± 2 | 1041 ± 3 | 1.97 | 1.46 | 1.37 | 0.28 | |
| 085 | ND | ND | ND | ND | 1.76 | 1.53 | 1.42 | ND | X |
| 087 | 3070 ± 20 | 3310 ± 90 | 7100 ± 300 | ND | 1.22 | 2.95 | ND | 0.39 | |
| 088 | 155 ± 1 | 820 ± 4 | 475 ± 2 | 1034 ± 2 | 1.82 | 1.59 | 2.06 | 0.44 | X |
| 092 | 3542 ± 9 | 3810 ± 20 | 3450 ± 8 | 2407 ± 4 | 1.10 | 1.23 | 1.04 | 0.26 | |
| 099 | 1342 ± 3 | 2010 ± 6 | 3037 ± 7 | 4750 ± 40 | 1.33 | 2.10 | 2.62 | 0.31 | X |
| 105 | ND | ND | ND | ND | ND | ND | ND | ND | |
| 109 | 896 ± 3 | 6300 ± 100 | 4230 ± 10 | ND | 4.02 | 3.16 | 2.07 | 0.30 | X |
| 119 | 3200 ± 10 | 3890 ± 60 | 3920 ± 10 | 5490 ± 20 | 1.30 | 1.38 | 1.51 | 0.27 | |
| 121 | 5030 ± 30 | 8900 ± 100 | 3055 ± 5 | 8240 ± 20 | 1.62 | 1.79 | 1.70 | 0.28 | |
| 125 | 571 ± 3 | 1460 ± 10 | 1384 ± 3 | 1890 ± 20 | 1.75 | 1.82 | 2.15 | 0.35 | X |
| 131 | 292 ± 2 | 1770 ± 10 | 865 ± 2 | 1121 ± 2 | 2.33 | 1.71 | 1.79 | 0.49 | X |
| 136 | ND | ND | ND | ND | 0.64 | 0.36 | 0.53 | ND | |
| 137 | 2701 ± 6 | 10,100 ± 200 | 8510 ± 40 | 9900 ± 200 | 2.96 | 2.96 | 3.27 | 0.30 | X |
| 141 | 8120 ± 20 | 6610 ± 60 | 3088 ± 6 | 10,140 ± 20 | 0.89 | 0.99 | 1.89 | 0.22 | |
| 143 | ND | ND | ND | ND | 2.68 | 2.68 | 1.62 | ND | |
| 162 | ND | ND | ND | ND | ND | ND | ND | ND | |
| 168 | 1830 ± 10 | 6200 ± 200 | 4490 ± 40 | 2910 ± 30 | 3.07 | 2.43 | 1.48 | 0.32 | |
| 181 | 709 ± 1 | 1682 ± 3 | 1240 ± 2 | 1029 ± 2 | 1.64 | 1.86 | 1.28 | 0.40 | |
| 183 | ND | ND | ND | ND | ND | ND | 1.37 | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarado, C.; Stahl, E.; Koessel, K.; Rivera, A.; Cherry, B.R.; Pulavarti, S.V.S.R.K.; Szyperski, T.; Cance, W.; Marlowe, T. Development of a Fragment-Based Screening Assay for the Focal Adhesion Targeting Domain Using SPR and NMR. Molecules 2019, 24, 3352. https://doi.org/10.3390/molecules24183352
Alvarado C, Stahl E, Koessel K, Rivera A, Cherry BR, Pulavarti SVSRK, Szyperski T, Cance W, Marlowe T. Development of a Fragment-Based Screening Assay for the Focal Adhesion Targeting Domain Using SPR and NMR. Molecules. 2019; 24(18):3352. https://doi.org/10.3390/molecules24183352
Chicago/Turabian StyleAlvarado, Carlos, Erik Stahl, Karissa Koessel, Andrew Rivera, Brian R. Cherry, Surya V.S.R.K. Pulavarti, Thomas Szyperski, William Cance, and Timothy Marlowe. 2019. "Development of a Fragment-Based Screening Assay for the Focal Adhesion Targeting Domain Using SPR and NMR" Molecules 24, no. 18: 3352. https://doi.org/10.3390/molecules24183352
APA StyleAlvarado, C., Stahl, E., Koessel, K., Rivera, A., Cherry, B. R., Pulavarti, S. V. S. R. K., Szyperski, T., Cance, W., & Marlowe, T. (2019). Development of a Fragment-Based Screening Assay for the Focal Adhesion Targeting Domain Using SPR and NMR. Molecules, 24(18), 3352. https://doi.org/10.3390/molecules24183352