
Mechanochemical Synthesis and Isomerization of N-Substituted Indole-3-carboxaldehyde Oximes †

 ,
,  ,
,  ,
,
Abstract
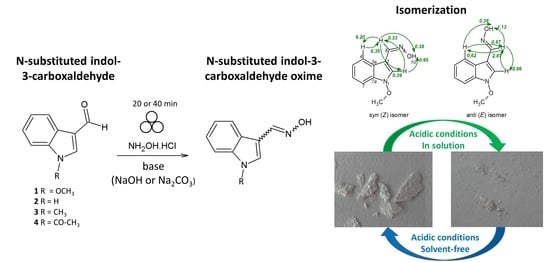
1. Introduction
2. Results and Discussion
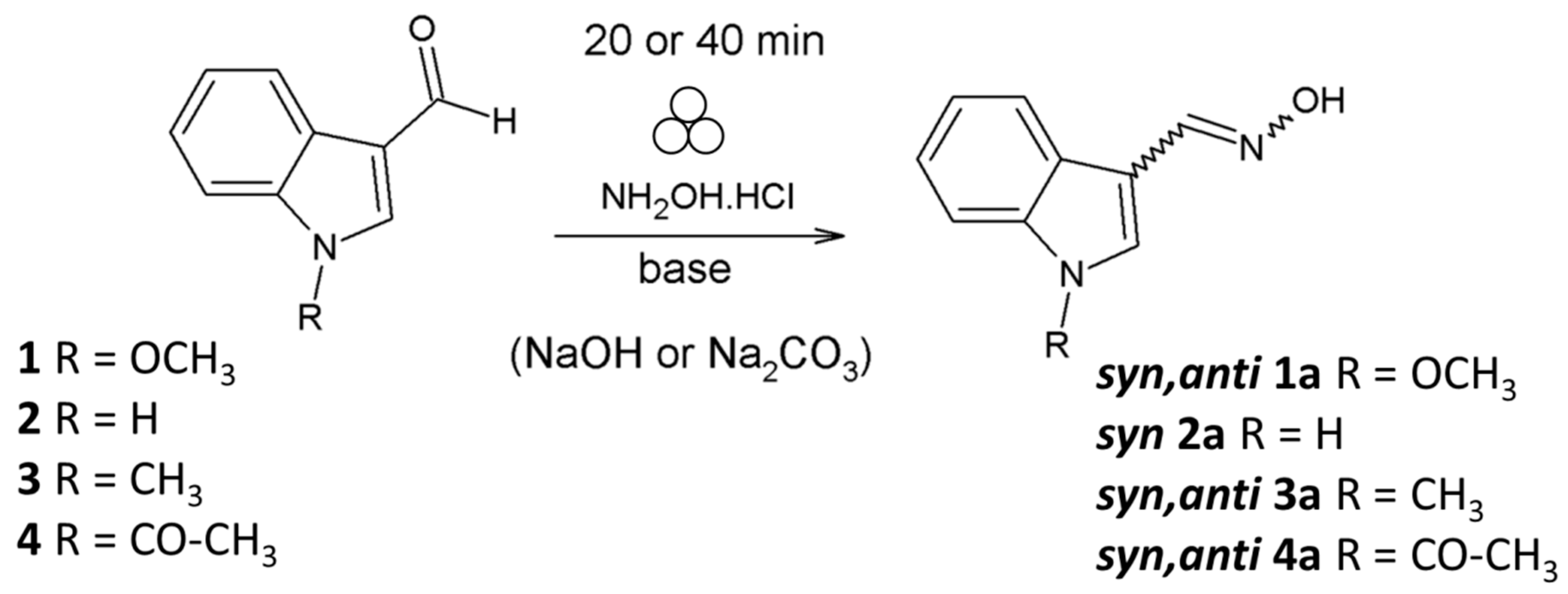
2.1. Mechanochemical Synthesis of 1-Methoxyindole-3-carboxaldehyde Oxime (Syn, Anti-1)
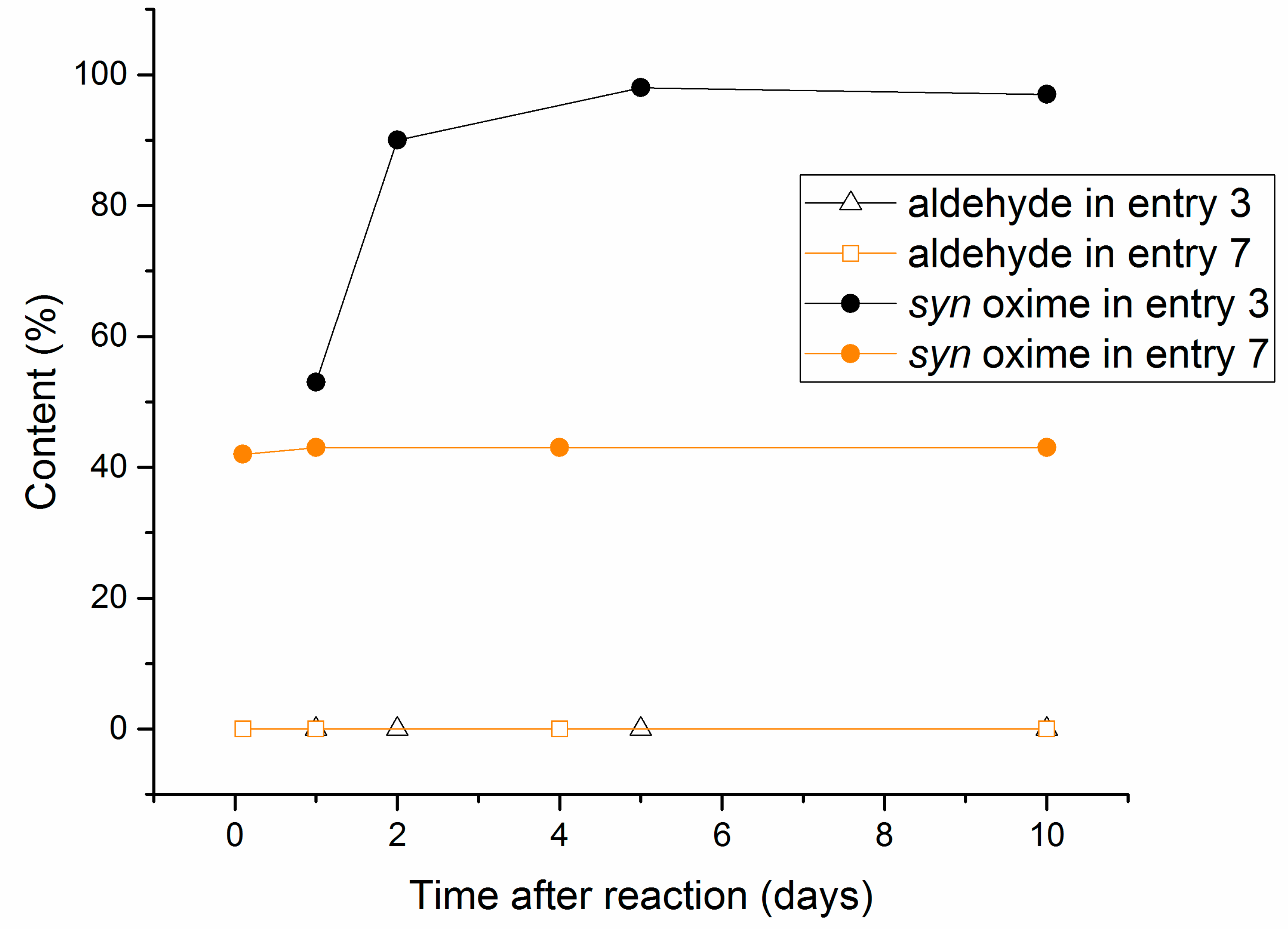
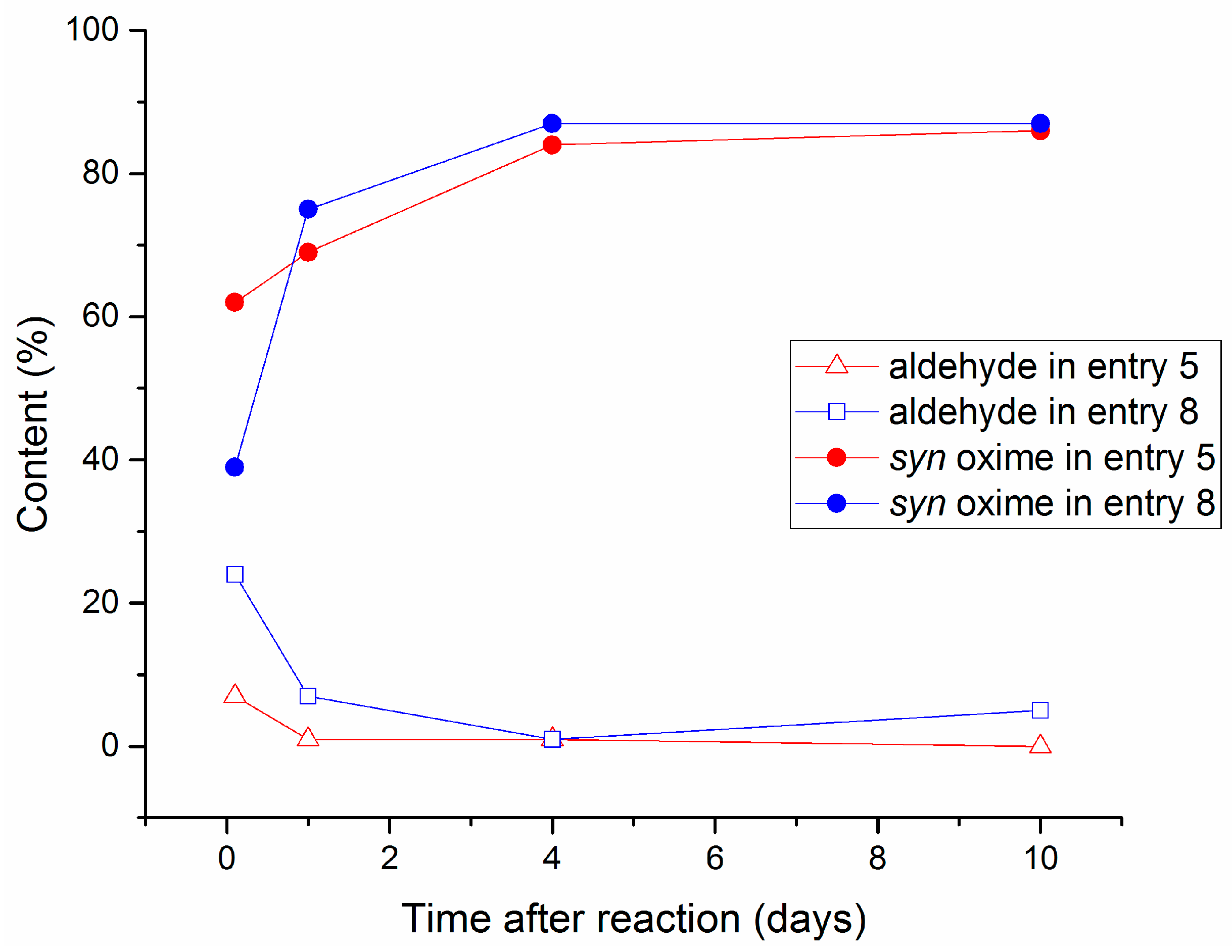
2.2. Aging Experiments for the Synthesis of 1-Methoxyindole-3-carboxaldehyde Oxime (Syn, Anti-1a)
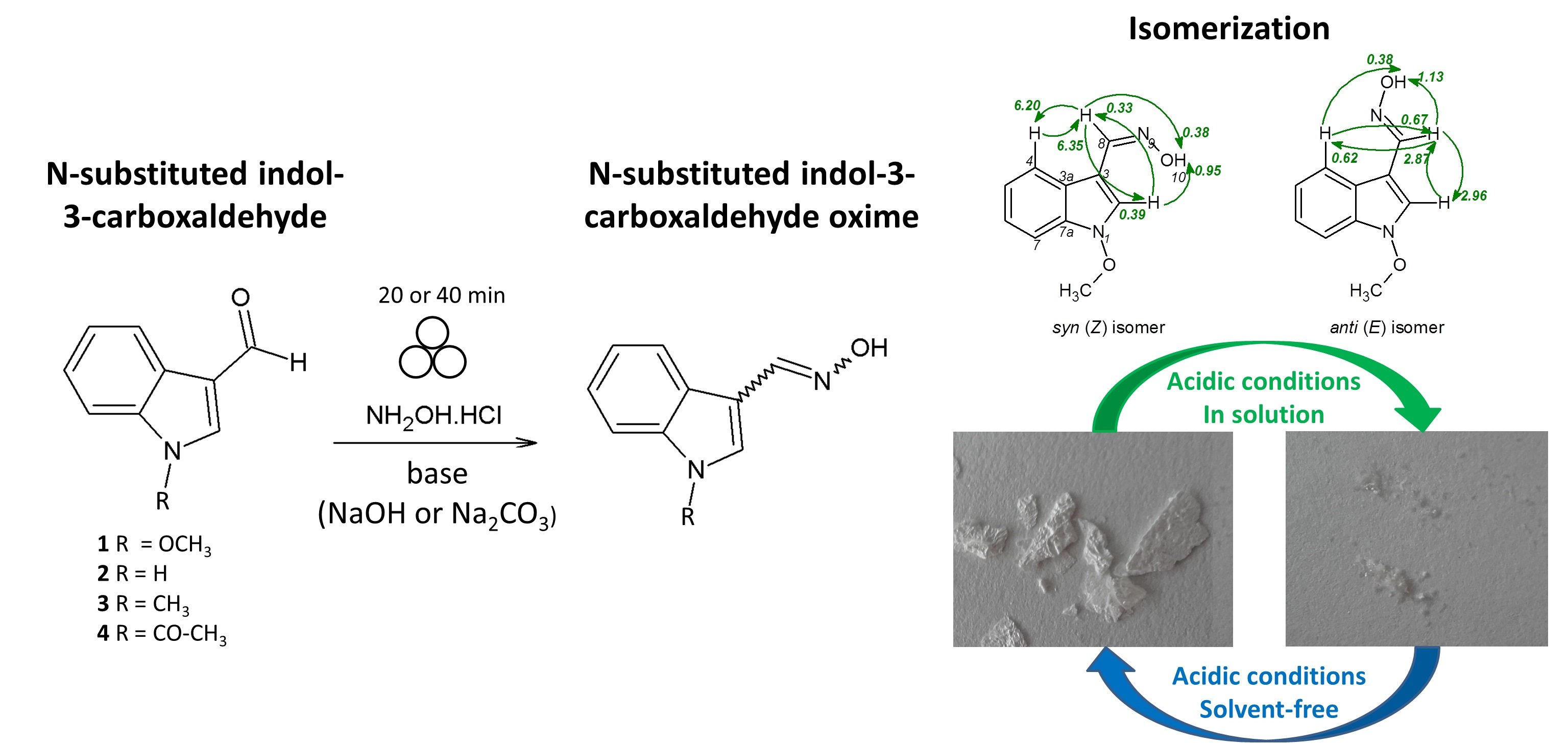
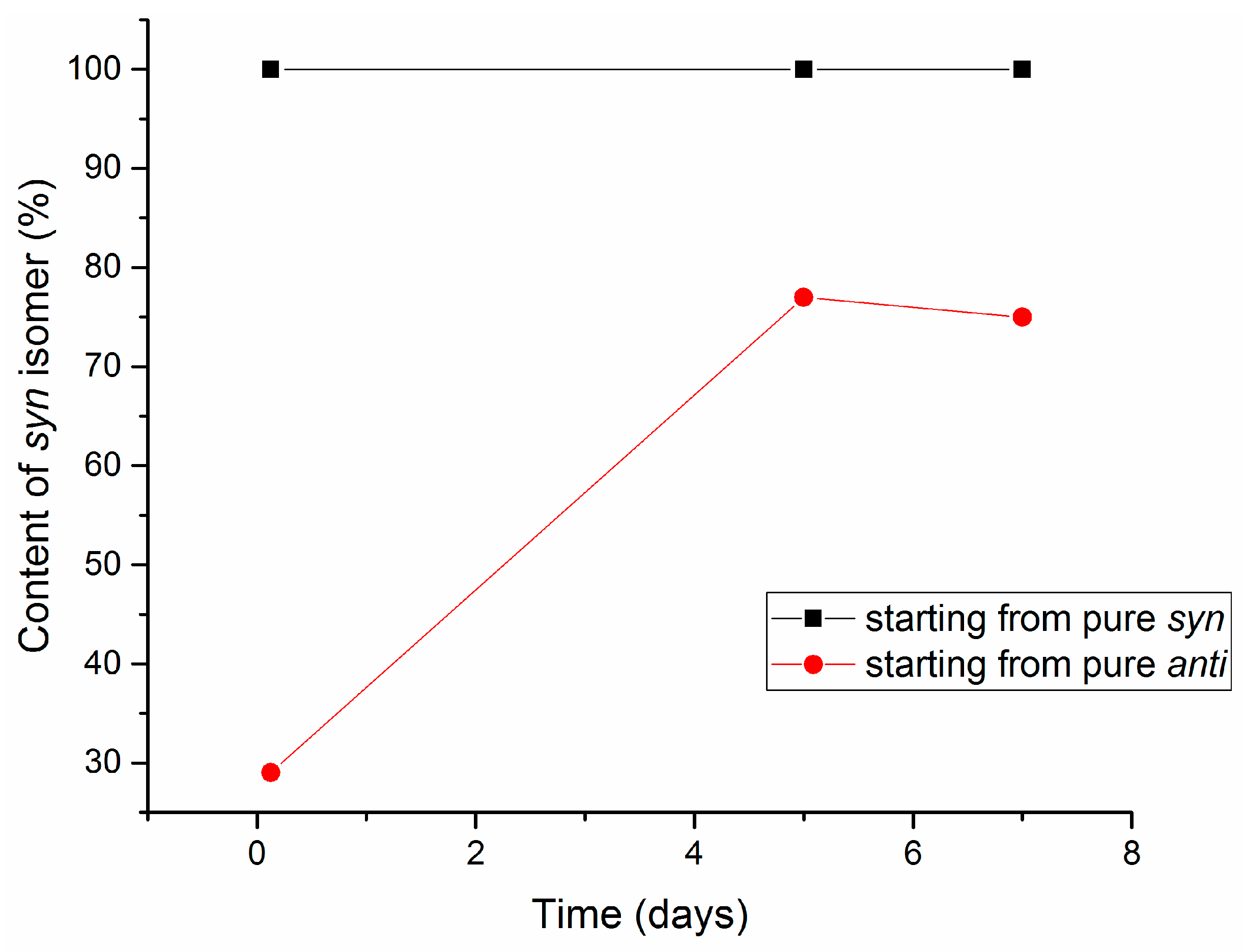
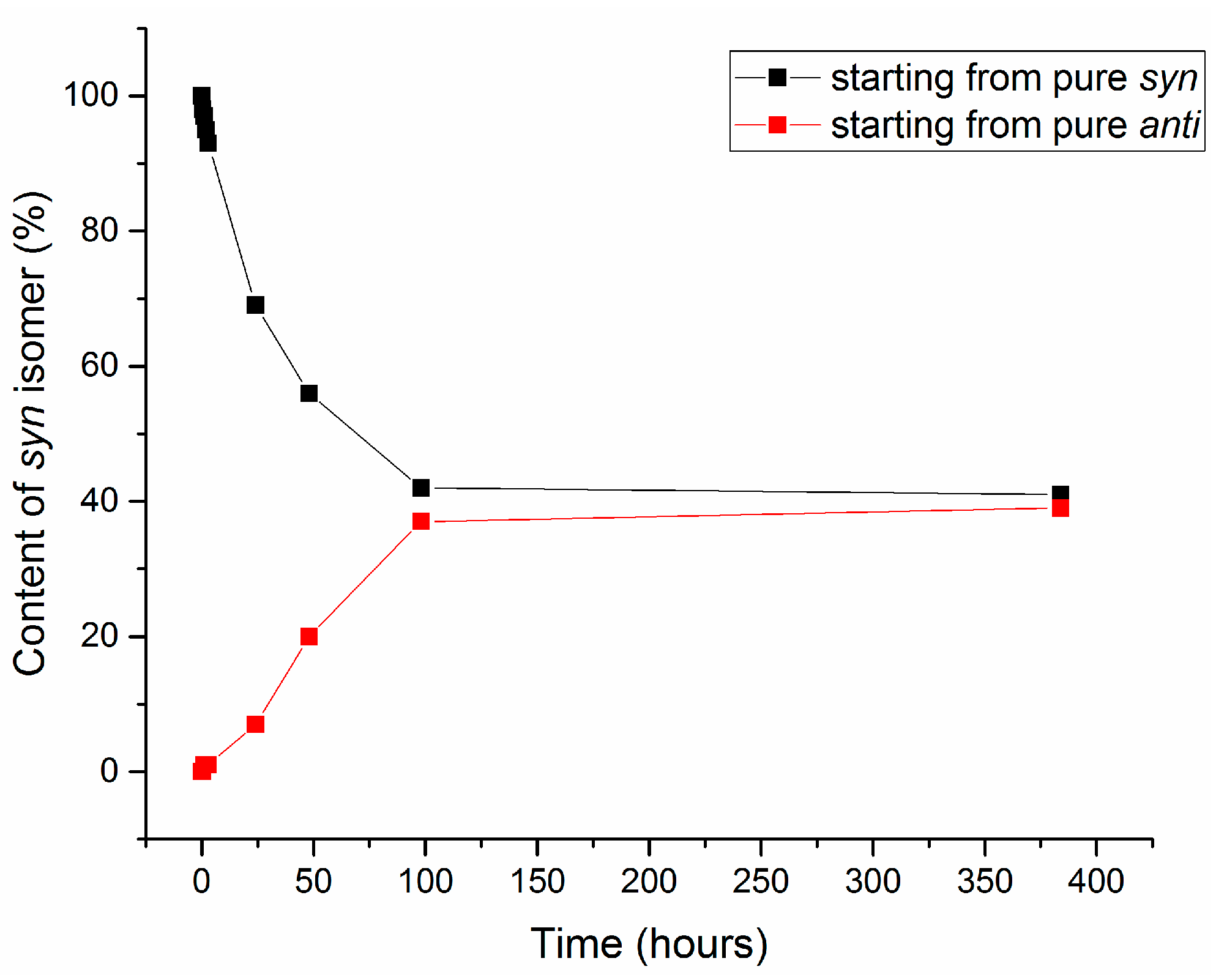
2.3. Isomerization of 1-Methoxyindole-3-carboxaldehyde Oxime (Syn, Anti-1a)
2.4. Substrate Scope in the Oximation-Isomerization Reactions
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Aldehydes
3.3. Experimental Procedure for Mechanochemical Synthesis
3.4. Experimental Procedure for Solution-Based Synthesis
3.5. Isolated Yield Calculation
3.6. Spectral Data for the Products
3.6.1. Anti-1-methoxyindole-3-carboxaldehyde Oxime (anti-1a)
3.6.2. Syn-1-methoxyindole-3-carboxyaldehyde Oxime (syn-1a)
3.6.3. Syn-indole-3-carboxyaldehyde Oxime (syn-2a)
3.6.4. Syn and anti-1-methylindole-3-carboxyaldehyde Oxime (3a)
3.6.5. Anti and syn-1-acetylindole-3-carboxyaldehyde Oxime (4a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frutos, R.P.; Spero, D.M. Synthesis of protected, chiral alpha,alpha-disubstituted alpha-amino acids via a Beckmann rearrangement. Tetrahedron Lett. 1998, 39, 2475–2478. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Gopalaiah, K. Beckmann rearrangement in the solid state: Reaction of oxime hydrochlorides. Tetrahedron Lett. 2001, 42, 8123–8125. [Google Scholar] [CrossRef]
- Mendelsohn, B.A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V.S.; Ciufolini, M.A. Oxidation of Oximes to Nitrile Oxides with Hypervalent Iodine Reagents. Org. Lett. 2009, 11, 1539–1542. [Google Scholar] [CrossRef]
- Dave, P.R.; Forohar, F.; Axenrod, T.; Das, K.K.; Qi, L.; Watnick, C.; Yazdekhasti, H. Facile preparation of 3,7-diazabicyclo [3.3.0]octane and 3,7,10-triheterocyclic [3.3.3]propellane ring systems from 1,5-diazacyclooctane 3,7-derivatives. J. Org. Chem. 1996, 61, 8897–8903. [Google Scholar] [CrossRef]
- Negi, S.; Matsukura, M.; Mizuno, M.; Miyake, K.; Minami, N. Synthesis of (2R)-1-(4-chloro-2-pyridyl)-2-(2-pyridyl)ethylamine: A selective oxime reduction and crystallization-induced asymmetric transformation. Synthesis 1996, 1996, 991–996. [Google Scholar] [CrossRef]
- Iwata, Y.; Koseki, H.; Hosoya, F. Study on decomposition of hydroxylamine/water solution. J. Loss Prev. Process Ind. 2003, 16, 41–53. [Google Scholar] [CrossRef]
- Raja, R.; Sankar, G.; Thomas, J.M. Bifunctional molecular sieve catalysts for the benign ammoximation of cyclohexanone: One-step, solvent-free production of oxime and epsilon-caprolactam with a mixture of air and ammonia. J. Am. Chem. Soc. 2001, 123, 8153–8154. [Google Scholar] [CrossRef]
- Raja, R.; Thomas, J.M. Engineering active sites in bifunctional nanopore and bimetallic nanoparticle catalysts for one-step, solvent-free processes. In Nanoporous Materials III; Elsevier: Amsterdam, The Netherlands, 2002; Volume 141, pp. 317–328. [Google Scholar]
- Bigdeli, M.A.; Nikje, M.M.A.; Jafari, S.; Heravi, M.M. Regioselective synthesis of syn-oximes using 3 angstrom molecular sieves in a solventless system. J. Chem. Res.-S 2002, 20–21. [Google Scholar]
- Mohammed, A.H.A.; Nagendrappa, G. A remarkably simple alpha-oximation of ketones to 1,2-dione monooximes using the chlorotrimethylsilane-isoamyl nitrite combination. Tetrahedron Lett. 2003, 44, 2753–2755. [Google Scholar] [CrossRef]
- Kiasat, A.R.; Kazemi, F.; Nourbakhsh, K. A clean conversion of carbonyl compounds to oximes using silica gel supported hydroxylamine hydrochloride. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1193–1196. [Google Scholar] [CrossRef]
- Kiasat, A.R.; Kazemi, F.; Nourbakhsh, K. A convenient one-pot method of converting alcohols into oximes. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1809–1812. [Google Scholar] [CrossRef]
- Serna, P.; Lopez-Haro, M.; Calvino, J.J.; Corma, A. Selective hydrogenation of nitrocyclohexane to cyclohexanone oxime with H-2 on decorated Pt nanoparticles. J. Catal. 2009, 263, 328–334. [Google Scholar] [CrossRef]
- Fazaeli, R.; Aliyan, H. AlPW12O40 and AlPMo12O40 as Highly Effective and Eco-friendly Catalysts for Aldoximation of Aldehydes Under Solvent-Free Conditions. Asian J. Chem. 2010, 22, 855–858. [Google Scholar]
- Aghapour, G.; Mohamadian, S. Selective Tandem Synthesis of Oximes from Benzylic Alcohols Catalyzed with 2, 3-Dichloro-5, 6-dicyanobenzoquinone. Bull. Korean Chem. Soc. 2012, 33, 1209–1212. [Google Scholar] [CrossRef]
- Kong, D.L.; Liu, R.D.; Li, G.Z.; Zhang, P.W.; Wu, M.S. A Rapid, Convenient, Solventless Green Approach for the Synthesis of alpha-Hydroxyphosphonates by Grinding. Asian J. Chem. 2014, 26, 1246–1248. [Google Scholar] [CrossRef]
- Yadav, P.; Lal, K.; Rani, P.; Mor, S.; Kumar, A.; Kumar, A. Efficient synthesis and antimicrobial evaluation of 2-((1-substituted-1H-1,2,3-triazol-4-yl)-1-naphthaldehydes and their oxime derivatives. Med. Chem. Res. 2017, 26, 1469–1480. [Google Scholar] [CrossRef]
- Alam, M.; Lee, D.U. Green synthesis, biochemical and quantum chemical studies of steroidal oximes. Korean J. Chem. Eng. 2015, 32, 1142–1150. [Google Scholar] [CrossRef]
- Hoelz, L.V.B.; Goncalves, B.T.; Barros, J.C.; da Silva, J.F.M. Solvent Free, Microwave Assisted Conversion of Aldehydes into Nitriles and Oximes in the Presence of NH2OH center dot HCl and TiO2. Molecules 2010, 15, 94–99. [Google Scholar] [CrossRef]
- Hong, Z.; Li, J.J.; Chen, G.; Jiang, H.J.; Yang, X.F.; Pan, H.; Su, W.K. Solvent-free mechanochemical synthesis of arylcyanomethylenequinone oximes from phenylacetonitriles and 4-unsubstituted nitroaromatic compounds using KF/nano-gamma-Al2O3 as catalyst. RSC Adv. 2016, 6, 13581–13588. [Google Scholar] [CrossRef]
- Primozic, I.; Hrenar, T.; Baumann, K.; Kristo, L.; Krizic, I.; Tomic, S. Mechanochemical and Conformational Study of N-heterocyclic Carbonyl-Oxime Transformations. Croat. Chem. Acta 2014, 87, 153–160. [Google Scholar] [CrossRef]
- Mokhtari, J.; Naimi-Jamal, M.R.; Hamzeali, H.; Dekamin, M.G.; Kaupp, G. Kneading Ball-Milling and Stoichiometric Melts for the Quantitative Derivatization of Carbonyl Compounds with Gas-Solid Recovery. Chemsuschem 2009, 2, 248–254. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Sinha, A.S.; Epa, K.N.; Spartz, C.L.; Desper, J. A versatile and green mechanochemical route for aldehyde-oxime conversions. Chem. Commun. 2012, 48, 11289–11291. [Google Scholar] [CrossRef]
- Hernandez, J.G.; Bolm, C. Altering Product Selectivity by Mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef]
- Baláž, P.; Achimovičová, M.; Baláž, M.; Billik, P.; Cherkezova-Zheleva, Z.; Criado, J.M.; Delogu, F.; Dutková, E.; Gaffet, E.; Gotor, F.J.; et al. Hallmarks of mechanochemistry: From nanoparticles to technology. Chem. Soc. Rev. 2013, 42, 7571–7637. [Google Scholar] [CrossRef]
- Jones, W.; Eddleston, M.D. Introductory lecture: Mechanochemistry, a versatile synthesis strategy for new materials. Faraday Discuss. 2014, 170, 9–34. [Google Scholar] [CrossRef]
- Rightmire, N.R.; Hanusa, T.P. Advances in organometallic synthesis with mechanochemical methods. Dalton Trans. 2016, 45, 2352–2362. [Google Scholar] [CrossRef]
- Moores, A. Bottom up, solid-phase syntheses of inorganic nanomaterials by mechanochemistry and aging. Curr. Opin. Green Sust. Chem. 2018, 12, 33–37. [Google Scholar] [CrossRef]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef]
- Tan, D.; Friscic, T. Mechanochemistry for Organic Chemists: An Update. Eur. J. Org. Chem. 2018, 18–33. [Google Scholar] [CrossRef]
- Avila-Ortiz, C.G.; Péréz-Venegas, M.; Vargas-Carporali, J.; Juaristi, E. Recent applications of mechanochemistry in enantioselective synthesis. Tetrahedron Lett. 2019, 60, 1749–1757. [Google Scholar] [CrossRef]
- Bolm, C.; Hernandez, J.G. Mechanochemistry of Gaseous Reactants. Angew. Chem. Int. Ed. 2019, 58, 3285–3299. [Google Scholar] [CrossRef]
- Qu, J.; Sha, L.; Wu, C.J.; Zhang, Q.W. Applications of Mechanochemically Prepared Layered Double Hydroxides as Adsorbents and Catalysts: A Mini-Review. Nanomaterials 2019, 9, 80. [Google Scholar] [CrossRef]
- Tan, D.; García, F. Main group mechanochemistry: From curiosity to established protocols. Chem. Soc. Rev. 2019, 48, 2274–2292. [Google Scholar] [CrossRef]
- Hernandez, J.G.; Juaristi, E. Recent efforts directed to the development of more sustainable asymmetric organocatalysis. Chem. Commun. 2012, 48, 5396–5409. [Google Scholar] [CrossRef]
- Wang, G.W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Do, J.L.; Friščić, T. Mechanochemistry: A force of synthesis. ACS Central Sci. 2017, 3, 13–19. [Google Scholar] [CrossRef]
- Margetic, D.; Strukil, V. Mechanochemical Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 2016; p. 386. [Google Scholar]
- Damljanovic, I.; Vukicevic, M.; Vukicevic, R.D. A simple synthesis of oximes. Monatsh. Chem. 2006, 137, 301–305. [Google Scholar] [CrossRef]
- Saikia, L.; Baruah, J.M.; Thakur, A.J. A rapid, convenient, solventless green approach for the synthesis of oximes using grindstone chemistry. Org. Med. Chem. Lett. 2011, 1, 12. [Google Scholar] [CrossRef]
- Somei, M.; Ohnishi, H.; Shoken, Y. The Chemistry of Indoles. XXVII.: A Practical Synthesis of the 1-Methoxy Analog of an Ergot Alkaloid, (±)-1-Methoxy-6, 7-secoagroclavine. Chem. Pharm. Bull. 1986, 34, 677–681. [Google Scholar] [CrossRef]
- Somei, M.; Kawasaki, T. A New and Simple Synthesis of 1-Hydroxyindole Derivatives. Heterocycles 1989, 29, 1251–1254. [Google Scholar] [CrossRef]
- Somei, M. 1-Hydroxyindoles. Heterocycles 1999, 50, 1157–1211. [Google Scholar] [CrossRef]
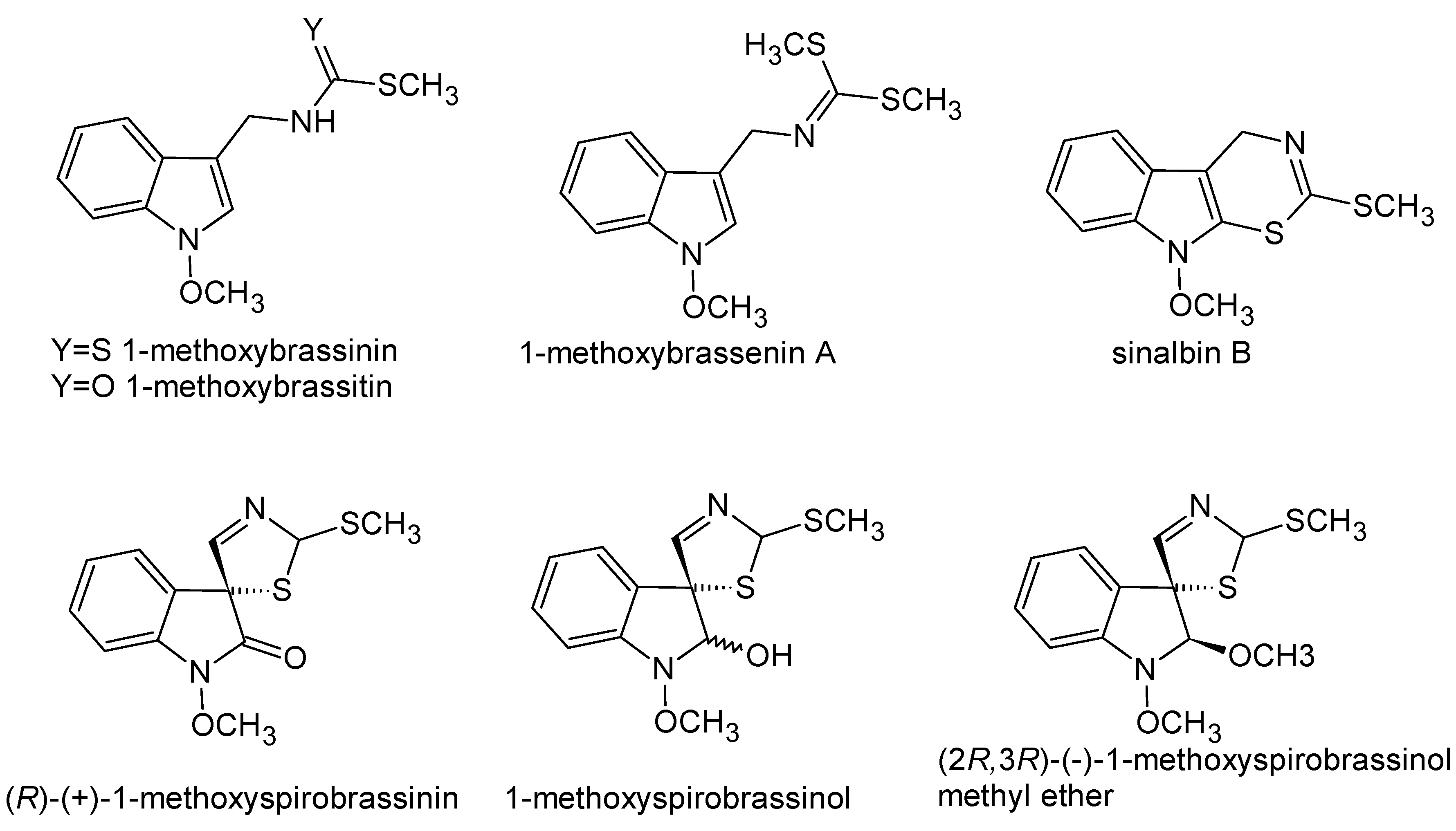
- Pedras, M.S.C.; Yaya, E.E.; Glawischnig, E. The phytoalexins from cultivated and wild crucifers: Chemistry and biology. Nat. Prod. Rep. 2011, 28, 1381–1405. [Google Scholar] [CrossRef] [PubMed]
- Chripkova, M.; Zigo, F.; Mojzis, J. Antiproliferative Effect of Indole Phytoalexins. Molecules 2016, 21, 1626. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Sarwar, M.G.; Suchy, M.; Adio, A.M. The phytoalexins from cauliflower, caulilexins A, B and C: Isolation, structure determination, syntheses and antifungal activity. Phytochemistry 2006, 67, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Zaharia, I.L. Sinalbins A and B, phytoalexins from Sinapis alba: Elicitation, isolation, and synthesis. Phytochemistry 2000, 55, 213–216. [Google Scholar] [CrossRef]
- Chakrabarty, M.; Sarkar, S.; Khasnobis, S.; Harigaya, Y.; Sato, N.; Arima, S. Study of the reactions of four indolic 1-azadienes with a few enoic, ynoic, and azo dienophiles. Synth. Commun. 2002, 32, 2295–2306. [Google Scholar] [CrossRef]
- Ramon, R.S.; Bosson, J.; Diez-Gonzalez, S.; Marion, N.; Nolan, S.P. Au/Ag-Cocatalyzed Aldoximes to Amides Rearrangement under Solvent- and Acid-Free Conditions. J. Org. Chem. 2010, 75, 1197–1202. [Google Scholar] [CrossRef]
- Goyard, D.; Konya, B.; Chajistamatiou, A.S.; Chrysina, E.D.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; Duret, C.; et al. Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur. J. Med. Chem. 2016, 108, 444–454. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, N.; Xu, J.X. A concise synthesis of cyclobrassinin and its analogues via a thiyl radical aromatic substitution. New J. Chem. 2018, 42, 13549–13557. [Google Scholar] [CrossRef]
- Budovska, M.; Pilatova, M.B.; Tischlerova, V.; Mojzis, J. Spirocyclization reactions and antiproliferative activity of indole phytoalexins 1-methoxybrassinin and its 1-substituted derivatives. Arkivoc 2016, 198–234. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Okinyo-Owiti, D.P.; Thoms, K.; Adio, A.M. The biosynthetic pathway of crucifer phytoalexins and phytoanticipins: De novo incorporation of deuterated tryptophans and quasi-natural compounds. Phytochemistry 2009, 70, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Hanley, A.B.; Parsley, K.R.; Lewis, J.A.; Fenwick, G.R. Chemistry of Indole Glucosinolates—Intermediacy of Indol-3-Ylmethyl Isothiocyanates in the Enzymatic-Hydrolysis of Indole Glucosinolates. J. Chem. Soc. Perkin. Trans. 1 1990, 1, 2273–2276. [Google Scholar] [CrossRef]
- Kawasaki, T.; Somei, M. The first total syntheses of 9-methoxycarbazole-3-carboxaldehyde and methoxybrassinin. Heterocycles 1990, 31, 1605–1607. [Google Scholar]
- Pedras, M.S.C.; Okinyo, D.P.O. Syntheses of perdeuterated indoles and derivatives as probes for the biosyntheses of crucifer phytoalexins. J. Labelled Compd. Radiopharm. 2006, 49, 33–45. [Google Scholar] [CrossRef]
- Kutschy, P.; Salayova, A.; Curillova, Z.; Kozar, T.; Mezencev, R.; Mojzis, J.; Pilatova, M.; Balentova, E.; Pazdera, P.; Sabol, M.; et al. 2-(Substituted phenyl)amino analogs of 1-methoxyspirobrassinol methyl ether: Synthesis and anticancer activity. Bioorg. Med. Chem. 2009, 17, 3698–3712. [Google Scholar] [CrossRef] [PubMed]
- Budovska, M. A novel palladium-catalyzed cyclization of indole phytoalexin brassinin and its 1-substituted derivatives. RSC Adv. 2014, 4, 5575–5582. [Google Scholar] [CrossRef]
- Sundberg, R.J. The Chemistry of Indoles in Organic Chemistry; Academic Press: New York, NY, USA, 1970; p. 405. [Google Scholar]
- Pankrushina, N.; Nikitina, I.; Chernjak, E.; Myz, C.; Shakhtshneider, T.; Boldyrev, V. Solvent-free mechanochemical modification of lappaconitine and piroxicam. Mater. Manuf. Processes 2008, 23, 561–565. [Google Scholar] [CrossRef]
- Fulmer, D.A.; Shearouse, W.C.; Medonza, S.T.; Mack, J. Solvent-free Sonogashira coupling reaction via high speed ball milling. Green Chem. 2009, 11, 1821–1825. [Google Scholar] [CrossRef]
- Cook, T.L.; Walker, J.A.; Mack, J. Scratching the catalytic surface of mechanochemistry: A multi-component CuAAC reaction using a copper reaction vial. Green Chem. 2013, 15, 617–619. [Google Scholar] [CrossRef]
- Stefanic, G.; Krehula, S.; Stefanic, I. The high impact of a milling atmosphere on steel contamination. Chem. Commun. 2013, 49, 9245–9247. [Google Scholar] [CrossRef] [PubMed]
- Miklos, F.; Hum, V.; Fulop, F. Eco-friendly syntheses of 2,2-disubstituted- and 2-spiroquinazolinones. Arkivoc 2014, 2014, 25–37. [Google Scholar]
- Do, J.L.; Mottillo, C.; Tan, D.; Strukil, V.; Friscic, T. Mechanochemical Ruthenium-Catalyzed Olefin Metathesis. J. Am. Chem. Soc. 2015, 137, 2476–2479. [Google Scholar] [CrossRef] [PubMed]
- Metro, T.X.; Bonnamour, J.; Reidon, T.; Duprez, A.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Comprehensive Study of the Organic-Solvent-Free CDI-Mediated Acylation of Various Nucleophiles by Mechanochemistry. Chem. Eur. J. 2015, 21, 12787–12796. [Google Scholar] [CrossRef] [PubMed]
- Friscic, T.; Reid, D.G.; Halasz, I.; Stein, R.S.; Dinnebier, R.E.; Duer, M.J. Ion- and Liquid-Assisted Grinding: Improved Mechanochemical Synthesis of Metal-Organic Frameworks Reveals Salt Inclusion and Anion Templating. Angew. Chem. Int. Ed. 2010, 49, 712–715. [Google Scholar] [CrossRef]
- Jiang, Z.J.; Li, Z.H.; Yu, J.B.; Su, W.K. Liquid-Assisted Grinding Accelerating: Suzuki-Miyaura Reaction of Aryl Chlorides under High-Speed Ball-Milling Conditions. J. Org. Chem. 2016, 81, 10049–10055. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D.L. Controlling reactivity through liquid assisted grinding: The curious case of mechanochemical fluorination. Green Chem. 2017, 19, 2798–2802. [Google Scholar] [CrossRef]
- Nsikabaka, S.; Harb, W.; Ruiz-Lopez, M.F. The role of water on the acid-promoted E/Z isomerization of oximes in aqueous solution. J. Mol. Struct. THEOCHEM 2006, 764, 161–166. [Google Scholar] [CrossRef]
- Vasil’tsov, A.M.; Zhang, K.; Ivanov, A.V.; Ushakov, I.A.; Afonin, A.V.; Petrushenko, K.B.; Li, S.Y.; Ma, J.S.; Mikhaleva, A.I.; Trofimov, B.A.; et al. 1-Vinylpyrrole-2-carbaldehyde oximes: Synthesis, isomerization, and spectral properties. Mon. Chem. 2009, 140, 1475–1480. [Google Scholar] [CrossRef]
- O’Ferrall, R.A.M.; O’Brien, D. Rate and equilibrium constants for hydrolysis and isomerization of (E)- and (Z)-p-methoxybenzaldehyde oximes. J. Phys. Org. Chem. 2004, 17, 631–640. [Google Scholar] [CrossRef]
- Liu, J.X.; Ma, S.M. Aerobic oxidation of indole carbinols using Fe(NO3)3.9H2O/TEMPO/NaCl as catalysts. Org. Biomol. Chem. 2013, 11, 4186–4193. [Google Scholar] [CrossRef]
- Fei, H.Y.; Yu, J.T.; Jiang, Y.; Guo, H.; Cheng, J. The ammonium-promoted formylation of indoles by DMSO and H2O. Org. Biomol. Chem. 2013, 11, 7092–7095. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, N.; Rajulu, G.G. Efficient total syntheses of phytoalexin and (+/−)-paniculidine B and C based on the novel methodology for the preparation of 1-methoxyindoles. J. Org. Chem. 2004, 69, 4429–4432. [Google Scholar] [CrossRef] [PubMed]
- Suzdalev, K.F.; Babakova, M.N. Synthesis of analogues of indole alkaloids from sea sponges—aplysinopsins by the reaction of amines with (4Z)-4-[(1H-indol-3-yl)-methylene]-1,3-oxazol-5(4H)-ones. J. Heterocycl. Chem. 2016, 53, 1200–1206. [Google Scholar] [CrossRef]
- Contreras, R.H.; Peralta, J.E. Angular dependence of spin-spin coupling constants. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 37, 321–425. [Google Scholar] [CrossRef]
- Afonin, A.V.; Ushakov, I.A.; Vashchenko, A.V.; Simonenko, D.E.; Ivanov, A.V.; Vasil’tsov, A.M.; Mikhaleva, A.I.; Trofimov, B.A. C–H...N and C–H...O intramolecular hydrogen bonding effects in the 1H, 13C and 15N NMR spectra of the configurational isomers of 1-vinylpyrrole-2-carbaldehyde oxime substantiated by DFT calculations. Magn. Reson. Chem. 2009, 47, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Palm, A.; Werbin, H. The infrared spectra of alpha and beta oximes. Can. J. Chem. 1953, 31, 1004–1008. [Google Scholar] [CrossRef]
Sample Availability: Samples of the final compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
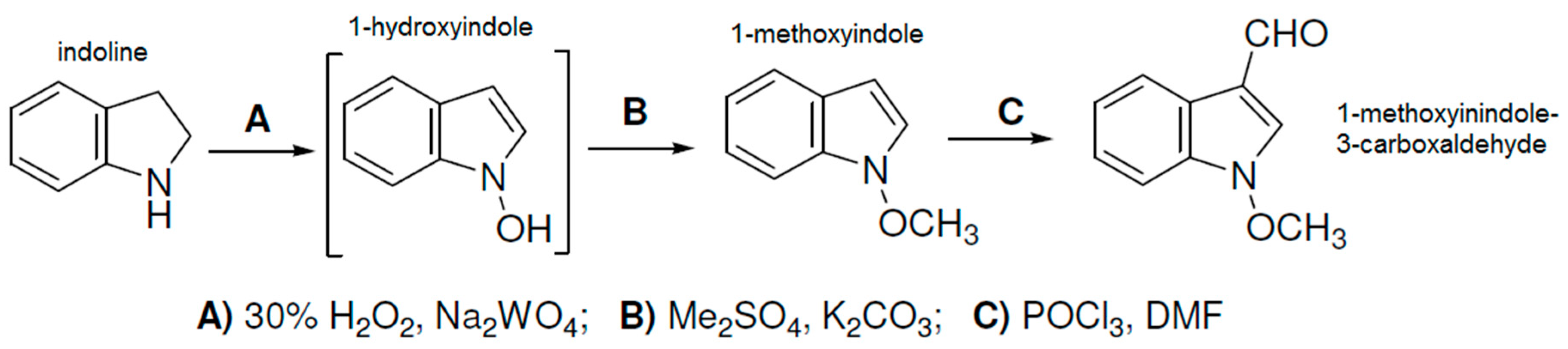{kind=link}
| Entry | Equivalents | Milling/Stirring Time (min) | Conversion from NMR | Syn:anti Oxime Ratio | Isolated Yield | |
|---|---|---|---|---|---|---|
| NH2OH·HCl | NaOH | |||||
| 1 | 3.1 | 2.5 | 30 | 100 | 35:65 | 78 |
| 2 | 5 | 2.5 | 20 | 100 | 98:2 | 72 |
| 3 a | 5 | 2 | 20 | 100 | 97:3 | 94 |
| 4 a | 5 | 5 | 20 | 100 | 43:57 | 93 |
| 5 b | 5 | 2 | 30 | 100 | 87:13 | 20 |
| 6 ac | 5 | 2 | 40 | 100 | 91:9 | 80 |
| 7 d | 5.2 | 2.7 | 40 | 100 | 30:70 | 45 |
| Aging experiments | ||||||
| 8 | 5 | 2 | 5 | 97 e | 95:5 | 25 |
| 9 | 5 | 2 | 0.25 | 85 e | 89:11 | 65 |
| N-R | Conversion from NMR | Syn:anti Ratio | Isolated Yield | Reference | ||
|---|---|---|---|---|---|---|
| Base | After Reaction | After Isomerization | ||||
| OCH3 | NaOH | 99 | 81:19 | 97:3 | 78 | Figure S26 |
| Na2CO3 | 99 | 43:57 | 43:57 | 67 | ||
| Na2CO3 | 50 | [55] | ||||
| pyridine | 60:40 a | 98 | [56] | |||
| Na2CO3 | 99 | [57] | ||||
| H | NaOH | 100 | 100:0 | 100:0 | 95 | Figure S27–S29 |
| Na2CO3 | 100 | 100:0 | 100:0 | 76 | ||
| NaOH | 91 | [49] | ||||
| NaOH | 62 | [50] | ||||
| Na2CO3 | 78 | [51] | ||||
| Na2CO3 | 98 | [52] | ||||
| CH3 | NaOH | 98 | 53:47 | 74:26 | 85 | Figure S30–S32 |
| Na2CO3 | 100 | 32:68 | 33:67 | 71 | ||
| NaOH | 68:32 b | 86 | [50] | |||
| Na2CO3 | 100 | [52] | ||||
| CO-CH3 | NaOH | 100 | 19:70:12 c | 34:45:21 c | 96 | Figure S33–S35 |
| Na2CO3 | 100 | 2:50:48 c | 0:0:100 c | 71 | ||
| NaOAc | 88 | [53] | ||||
| NaOAc | 84 | [54] | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baláž, M.; Kudličková, Z.; Vilková, M.; Imrich, J.; Balážová, Ľ.; Daneu, N.
Mechanochemical Synthesis and Isomerization of N-Substituted Indole-3-carboxaldehyde Oximes
Baláž M, Kudličková Z, Vilková M, Imrich J, Balážová Ľ, Daneu N.
Mechanochemical Synthesis and Isomerization of N-Substituted Indole-3-carboxaldehyde Oximes
Baláž, Matej, Zuzana Kudličková, Mária Vilková, Ján Imrich, Ľudmila Balážová, and Nina Daneu.
2019. "Mechanochemical Synthesis and Isomerization of N-Substituted Indole-3-carboxaldehyde Oximes
Baláž, M., Kudličková, Z., Vilková, M., Imrich, J., Balážová, Ľ., & Daneu, N.
(2019). Mechanochemical Synthesis and Isomerization of N-Substituted Indole-3-carboxaldehyde Oximes