
Site Selective Antibody-Oligonucleotide Conjugation via Microbial Transglutaminase

Abstract
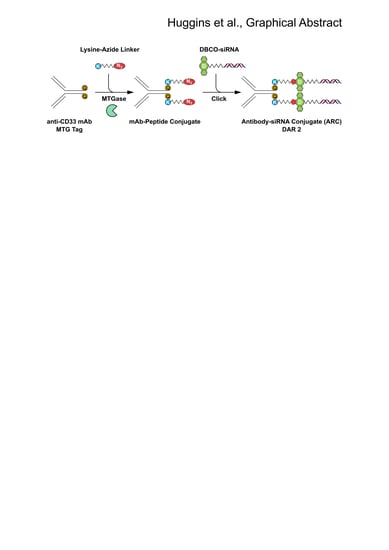
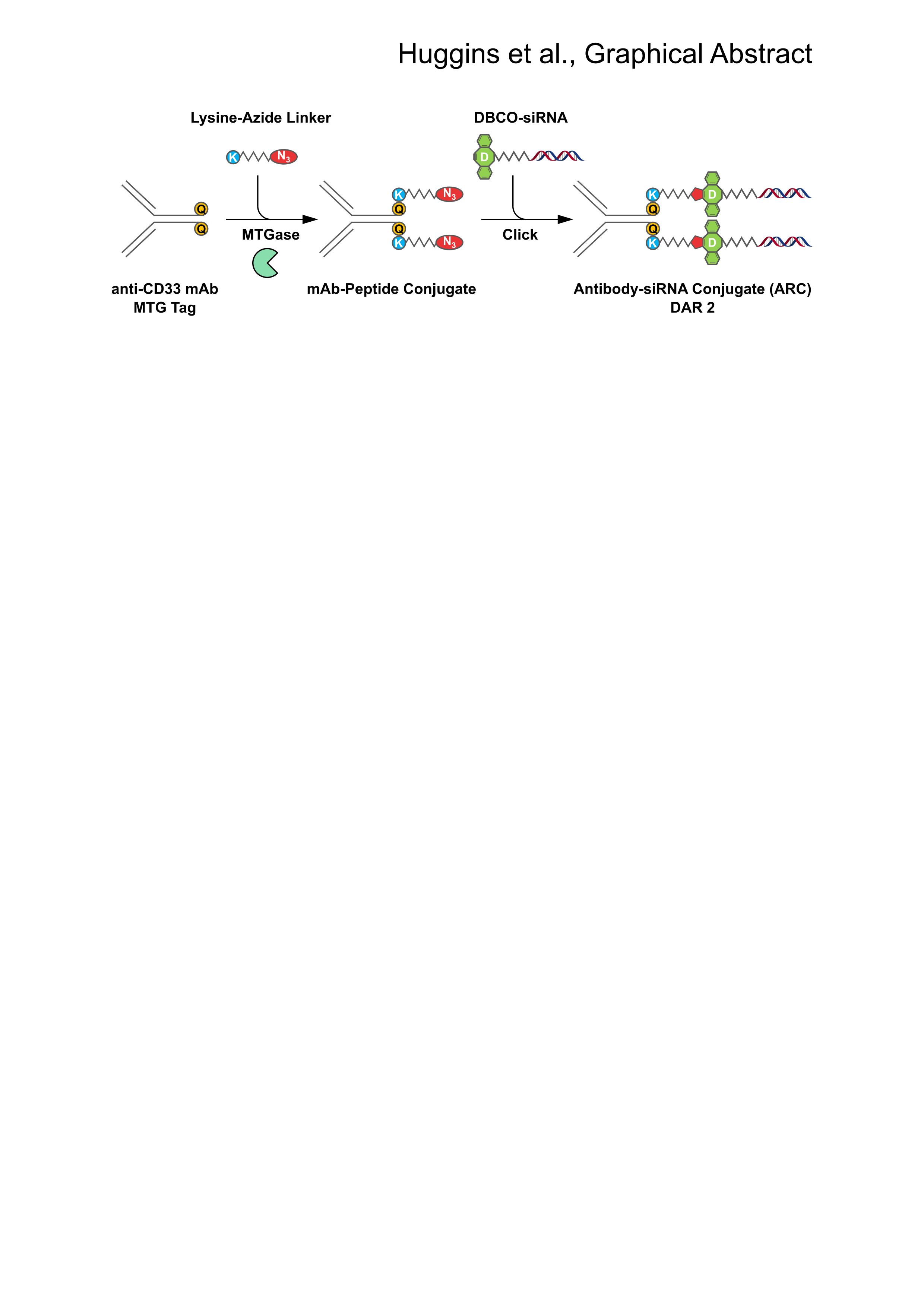{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
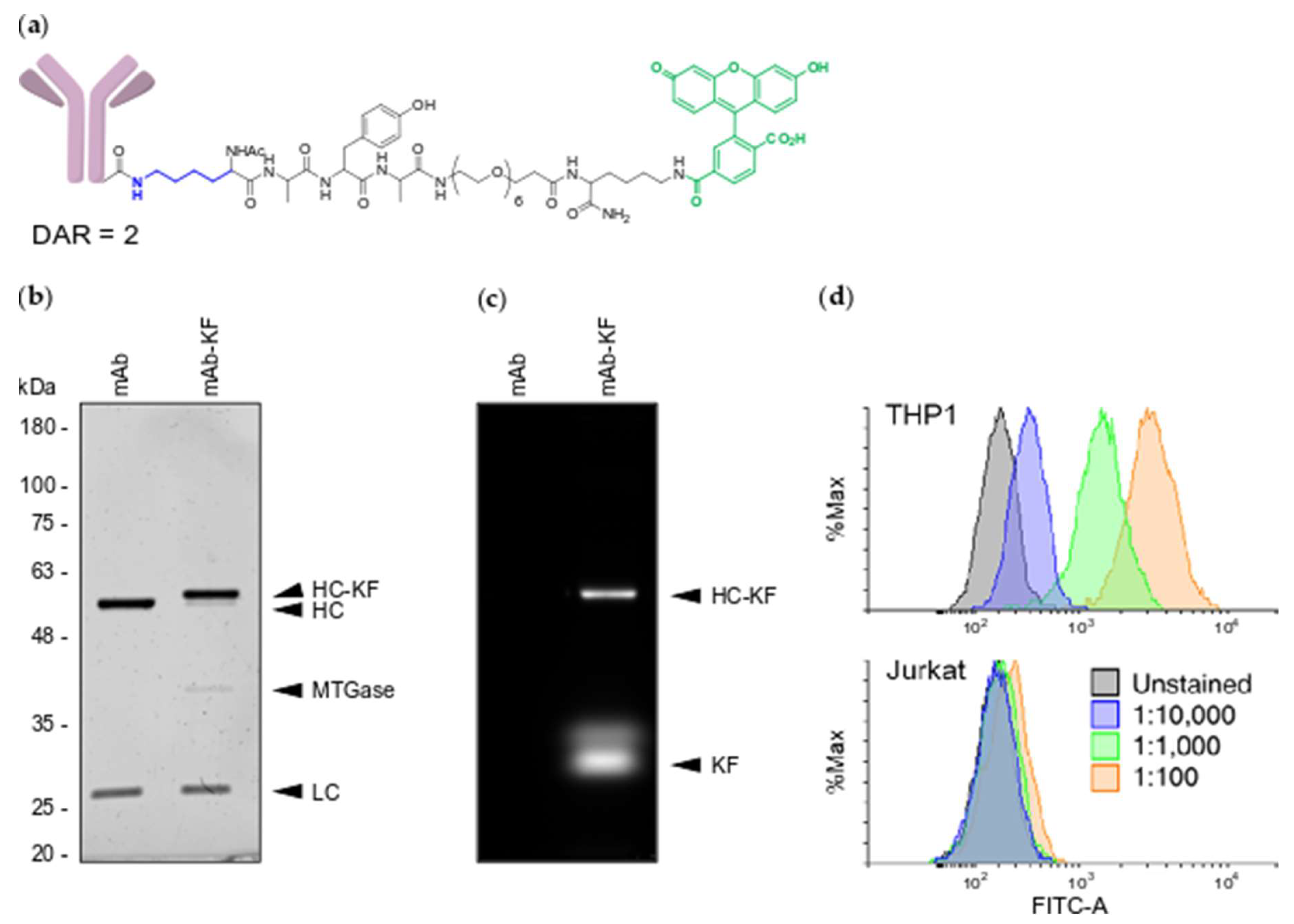
2.1. Generation of MTGase Engineered mAbs
2.2. MTGase Generation of ARCs
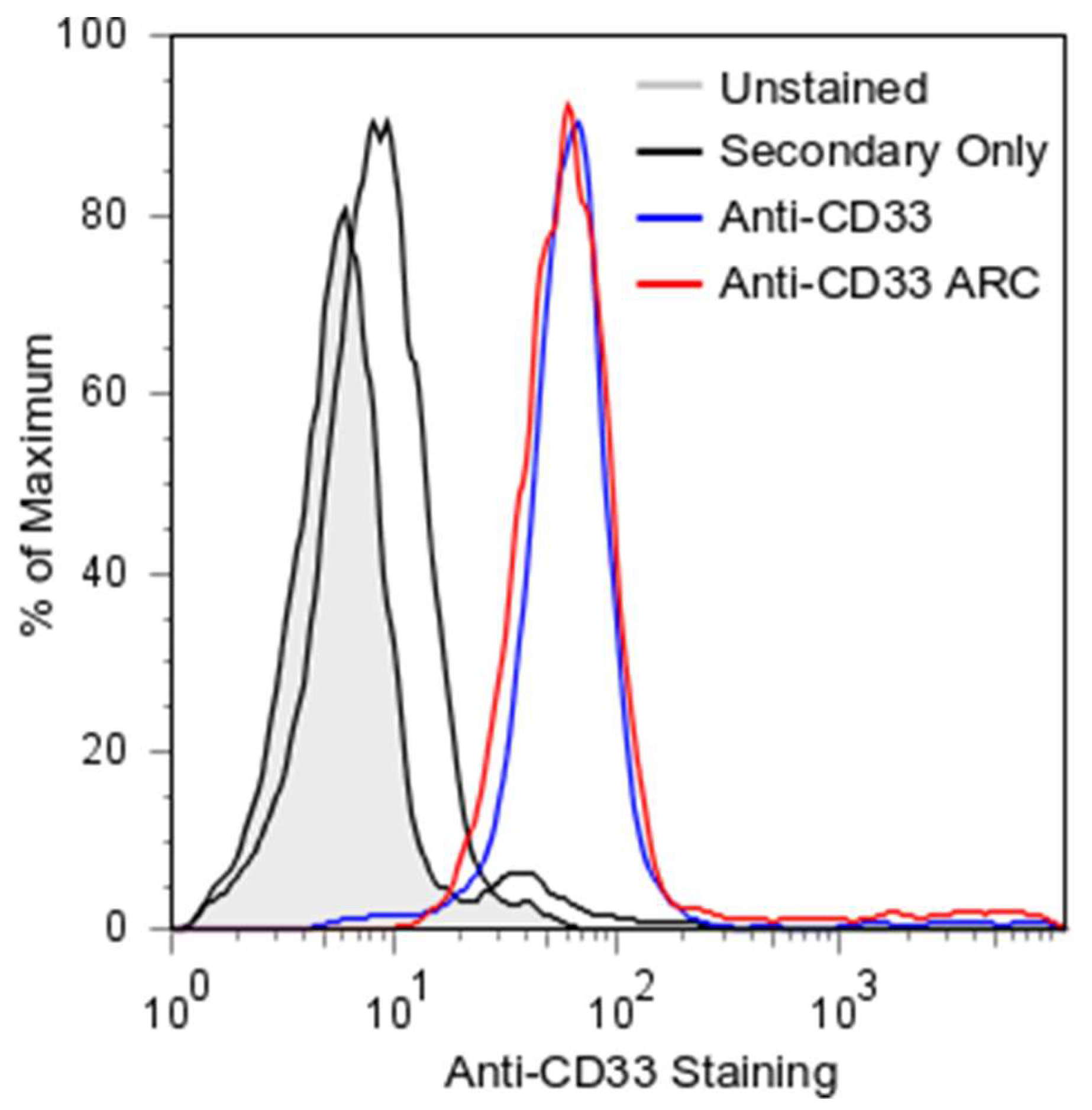
2.3. ARC Binding to CD33-Positive Cells
3. Discussion
4. Materials and Methods
4.1. Peptide Sequences and Synthesis
4.2. Oligonucleotide Sequences and Synthesis
4.3. Monoclonal Antibody Production and Purification
4.4. MTGase Conjugation
4.5. Azide-Alkyne Click Conjugation
4.6. FPLC Purification of Conjugates
4.7. Cell Culture
4.8. Cell Staining and Flow Cytometry
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-CHCA | Alpha-Cyano-4-Hydroxycinnamic Acid |
| ADC | Antibody-Drug Conjugate |
| ARC | Antibody-siRNA Conjugate |
| ASGPR | Asialoglycoprotein Receptor |
| ASO | Antisense Oligonucleotide |
| DAR | Drug-Antibody Ratio |
| DBCO | Dibenzylcyclooctyne |
| DCM | Dichloromethane |
| DIA | Diisopropylamine |
| DIPEA | N,N-Diisopropylethylamine |
| DMF | Dimethyl Formamide |
| EDTA | Ethylenediaminetetraacetic Acid |
| FACS | Fluorescence Activated Cell Sorting |
| FITC | Fluorescein Isothiocyanate |
| FPLC | Fast Protein Liquid Chromatography |
| GalNAc | N-Acetylgalactosamine |
| GMP | Good Manufacturing Practice |
| HC | Antibody Heavy Chain |
| kDa | Kilodalton |
| KF | KAYA-PEG6-K-Fluorescein |
| KN3 | K-PEG6-SG-K-N3 |
| LC | Antibody Light Chain |
| LNP | Lipid Nanoparticle |
| mAb | Monoclonal Antibody |
| MALDI-TOF | Matrix Assisted Laser Desorption Ionization Time-of-Flight |
| MTGase | Microbial Transglutaminase |
| MWCO | Molecular Weight Cutoff |
| NATs | Nucleic Acid Therapeutics |
| PBS | Phosphate Buffered Saline |
| PCR | Polymerase Chain Reaction |
| PD | Pharmacodynamic |
| PK | Pharmacokinetic |
| PS | Phosphorothioate |
| RP-HPLC | Reverse Phase High Performance Liquid Chromatography |
| SDS-PAGE | Sodium Dodecylsulfate Polyacrylamide Gel Electrophoresis |
| SEC | Size Exclusion Chromatography |
| siRNA | Small Interfering Ribonucleic Acid |
| SPAAC | Strain-Promoted Azide-Alkyne Cycloaddition |
| TEG | Triethylene Glycol |
References
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Taubel, J.; Zimmermann, T.; Karsten, V.; Martinez, C.; Chan, A.; Wang, Y.; Attarwala, H.; Gollob, J.; Vest, J. Phase 1 Study of ALN-TTRsc02, a Subcutaneously Administered Investigational RNAi Therapeutic for the Treatment of Transthyretin-Mediated Amyloidosis. 2018. Available online: http://alnylam.com/wp-content/uploads/2018/03/10.-TTR-SCO2_FINAL.pdf (accessed on 29 July 2019).
- Balwani, M.; Gouya, L.; Rees, D.; Stein, P.; Stölzel, U.; Aguilera, P.; Bissell, D.M.; Bonkovsky, H.; Keel, S.; Parker, C.; et al. ENVISION, a Phase 3 Study to Evaluate the Efficacy and Safety of Givosiran, an Investigational RNAi Therapeutic Targeting Aminolevulinic Acid Synthase 1, in Acute Hepatic Porphyria Patients. 2019. Available online: https://www.alnylam.com/wp-content/uploads/2019/04/Balwani_ENVISION_EASL_FINAL2-2.pdf (accessed on 29 July 2019).
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody-Drug Conjugate for Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. TR1801-ADC in Patients With Tumors That Express c-Met. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03859752 (accessed on 29 July 2019).
- Strop, P. Versatility of microbial transglutaminase. Bioconjug. Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef]
- Farias, S.E.; Strop, P.; Delaria, K.; Galindo Casas, M.; Dorywalska, M.; Shelton, D.L.; Pons, J.; Rajpal, A. Mass spectrometric characterization of transglutaminase based site-specific antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 240–250. [Google Scholar] [CrossRef]
- ExpiCHO Expression System User Guide. 2018. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0014337_expicho_expression_system_UG.pdf (accessed on 25 May 2018).
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- 5′ DBCO-TEG Phosphoramidite. 2019. Available online: https://www.glenresearch.com/5-dbco-teg-phosphoramidite.html (accessed on 29 July 2019).
- McCormack, T.; O’Keeffe, G.; Mac Craith, B.; O’Kennedy, R. Assessment of the Effect of Increased Fluorophore Labelling on the Binding Ability of an Antibody. Anal. Lett. 1996, 29, 953–968. [Google Scholar] [CrossRef]
- Vira, S.; Mekhedov, E.; Humphrey, G.; Blank, P.S. Fluorescent-labeled antibodies: Balancing functionality and degree of labeling. Anal. Biochem. 2010, 402, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Bagosi, A.; Szöllősi, J.; Jenei, A. Comparative study of the three different fluorophore antibody conjugation strategies. Anal. Bioanal. Chem. 2012, 404, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Szabó, Á.; Szendi-Szatmári, T.; Ujlaky-Nagy, L.; Rádi, I.; Vereb, G.; Szöllősi, J.; Nagy, P. The Effect of Fluorophore Conjugation on Antibody Affinity and the Photophysical Properties of Dyes. Biophys. J. 2018, 114, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Rickert, M.; Strop, P.; Lui, V.; Melton-Witt, J.; Farias, S.E.; Foletti, D.; Shelton, D.; Pons, J.; Rajpal, A. Production of soluble and active microbial transglutaminase in Escherichia coli for site-specific antibody drug conjugation. Protein Sci. 2016, 25, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Hickerson, R.P.; Vlassov, A.V.; Wang, Q.; Leake, D.; Ilves, H.; Gonzalez-Gonzalez, E.; Contag, C.H.; Johnston, B.H.; Kaspar, R.L. Stability study of unmodified siRNA and relevance to clinical use. Oligonucleotides 2008, 18, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Attarwala, H.; Sehgal, A.; Wang, Q.; Aluri, K.; Zhang, X.; Gao, M.; Liu, J.; Indrakanti, R.; Schofield, S.; et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017, 45, 10969–10977. [Google Scholar] [CrossRef]
- Ora, M.; Peltomäki, M.; Oivanen, M.; Lönnberg, H. Metal-ion-promoted cleavage, isomerization, and desulfurization of the diastereomeric phosphoromonothioate analogues of uridylyl(3 ‘,5 ‘)uridine. J. Org. Chem. 1998, 63, 2939–2947. [Google Scholar] [CrossRef]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huggins, I.J.; Medina, C.A.; Springer, A.D.; van den Berg, A.; Jadhav, S.; Cui, X.; Dowdy, S.F. Site Selective Antibody-Oligonucleotide Conjugation via Microbial Transglutaminase. Molecules 2019, 24, 3287. https://doi.org/10.3390/molecules24183287
Huggins IJ, Medina CA, Springer AD, van den Berg A, Jadhav S, Cui X, Dowdy SF. Site Selective Antibody-Oligonucleotide Conjugation via Microbial Transglutaminase. Molecules. 2019; 24(18):3287. https://doi.org/10.3390/molecules24183287
Chicago/Turabian StyleHuggins, Ian J., Carlos A. Medina, Aaron D. Springer, Arjen van den Berg, Satish Jadhav, Xianshu Cui, and Steven F. Dowdy. 2019. "Site Selective Antibody-Oligonucleotide Conjugation via Microbial Transglutaminase" Molecules 24, no. 18: 3287. https://doi.org/10.3390/molecules24183287
APA StyleHuggins, I. J., Medina, C. A., Springer, A. D., van den Berg, A., Jadhav, S., Cui, X., & Dowdy, S. F. (2019). Site Selective Antibody-Oligonucleotide Conjugation via Microbial Transglutaminase. Molecules, 24(18), 3287. https://doi.org/10.3390/molecules24183287