
Mechanistic and Structural Insights on the IL-15 System through Molecular Dynamics Simulations

 , , ,
, , ,
Abstract
1. Introduction
2. Methods
2.1. Structure Preparation
2.2. Molecular Dynamics Simulations
2.3. MD Analyses
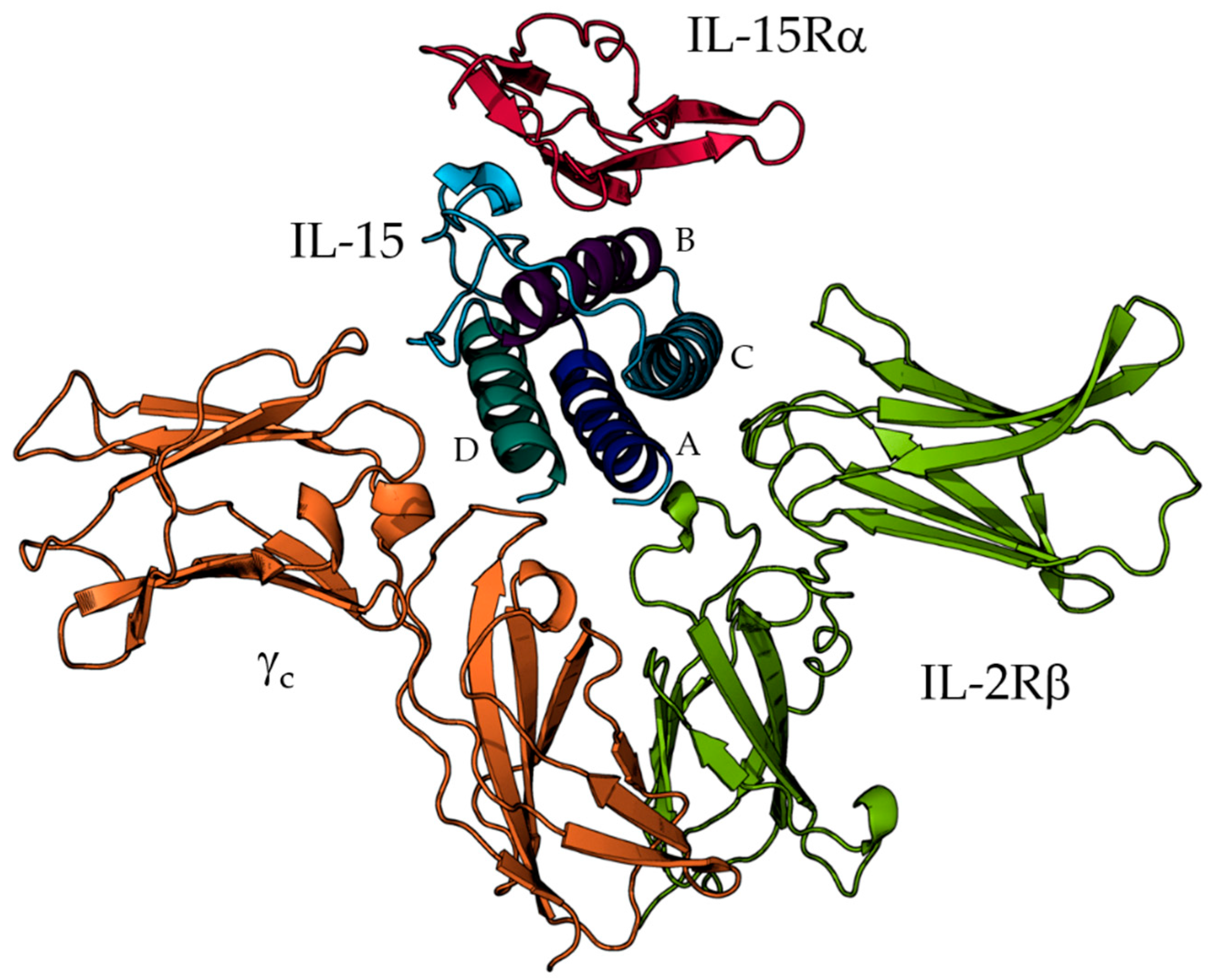
3. Results and Discussion
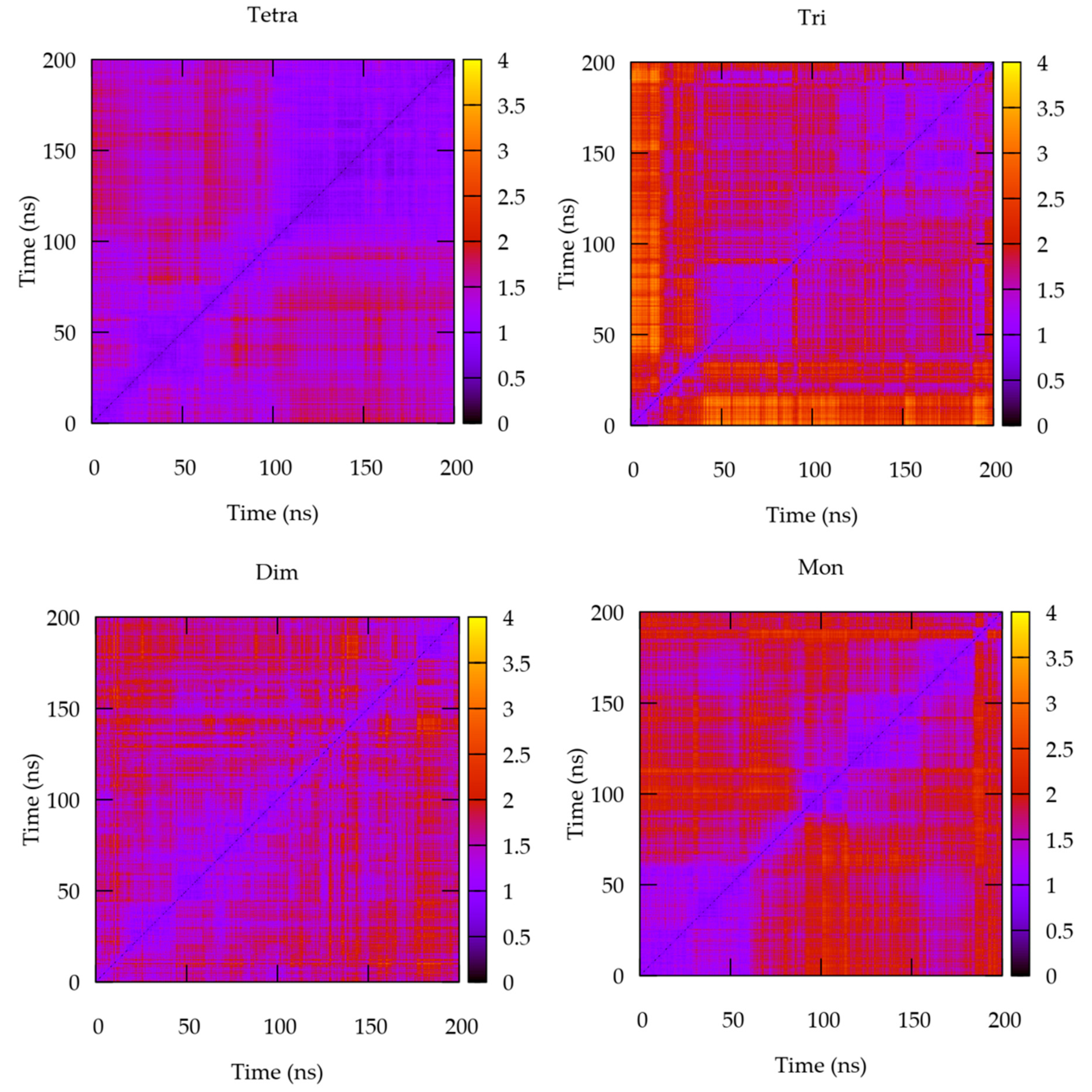
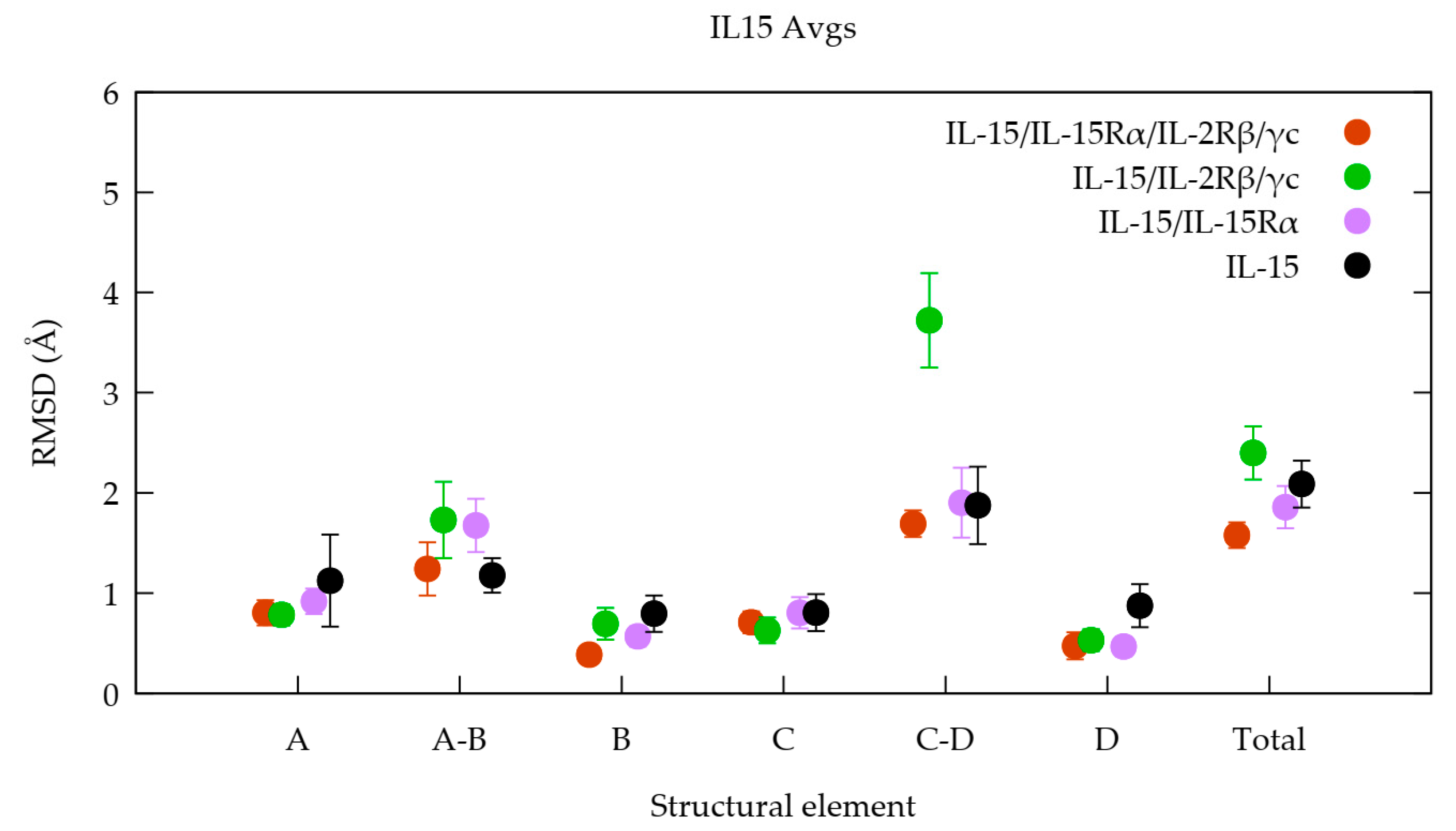
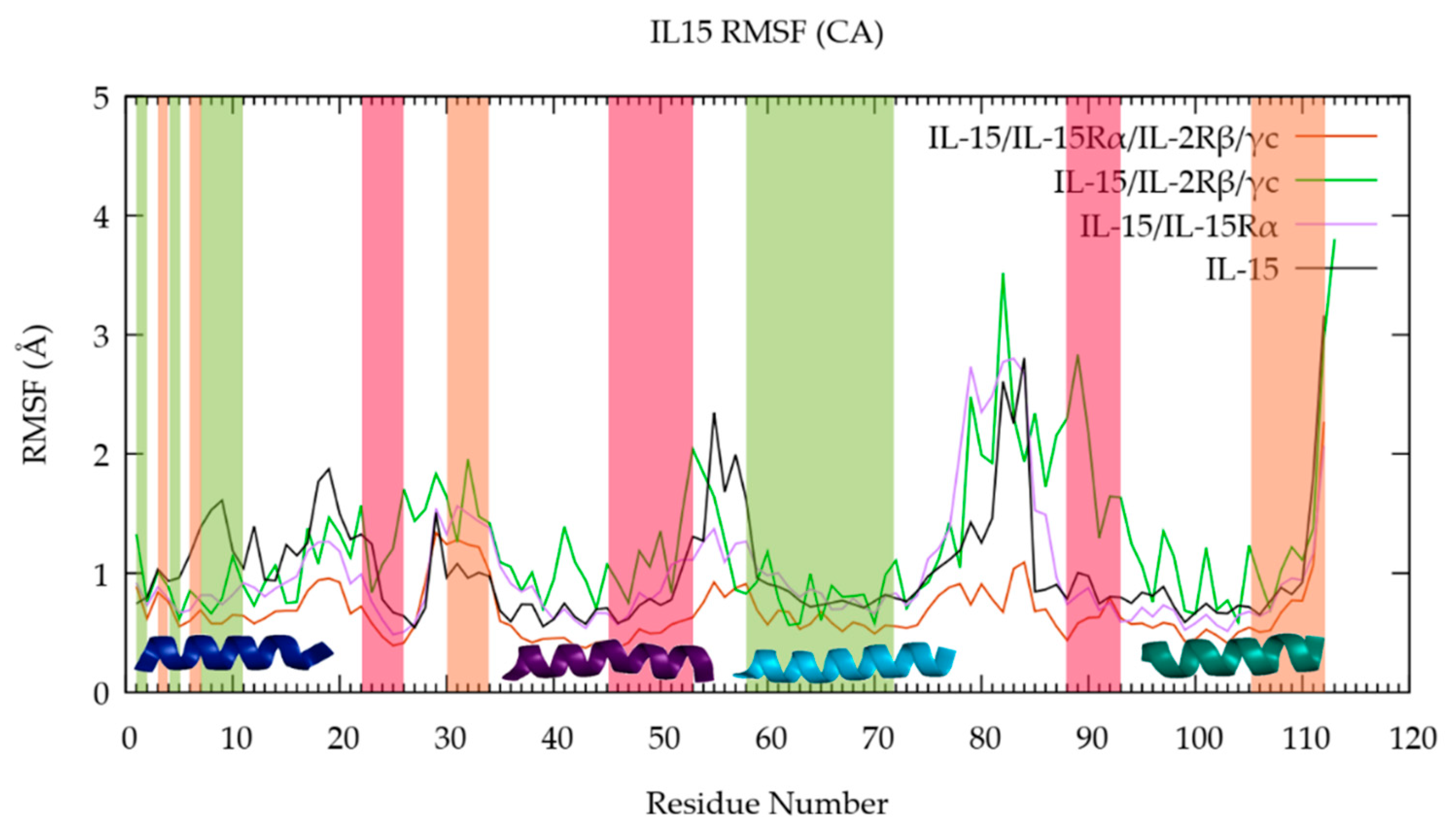
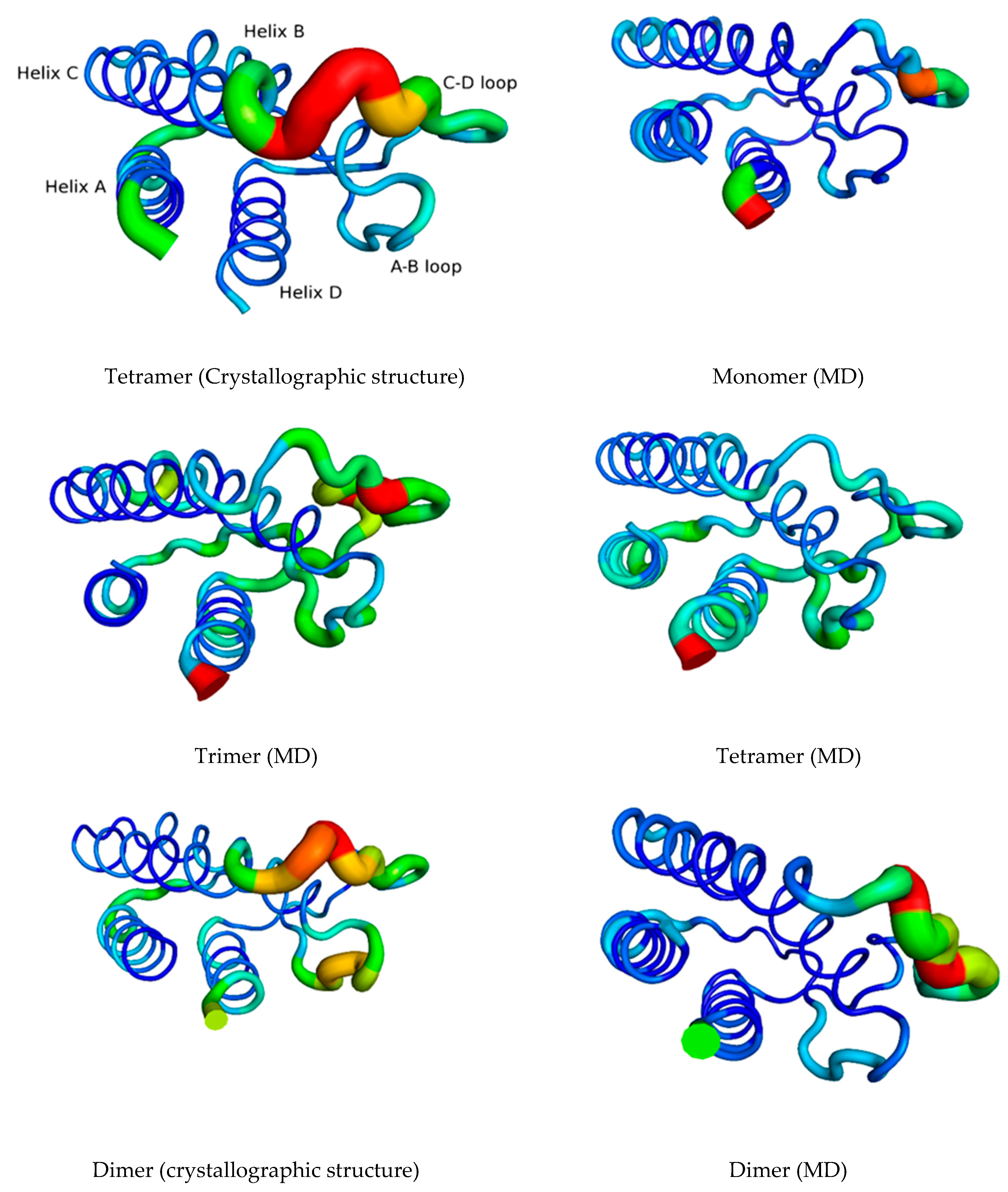
3.1. The Quaternary Structure Impacts the Flexibility of the IL-15 Receptor Interfaces
RMSF
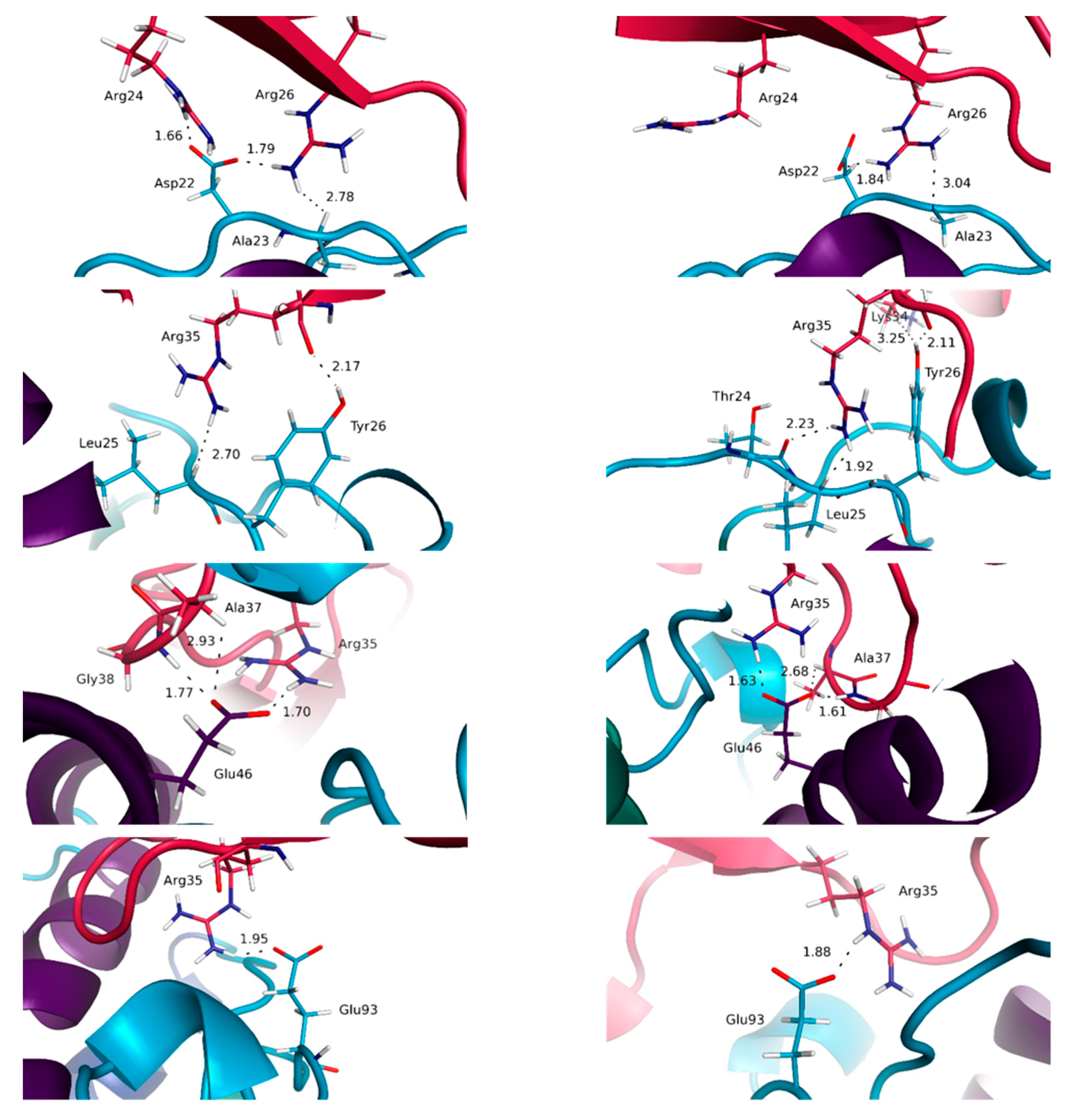
3.2. The Quaternary Structure Impacts the Structural and Energetic Features of the Various Interfaces
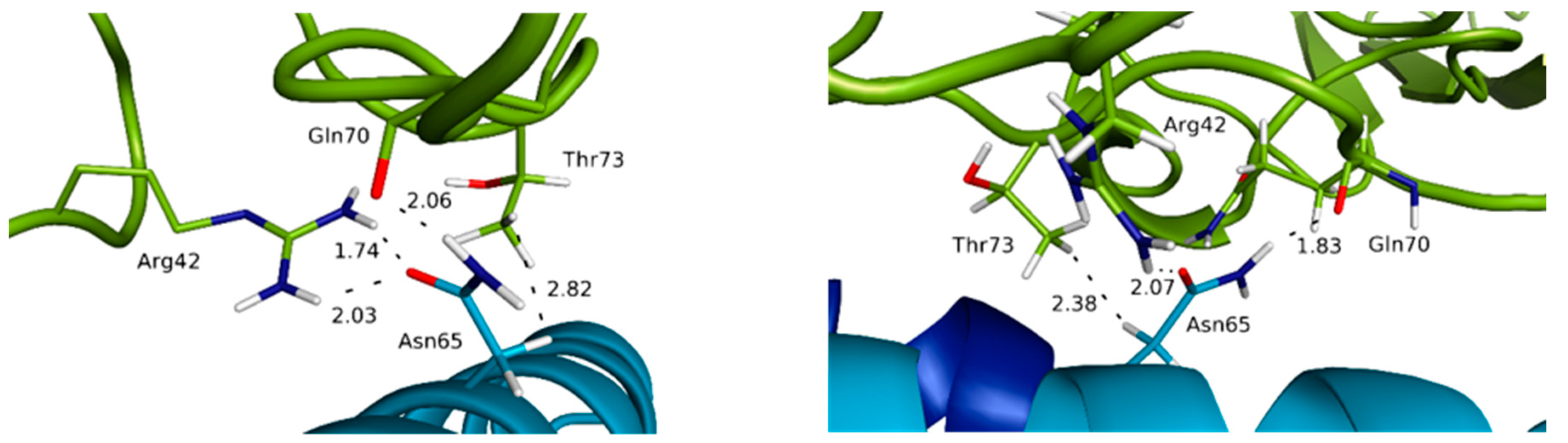
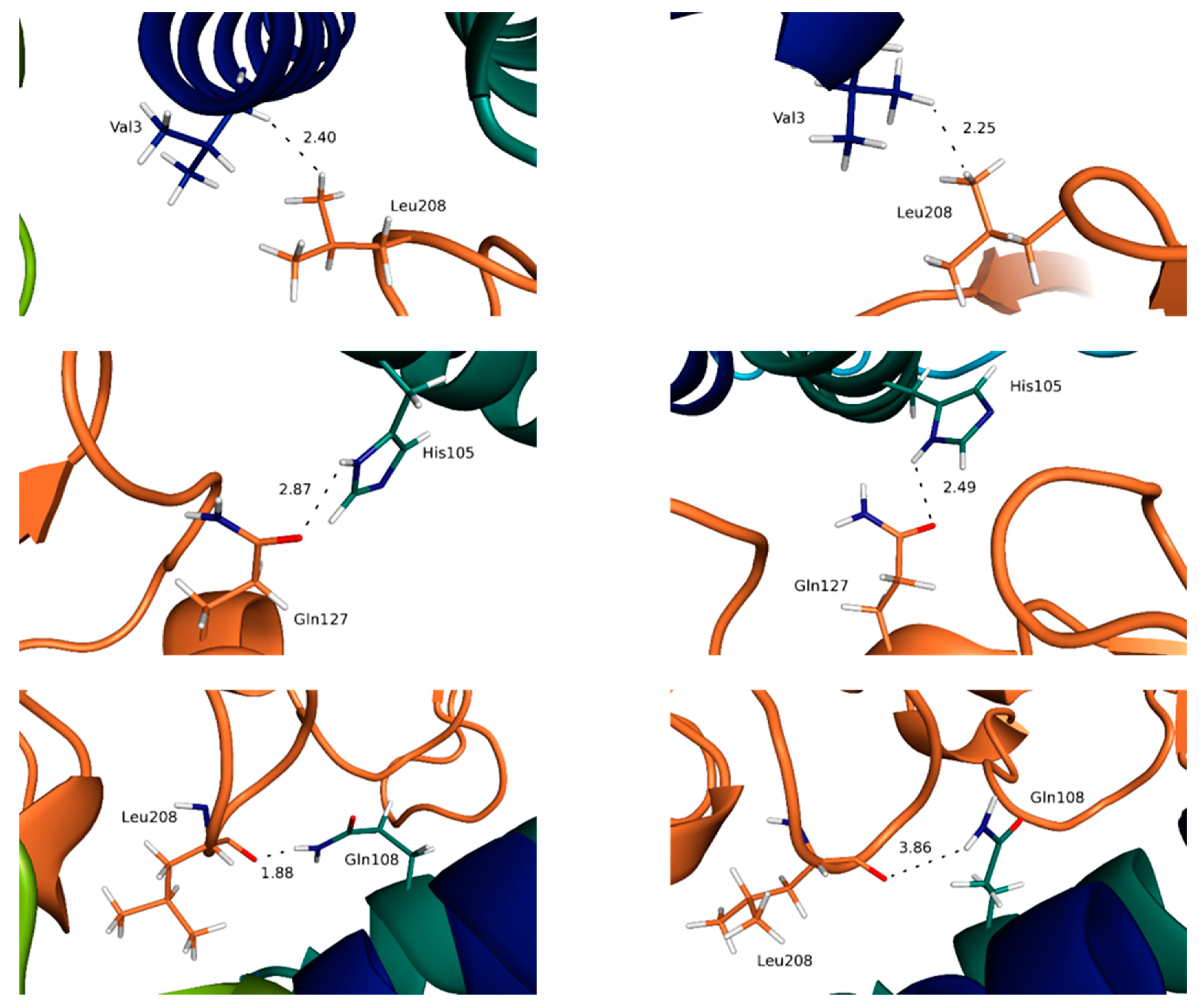
3.3. Highlighting Novel Key Structural Features at IL-15 Interfaces
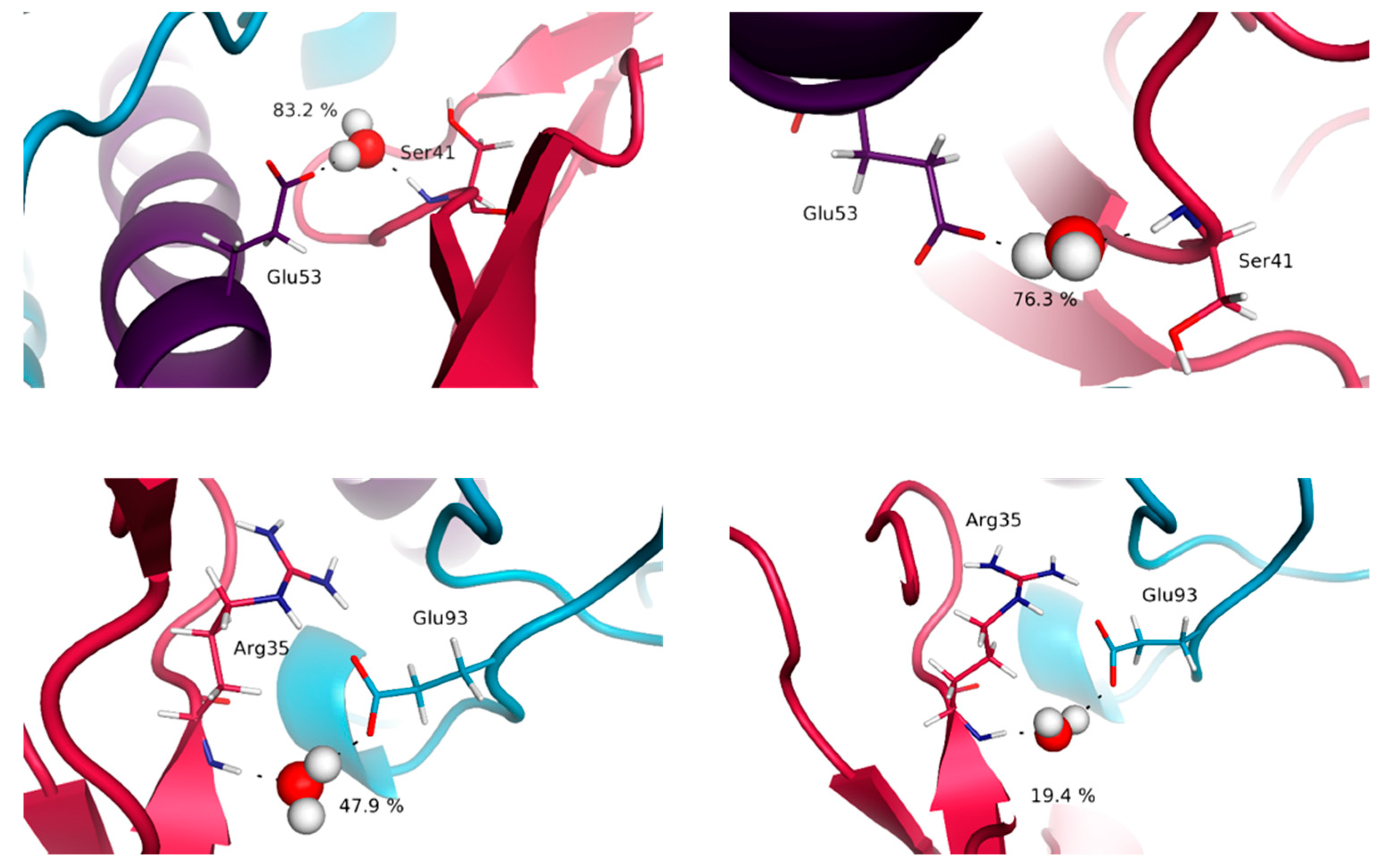
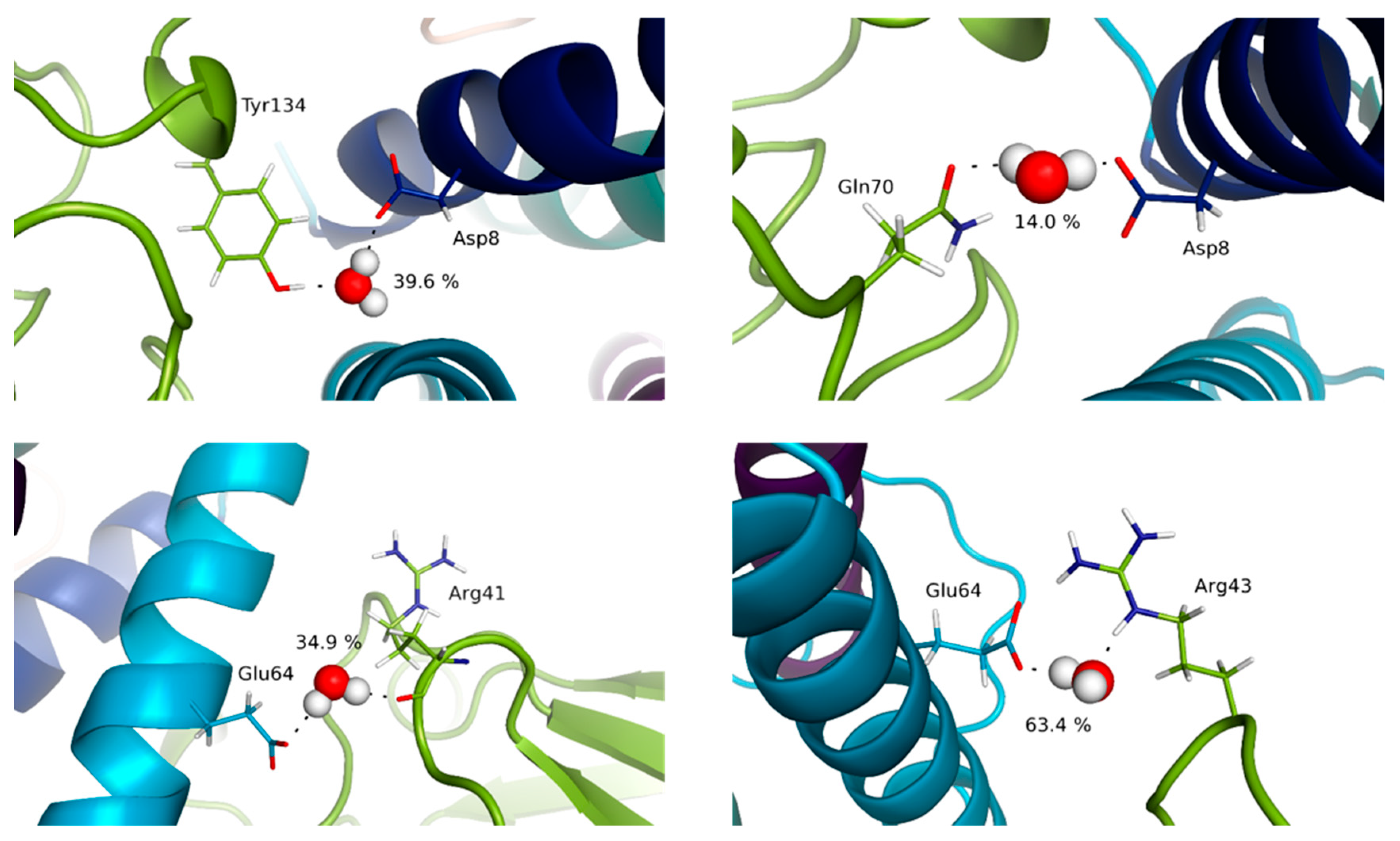
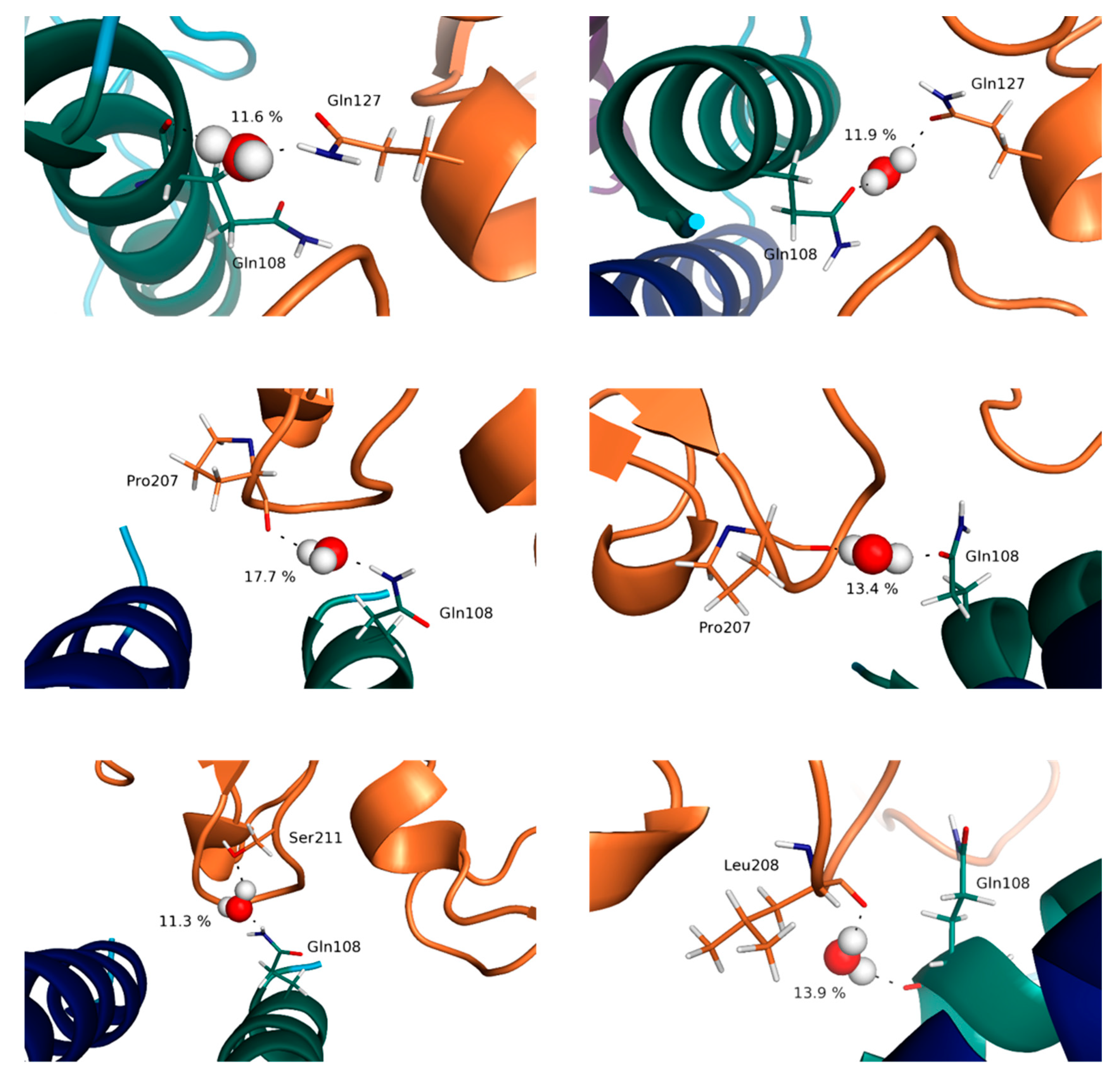
3.4. Water Molecules Stabilize the Interfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burton, J.D.; Bamford, R.N.; Peters, C.; Grant, A.J.; Kurys, G.; Goldman, C.K.; Brennan, J.; Roessler, E.; Waldmann, T.A. A lymphokine, provisionally designated interleukin T and produced by a human adult T-cell leukemia line, stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4935–4939. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Koka, R.; Burkett, P. Diverse Functions of Il-2, Il-15, And Il-7 In Lymphoid Homeostasis. Annu. Rev. Immunol. 2006, 24, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.-X.; Leonard, W.J. IL-2 Family Cytokines: New Insights into the Complex Roles of IL-2 as a Broad Regulator of T helper Cell Differentiation. Curr. Opin. Immunol. 2011, 23, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Marks-Konczalik, J.; Dubois, S.; Losi, J.M.; Sabzevari, H.; Yamada, N.; Feigenbaum, L.; Waldmann, T.A.; Tagaya, Y. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.C.; Murakami, M.; Sakamato, A.; Kappler, J.; Marrack, P. Control of homeostasis of CD8(+) memory T cells by opposing cytokines. Science 2000, 288, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef] [PubMed]
- van de Vosse, E.; van Agtmael, M.A. Targets of anticytokine therapy and the risk of infections in humans and mice. Curr. Opin. Rheumatol. 2007, 19, 626–635. [Google Scholar] [CrossRef]
- Johnston, J.A.; Bacon, C.M.; Finbloom, D.S.; Rees, R.C.; Kaplan, D.; Shibuya, K.; Ortaldo, J.R.; Gupta, S.; Chen, Y.Q.; Giri, J.D.; et al. Tyrosine Phosphorylation and Activation of Stat5, Stat3, And Janus Kinases by Interleukin-2 And Interleukin-15. Proc. Natl. Acad. Sci. USA 1995, 92, 8705–8709. [Google Scholar] [CrossRef]
- Xq, W.; Orchardson, M.; A Gracie, J.; Leung, B.P.; Bm, G.; Guan, H.; Niedbala, W.; Paterson, G.K.; McInnes, I.B.; Liew, F.Y. The Sushi domain of soluble IL-15 receptor alpha is essential for binding IL-15 and inhibiting inflammatory and allogenic responses in vitro and in vivo. J. Immunol. 2001, 167, 277–282. [Google Scholar]
- Chirifu, M.; Hayashi, C.; Nakamura, T.; Toma, S.; Shuto, T.; Kai, H.; Yamagata, Y.; Davis, S.J.; Ikemizu, S. Crystal structure of the IL-15-IL-15R alpha complex, a cytokine-receptor unit presented in trans. Nat. Immunol. 2007, 8, 1001–1007. [Google Scholar] [CrossRef]
- Ring, A.M.; Lin, J.X.; Feng, D.; Mitra, S.; Rickert, M.; Bowman, G.R.; Pande, V.S.; Li, P.; Moraga, I.; Spolski, R.; et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat. Immunol. 2012, 13, 1187. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.M.; Patterson, A.; Johnson, C.B.; Neitzke, D.J.; Mehrotra, S.; Denlinger, C.E.; Paulos, C.M.; Li, Z.; Cole, D.J.; Rubinstein, M.P. IL-2 and Beyond in Cancer Immunotherapy. J. Interf. Cytokine Res. 2018, 38, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.M.; StGallay, S.; Kleywegt, G.J.; A St-Gallay, S. Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov. Today 2008, 13, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynska, M.; Ottmann, C. Protein–protein interactions as drug targets. Future Med. Chem. 2015, 7, 2195–2219. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J. Inhibition of protein–protein interactions using designed molecules. Chem. Soc. Rev. 2009, 38, 3289. [Google Scholar] [CrossRef] [PubMed]
- Schillinger, O.; Panwalkar, V.; Strodel, B.; Dingley, A.J. Molecular Dynamics Simulations Reveal Key Roles of the Interleukin-6 Alpha Receptor in the Assembly of the Human Interleukin-6 Receptor Complex. J. Phys. Chem. B 2017, 121, 8113–8122. [Google Scholar] [CrossRef]
- Bobby, R.; Robustelli, P.; Kralicek, A.V.; Mobli, M.; King, G.F.; Grötzinger, J.; Dingley, A.J. Functional implications of large backbone amplitude motions of the glycoprotein 130-binding epitope of interleukin-6. FEBS J. 2014, 281, 2471–2483. [Google Scholar] [CrossRef]
- Silva, D.-A.; Yu, S.; Ulge, U.Y.; Spangler, J.B.; Jude, K.M.; Labão-Almeida, C.; Ali, L.R.; Quijano-Rubio, A.; Ruterbusch, M.; Leung, I.; et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 2019, 565, 186–191. [Google Scholar] [CrossRef]
- Quéméner, A.; Maillasson, M.; Arzel, L.; Sicard, B.; Vomiandry, R.; Mortier, E.; Dubreuil, D.; Jacques, Y.; Lebreton, J.; Mathé-Allainmat, M. Discovery of a Small-Molecule Inhibitor of Interleukin 15: Pharmacophore-Based Virtual Screening and Hit Optimization. J. Med. Chem. 2017, 60, 6249–6272. [Google Scholar] [CrossRef]
- Żyżyńska-Granica, B.; Trzaskowski, B.; Niewieczerzał, S.; Filipek, S.; Zegrocka-Stendel, O.; Dutkiewicz, M.; Krzeczyński, P.; Kowalewska, M.; Koziak, K. Pharmacophore guided discovery of small-molecule interleukin 15 inhibitors. Eur. J. Med. Chem. 2017, 136, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Bioinformat. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, J.; Impey, R.W.; Jorgensen, W.L.; Madura, J.D.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The Amber Home Page. Available online: http://ambermd.org/index.php (accessed on 2 September 2019).
- Stella, L.; Melchionna, S. Equilibration and sampling in molecular dynamics simulations of biomolecules. J. Chem. Phys. 1998, 109, 10115–10117. [Google Scholar] [CrossRef]
- Anderson, D.M.; Kumaki, S.; Ahdieh, M.; Bertles, J.; Tometsko, M.; Loomis, A.; Giri, J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. Functional Characterization of the Human Interleukin-15 Receptor αChain and Close Linkage of IL15RA and IL2RA Genes. J. Biol. Chem. 1995, 270, 29862–29869. [Google Scholar]
- Sakamoto, S.; Caaveiro, J.M.; Sano, E.; Tanaka, Y.; Kudou, M.; Tsumoto, K. Contributions of Interfacial Residues of Human Interleukin15 to the Specificity and Affinity for Its Private α-Receptor. J. Mol. Biol. 2009, 389, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Mortier, E.; Quéméner, A.; Vusio, P.; Lorenzen, I.; Boublik, Y.; Grötzinger, J.; Plet, A.; Jacques, Y. Soluble Interleukin-15 Receptor α (IL-15Rα)-sushi as a Selective and Potent Agonist of IL-15 Action through IL-15Rβ/γ: Hyperagonist Il-15·Il-15rα Fusion Proteins. J. Biol. Chem. 2006, 281, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.; Harb, C.; Mortier, E.; Meloen, R.H.; Vermot-Desroches, C.; Wijdeness, J.; Van Dijken, P.; Slootstra, J.W.; Plet, A.; Jacques, Y.; et al. Identification of an Interleukin-15α Receptor-binding Site on Human Interleukin-15. J. Biol. Chem. 2004, 279, 24313–24322. [Google Scholar] [CrossRef] [PubMed]
- Quéméner, A.; Bernard, J.; Mortier, E.; Plet, A.; Jacques, Y.; Tran, V. Docking of human interleukin-15 to its specific receptor α chain: Correlation between molecular modeling and mutagenesis experimental data. Proteins Struct. Funct. Bioinformat. 2006, 65, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Chernov-Rogan, T.; Whitehorn, E.; Tate, E.; Bell, M.P.; Barrett, R.W.; Balasubramanian, S.; Davis, A.M.; Zurawski, G. Ligand binding kinetics of IL-2 and IL-15 to heteromers formed by extracellular domains of the three IL-2 receptor subunits. Int. Immunol. 1995, 7, 1839–1849. [Google Scholar]
- Flemming, C.L.; Russell, S.J.; Collins, M.K.L. Mutation of Asp20of human interleukin-2 reveals a dual role of the p55 α chain of the interleukin-2 receptor. Eur. J. Immunol. 1993, 23, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, A.B.; Lin, Y.; Shanafelt, M.-C.; Forte, C.P.; Dubois-Stringfellow, N.; Carter, C.; Gibbons, J.A.; Cheng, S.-L.; Delaria, K.A.; Fleischer, R.; et al. A T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat. Biotechnol. 2000, 18, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Bonnert, T.P. Structure-Function Studies of Interleukin 15using Site-specific Mutagenesis, Polyethylene Glycol Conjugation, and Homology Modeling. J. Biol. Chem. 1997, 272, 2312–2318. [Google Scholar]
- Collins, L.; Tsien, W.H.; Seals, C.; Hakimi, J.; Weber, D.; Bailon, P.; Hoskings, J.; Greene, W.C.; Toome, V.; Ju, G. Identification of specific residues of human interleukin 2 that affect binding to the 70-kDa subunit (p70) of the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 7709. [Google Scholar] [CrossRef]
- Wang, X.Q.; Rickert, M.; Garcia, K.C. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gamma(c) receptors. Science 2005, 310, 1159–1163. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Contacts | ΔGbind | ||||||
|---|---|---|---|---|---|---|---|
| Interface | Model | Dimer | Trimer | Tetramer | Dimer | Trimer | Tetramer |
| IL-15/IL-15Rα | 43 (4) | - | 41 (3) | −80.3 (6.6) | −83.8 (8.2) | ||
| IL-15/IL-2Rβ | 28 (4) | 26 (4) | −29.1 (5.6) | −27.2 (6.6) | |||
| IL-15/γc | 18 (5) | 18 (7) | −16.2 (7.7) | −17.3 (10.5) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, R.P.; Laurent, A.D.; Quéméner, A.; Mortier, E.; Le Questel, J.-Y. Mechanistic and Structural Insights on the IL-15 System through Molecular Dynamics Simulations. Molecules 2019, 24, 3261. https://doi.org/10.3390/molecules24183261
Sousa RP, Laurent AD, Quéméner A, Mortier E, Le Questel J-Y. Mechanistic and Structural Insights on the IL-15 System through Molecular Dynamics Simulations. Molecules. 2019; 24(18):3261. https://doi.org/10.3390/molecules24183261
Chicago/Turabian StyleSousa, Rui P., Adèle D. Laurent, Agnès Quéméner, Erwan Mortier, and Jean-Yves Le Questel. 2019. "Mechanistic and Structural Insights on the IL-15 System through Molecular Dynamics Simulations" Molecules 24, no. 18: 3261. https://doi.org/10.3390/molecules24183261
APA StyleSousa, R. P., Laurent, A. D., Quéméner, A., Mortier, E., & Le Questel, J.-Y. (2019). Mechanistic and Structural Insights on the IL-15 System through Molecular Dynamics Simulations. Molecules, 24(18), 3261. https://doi.org/10.3390/molecules24183261