
Simultaneous Quantification of Four Phenylethanoid Glycosides in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study of Acanthus Ilicifolius Herb

 ,
, 

Abstract
:1. Introduction
2. Results and Discussion
2.1. Establishment and Optimization of the UPLC-MS/MS Method
2.2. Contents of the Four PhGs in AIH
2.3. Method Validation
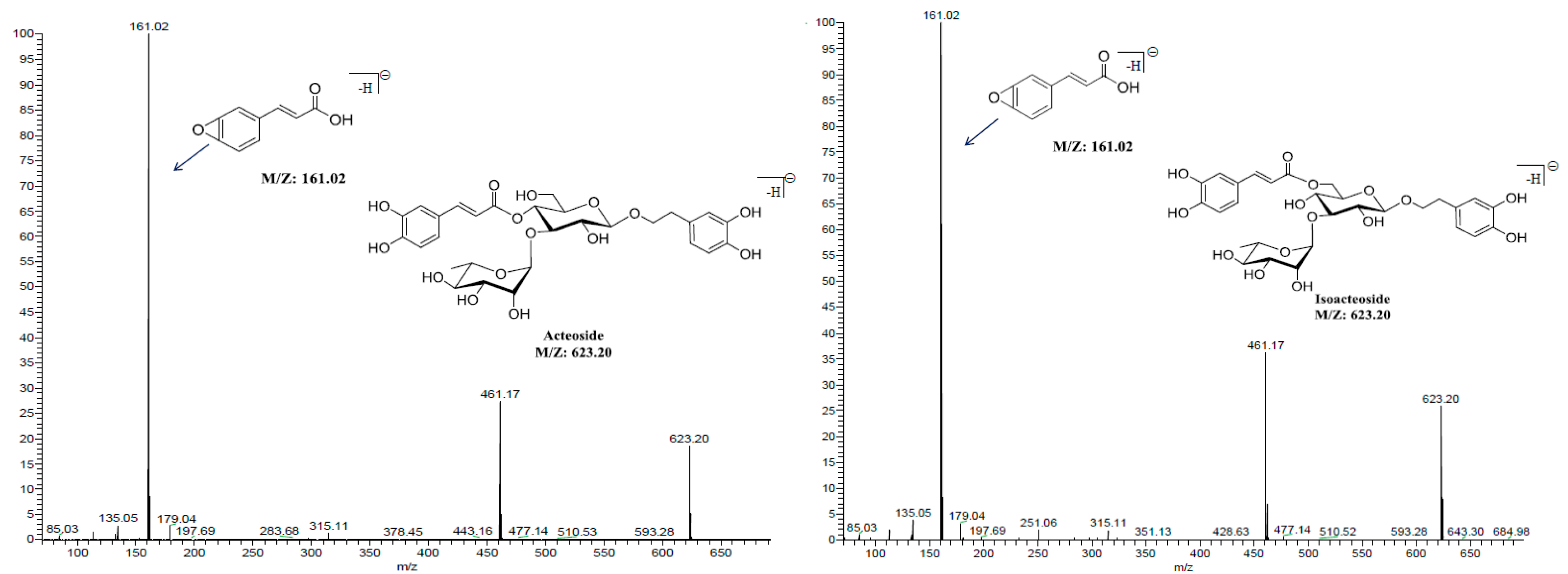
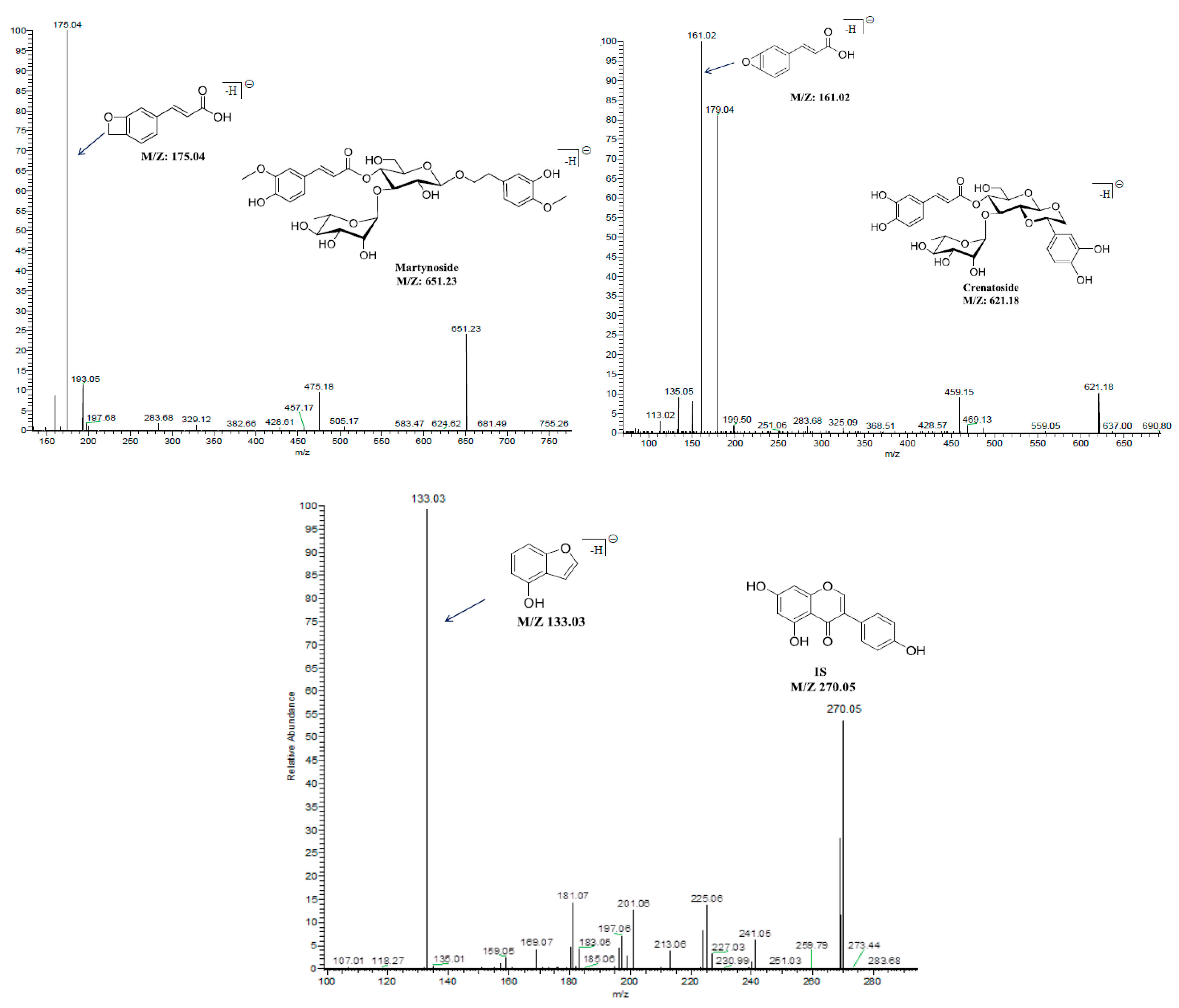
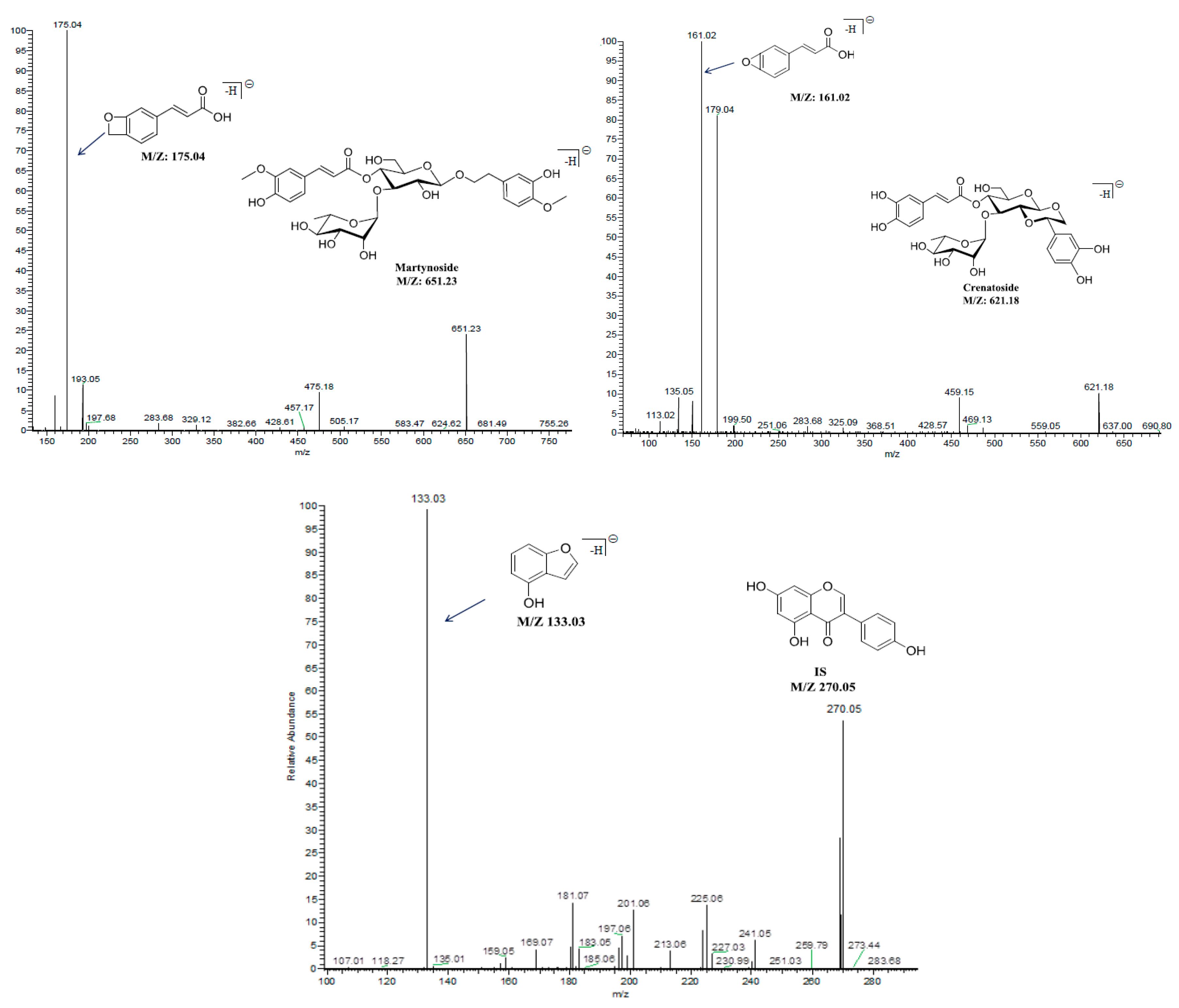
2.3.1. Selectivity
2.3.2. Linearity and LLOQ
2.3.3. Accuracy and Precision
2.3.4. Extraction Recovery and Matrix Effect
2.3.5. Stability
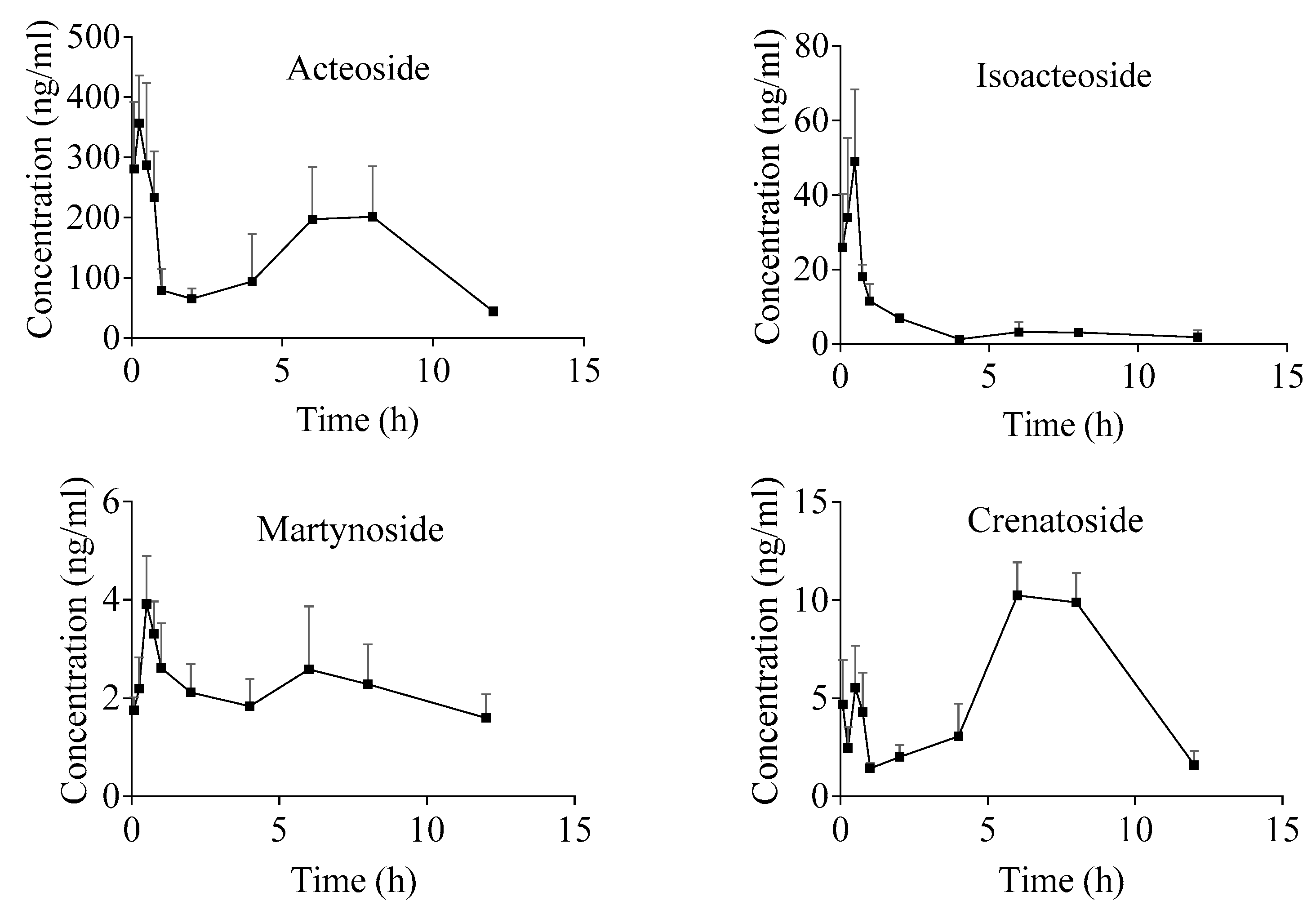
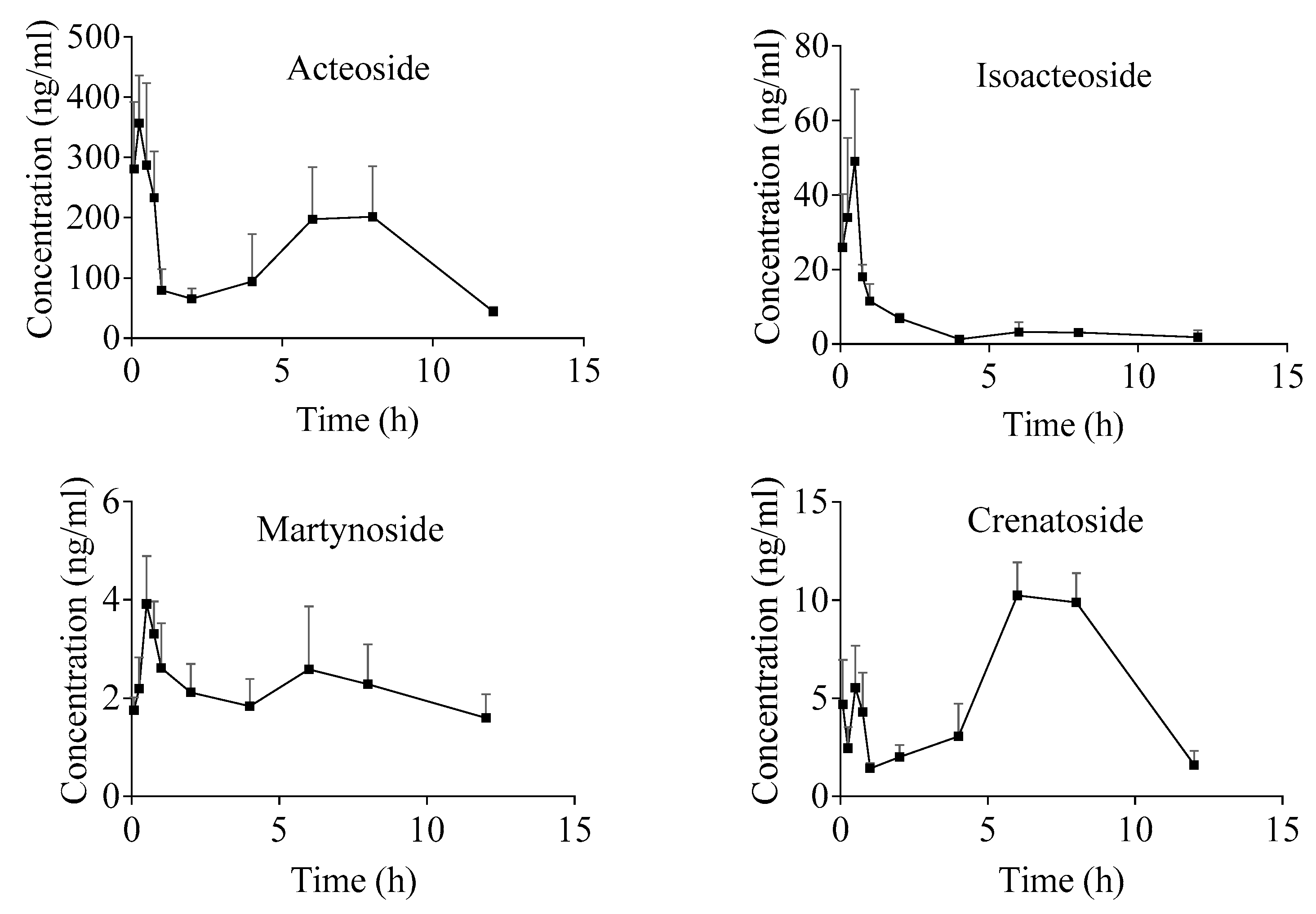
2.4. Pharmacokinetic Study
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of AIH Extracts
3.3. Animals
3.4. Preparation of Stock Solutions, Calibration Samples, and Quality Control Samples
3.5. Pretreatment of Calibration Samples and QC Samples
3.6. Instrumentation and Chromatographic Conditions
3.7. Method Validation
3.8. Pharmacokinetic Study
3.9. Data Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nanjing University of Chinese Medicine. Dictionary of Chinese Herbal Medicine; Shanghai Science and Technology Press: Shanghai, China, 1977; pp. 844–845. [Google Scholar]
- Guan, H.S.; Wang, S.G. Chinese Marine Materia Medica; Shanghai Scientific and Technical Publishers: Shanghai, China, 2009; Volume 2, pp. 371–372. [Google Scholar]
- Guan, H.S.; Wang, S.G. Selection of Chinese Marine Materia Medica; Shanghai Scientific and Technical Publishers: Shanghai, China, 2013; pp. 96–98. [Google Scholar]
- Guan, H.S.; Wang, S.G. Illustrated Handbook of Chinese Marine Materia Medica; Shanghai Scientific and Technical Publishers: Shanghai, China, 2015; Volume 1, pp. 228–231. [Google Scholar]
- Fu, X.M.; Zhang, M.Q.; Shao, C.L.; Li, G.Q.; Bai, H.; Dai, G.L.; Chen, Q.W.; Kong, W.; Fu, X.J.; Wang, C.Y. Chinese Marine Materia Medica Resources: Status and Potential. Mar. Drugs 2016, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Babu, B.H.; Shylesh, B.S.; Padikkala, J. Antioxidant and hepatoprotective effect of Acanthus ilicifolius. Fitoterapia 2001, 72, 272–277. [Google Scholar] [CrossRef]
- Babu, B.H.; Shylesh, B.S.; Padikkala, J. Tumour reducing and anticarcinogenic. activity of Acanthus ilicifolius in mice. J. Ethnopharmacol. 2002, 79, 27–33. [Google Scholar] [CrossRef]
- Kumar, K.T.M.S.; Gorain, B.; Roy, D.K.; Samanta, S.K.; Pal, M.; Biswas, P.; Roy, A.; Adhikari, D.; Karmakar, S.; Sen, T. Anti-inflammatory activity of Acanthus ilicifolius. J. Ethnopharmacol. 2008, 120, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Kalaskar, P.S.; Karande, V.V.; Bannalikar, A.S.; Gatne, M.M. Antifungal activity of leaves of mangroves plant Acanthus ilicifolius against Aspergillus fumigatus. Indian J. Pharm. Sci. 2012, 74, 575–579. [Google Scholar] [CrossRef]
- Firdaus, M.; Prihanto, A.A.; Nurdiani, R. Antioxidant and cytotoxic activity of Acanthus ilicifolius flower. Asian Pac. J. Trop. Biomed. 2013, 3, 17–21. [Google Scholar] [CrossRef]
- Wahidulla, S.; Bhattacharjee, J.J. Benzoxazinoids from Acanthus ilicifolius. J. Indian Inst. Sci. 2001, 81, 485–490. [Google Scholar]
- Kanchanapoom, T.; Kamel, M.S.; Kasai, R.; Yamasaki, K.; Picheansoonthon, C.; Hiraga, Y. Benzoxazinoid glucosides from Acanthus ilicifolius. Phytochemistry 2001, 58, 637–640. [Google Scholar] [CrossRef]
- Kanchanapoom, T.; Kamel, M.S.; Kasai, R.; Yamasaki, K.; Picheansoonthon, C.; Hiraga, Y. Lignan glucosides from Acanthus ilicifolius. Phytochemistry 2001, 56, 369–372. [Google Scholar] [CrossRef]
- Kanchanapoom, T.; Kasai, R.; Yamasaki, K. Flavonoid glycosides from Acanthus ilicifolius L. Nat. Med. 2002, 56, 122. [Google Scholar]
- Wu, J.; Zhang, S.; Huang, J.; Xiao, Q.; Li, Q.; Long, L. Phenylethanoid and aliphatic alcohol glycosides from Acanthus ilicifolius. Phytochemistry 2003, 63, 491–495. [Google Scholar] [CrossRef]
- Wu, J.; Huang, J.S.; Xiao, Q.; Zhang, S.; Xiao, Z.H.; Li, Q.X.; Long, L.J.; Huang, L.M. Complete assignments of 1H and 13C NMR data for 10 phenylethanoid glycosides. Magn. Reson. Chem. 2004, 42, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Van, K.P.; Quang, T.H.; Huong, T.T.; Nhung le, L.T.; Cuong, N.X.; Van, M.C. Chemical constituents of Acanthus ilicifolius L. and effect on osteoblastic MC3T3E1 cells. Arch. Pharm. Res. 2008, 31, 823–829. [Google Scholar]
- Morikawa, T.; Pan, Y.; Ninomiya, K.; Imura, K.; Matsuda, H.; Yoshikawa, M.; Yuan, D.; Muraoka, O. Acylated phenylethanoid oligoglycosides with hepatoprotective activity from the desert plant Cistanche tubulosa. Bioorg. Med. Chem. 2010, 18, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.N.; Morikawa, T.; Ninomiya, K.; Imura, K.; Yuan, D.; Yoshikawa, M.; Muraoka, O. Bioactive constituents from Chinese nature medicines: Four new acylated phenylethanoid oligoglycosides, kankanosides J1, J2, K1 and K2 from stems of Cistanche tubulosa. Chem. Pharm. Bull. 2010, 58, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Chen, Z.; Feng, Z.M.; Yang, Y.N.; Jiang, J.S.; Zhang, P.C. Hepatoprotective glycosides from Leonurus japonicus Houtt. Carbohydr. Res. 2012, 348, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.G.; Guo, Y.M.; Luo, B.M.; Liu, W.M.; Wei, R.R.; Yang, C.X.; Ding, C.H.; Xu, X.F.; He, M.H. Hepatoprotective phenylethanoid glycosides from Cirsium setosum. Nat. Prod. Res. 2016, 30, 1824–1829. [Google Scholar] [CrossRef]
- Shen, T.; Li, X.Q.; Hu, W.C.; Zhang, L.J.; Xu, X.D.; Wu, H.F.; Ji, L.L. Hepatoprotective effect of phenylethanoid glycosides from Incarvillea compacta against CCl4-induced cytotoxicity in HepG2 cells. J. Korean Soc. Appl. Biol. Chem. 2015, 58, 617–625. [Google Scholar] [CrossRef]
- Xue, Z.Z.; Yang, B. Phenylethanoid glycosides: Research advances in their phytochemistry, pharmacological activity and pharmacokinetics. Molecules 2016, 21, 991. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. (FDA) Guidance for Industry: Bioanalytical Method Validation. 2018. Available online: https://www.fda.gov/ucm/groups/fdagov-public/@fdagovdrugs-gen/documents/document/ucm070107.pdf (accessed on 24 May 2018).
- Gan, P.; Huo, S.X.; Bai, P.; Li, G.; Peng, X.M.; He, Y.; Yan, M. Pharmacokinetics and tissue distribution of acteoside in rats. Chin. Pharm. Bull. 2014, 30, 417–420. [Google Scholar]
- Feng, B.W.; Song, Y.G.; Xu, Q.M.; Xu, P.F.; Zeng, Q.; Shan, B.X.; Liu, K.Y.; Su, D. Simultaneous determination of savaside A, acteoside, and isoacteoside in rat plasma by UHPLC-MS/MS: Comparative pharmacokinetic and bioavailability characteristics of Monochasma savatieri via different routes of administration. J. Sep. Sci. 2018, 41, 4408–4418. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.K.; Chen, W.K.; Ma, S.C.; Shao, J.; Wang, J.; Luo, Y.H. Simultaneous determination of three phenolic glycosides in Callicarpa nudiflora by UHPLC-MS methods and analysis of their pharmacokinetics in plasma of rats. Chin. Tradit. Herb. Drugs 2015, 46, 3533–3538. [Google Scholar] [CrossRef]
- Zhao, M.; Qian, D.W.; Liu, P.; Shang, E.X.; Jiang, S.; Guo, J.M.; Su, S.L.; Duan, J.A.; Du, L.Y.; Tao, J.H. Comparative pharmacokinetics of catalpol and acteoside in normal and chronic kidney disease rats after oral administration of Rehmannia glutinosa extract. Biomed. Chromatogr. 2015, 29, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Gan, L.; Li, G.Q.; Deng, L.; Zhang, X.S.; Deng, Y.L. Pharmacokinetics of plantamajoside and acteoside from Plantago asiatica in rats by liquid chromatography-mass spectrometry. J. Pharm. Biomed. Ana. 2014, 89, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Huo, S.; Zhang, W.; Xing, H.; Qi, L.; Zhao, D.; Li, N.; Xu, J.; Yan, M.; Chen, X. Pharmacokinetics, biodistribution, excretion and plasma protein binding studies of acteoside in rats. Drug Res. 2016, 66, 148–153. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Retention Time (min) | MRM Transitions (precursor→product) | Collision Energy (v) | Cone Voltage (v) |
|---|---|---|---|---|
| Acteoside | 3.37 | 623.2→161.0 | 50 | 76 |
| Isoacteoside | 3.56 | 623.2→161.0 | 50 | 76 |
| Martynoside | 4.42 | 651.2→175.0 | 36 | 78 |
| Crenatoside | 3.54 | 621.2→161.0 | 44 | 72 |
| Genistein (IS) | 5.02 | 270.0→133.0 | 34 | 78 |
| Components | Contents (mg/g) |
|---|---|
| Acteoside | 6.245 ± 0.723 |
| Isoacteoside | 0.822 ± 0.102 |
| Martynoside | 0.071 ± 0.023 |
| Crenatoside | 0.023 ± 0.008 |
| Components | Linear Regression Equation | r2 | Range (ng/mL) | LLOQ (ng/mL) |
|---|---|---|---|---|
| Acteoside | y = 0.000655 x − 0.001804 | 0.9979 | 2.0–1000 | 2.0 |
| Isoacteoside | y = 0.000917 x − 0.000488 | 0.9935 | 0.2–100 | 0.2 |
| Martynoside | y = 0.005864 x − 0.001393 | 0.9990 | 0.4–200 | 0.4 |
| Crenatoside | y = 0.000592 x − 0.000034 | 0.9949 | 0.4–200 | 0.4 |
| Components | Concentration (ng/mL) | Accuracy (RE%) | Precision (RSD%) | ||
|---|---|---|---|---|---|
| Intra-day | Inter-day | Intra-day | Inter-day | ||
| Acteoside | 2.0 | 4.36 | 5.53 | 7.24 | 6.17 |
| 5.0 | −5.26 | −3.81 | 2.17 | 3.60 | |
| 50.0 | −7.25 | −4.89 | 7.32 | 2.37 | |
| 800.0 | 4.70 | 4.27 | 7.04 | 5.90 | |
| Isoacteoside | 0.2 | 9.88 | −4.86 | 6.42 | 3.80 |
| 0.5 | 2.98 | −0.98 | 5.53 | 8.46 | |
| 5.0 | 4.24 | −4.38 | 8.35 | 11.90 | |
| 80.0 | 9.26 | 7.27 | 8.27 | 9.47 | |
| Martynoside | 0.4 | −7.24 | 4.18 | 5.54 | 10.00 |
| 1.0 | 2.04 | 3.41 | 9.09 | 10.49 | |
| 10.0 | −1.02 | 1.54 | 2.70 | 1.80 | |
| 160.0 | 1.44 | −2.71 | 1.47 | 3.39 | |
| Crenatoside | 0.4 | 9.43 | 6.63 | 9.72 | 6.42 |
| 1.0 | −8.53 | −5.12 | 12.37 | 13.08 | |
| 10.0 | −5.32 | −8.92 | 2.08 | 8.10 | |
| 160.0 | −2.95 | −5.22 | 6.39 | 4.35 | |
| Components | Concentration (ng/mL) | Extraction Recovery | Absolute Matrix Effect | Relative Matrix Effect | IS Normalized MF | |||
|---|---|---|---|---|---|---|---|---|
| Mean (%) | RSD (%) | Mean (%) | RSD (%) | RSD (%) | Mean ± SD | RSD (%) | ||
| Acteoside | 5.0 | 75.51 | 8.03 | 99.00 | 12.97 | 9.41 | 0.95 ± 0.06 | 7.62 |
| 50.0 | 88.51 | 9.75 | 85.03 | 4.40 | 3.22 | 0.93 ± 0.03 | 3.11 | |
| 800.0 | 97.14 | 3.43 | 86.70 | 6.21 | 8.97 | 0.92 ± 0.02 | 6.23 | |
| Isoacteoside | 0.5 | 98.50 | 11.47 | 95.66 | 9.83 | 10.11 | 1.00 ± 0.10 | 7.96 |
| 5.0 | 70.56 | 1.48 | 89.52 | 6.38 | 3.98 | 1.02 ± 0.09 | 12.14 | |
| 80.0 | 71.01 | 2.76 | 88.11 | 5.34 | 2.32 | 1.04 ± 0.11 | 2.35 | |
| Martynoside | 1.0 | 104.54 | 12.41 | 89.27 | 12.66 | 4.78 | 0.96 ± 0.05 | 9.76 |
| 10.0 | 92.81 | 9.64 | 101.02 | 7.78 | 6.33 | 0.98 ± 0.09 | 4.12 | |
| 160.0 | 98.19 | 4.14 | 96.23 | 3.42 | 5.22 | 0.99 ± 0.08 | 10.35 | |
| Crenatoside | 1.0 | 82.96 | 11.42 | 85.31 | 2.34 | 10.90 | 1.00 ± 0.08 | 8.62 |
| 10.0 | 90.11 | 9.57 | 89.72 | 10.64 | 3.51 | 0.97 ± 0.04 | 10.69 | |
| 160.0 | 80.38 | 5.33 | 106.66 | 4.47 | 2.96 | 0.90 ± 0.03 | 7.62 | |
| Components | Concentration (ng/mL) | Freeze and Thaw | Short-Term | Long-Term | Post-Preparative | ||||
|---|---|---|---|---|---|---|---|---|---|
| RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | ||
| Acteoside | 5.0 | 2.92 | −1.58 | 2.17 | −5.26 | 2.51 | −2.45 | 4.68 | −2.72 |
| 50.0 | 4.60 | −5.42 | 7.32 | −7.25 | 8.01 | −8.44 | 11.78 | 1.40 | |
| 800.0 | 6.32 | 1.50 | 7.04 | 4.70 | 6.49 | −0.01 | 2.27 | −2.16 | |
| Isoacteoside | 0.5 | 6.59 | 0.61 | 7.53 | 2.98 | 7.43 | −1.42 | 9.61 | −2.70 |
| 5.0 | 9.90 | −1.65 | 11.69 | 4.24 | 10.32 | 2.20 | 10.68 | 1.88 | |
| 80.0 | 1.99 | 0.61 | 6.28 | 2.98 | 6.06 | −1.42 | 7.32 | −2.70 | |
| Martynoside | 1.0 | 9.46 | 3.03 | 9.09 | 2.04 | 8.74 | 1.27 | 7.47 | 2.27 |
| 10.0 | 3.13 | −0.19 | 2.70 | −1.02 | 2.82 | −0.79 | 1.82 | −0.66 | |
| 160.0 | 1.13 | −2.71 | 1.47 | 1.44 | 1.69 | −0.91 | 2.47 | −0.60 | |
| Crenatoside | 1.0 | 6.42 | 2.46 | 12.37 | −8.53 | 6.36 | 0.71 | 9.38 | −3.03 |
| 10.0 | 10.87 | −8.98 | 10.42 | −5.32 | 9.94 | −11.6 | 12.48 | −10.95 | |
| 160.0 | 5.27 | −3.37 | 6.39 | −2.95 | 3.80 | −4.97 | 6.02 | −1.40 | |
| Parameter | Acteoside | Isoacteoside | Martynoside | Crenatoside |
|---|---|---|---|---|
| AUC0-t/(µg/L × h) | 1826.3 ± 680.2 | 70.9 ± 26.9 | 23.6 ± 6.9 | 64.7 ± 14.5 |
| AUC0-∞/(µg/L × h) | 2243.1 ± 894.6 | 87.0 ± 40.0 | 39.5 ± 15.5 | 76.0 ± 30.0 |
| t1/2/(h) | 5.6 ± 3.4 | 4.6 ± 3.1 | 9.0 ± 2.7 | 3.4 ± 3.1 |
| tmax/(h) | 0.3 ± 0.1 | 0.4 ± 0.2 | 3.1 ± 3.6 | 6.8 ± 1.1 |
| Cmax/(µg/L) | 356.9 ± 64.2 | 58.2 ± 15.0 | 4.0 ± 0.9 | 10.9 ± 0.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Ren, X.; Yue, S.; Zhao, Q.; Shao, C.; Wang, C. Simultaneous Quantification of Four Phenylethanoid Glycosides in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study of Acanthus Ilicifolius Herb. Molecules 2019, 24, 3117. https://doi.org/10.3390/molecules24173117
Zhang M, Ren X, Yue S, Zhao Q, Shao C, Wang C. Simultaneous Quantification of Four Phenylethanoid Glycosides in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study of Acanthus Ilicifolius Herb. Molecules. 2019; 24(17):3117. https://doi.org/10.3390/molecules24173117
Chicago/Turabian StyleZhang, Mengqi, Xia Ren, Shijun Yue, Qing Zhao, Changlun Shao, and Changyun Wang. 2019. "Simultaneous Quantification of Four Phenylethanoid Glycosides in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study of Acanthus Ilicifolius Herb" Molecules 24, no. 17: 3117. https://doi.org/10.3390/molecules24173117
APA StyleZhang, M., Ren, X., Yue, S., Zhao, Q., Shao, C., & Wang, C. (2019). Simultaneous Quantification of Four Phenylethanoid Glycosides in Rat Plasma by UPLC-MS/MS and Its Application to a Pharmacokinetic Study of Acanthus Ilicifolius Herb. Molecules, 24(17), 3117. https://doi.org/10.3390/molecules24173117