l-Buthionine Sulfoximine Detection and Quantification in Polyurea Dendrimer Nanoformulations





Abstract
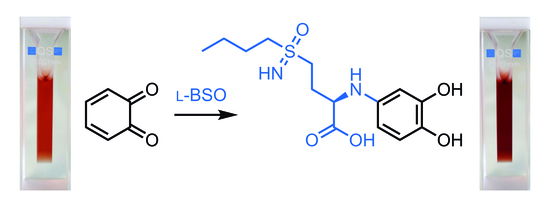
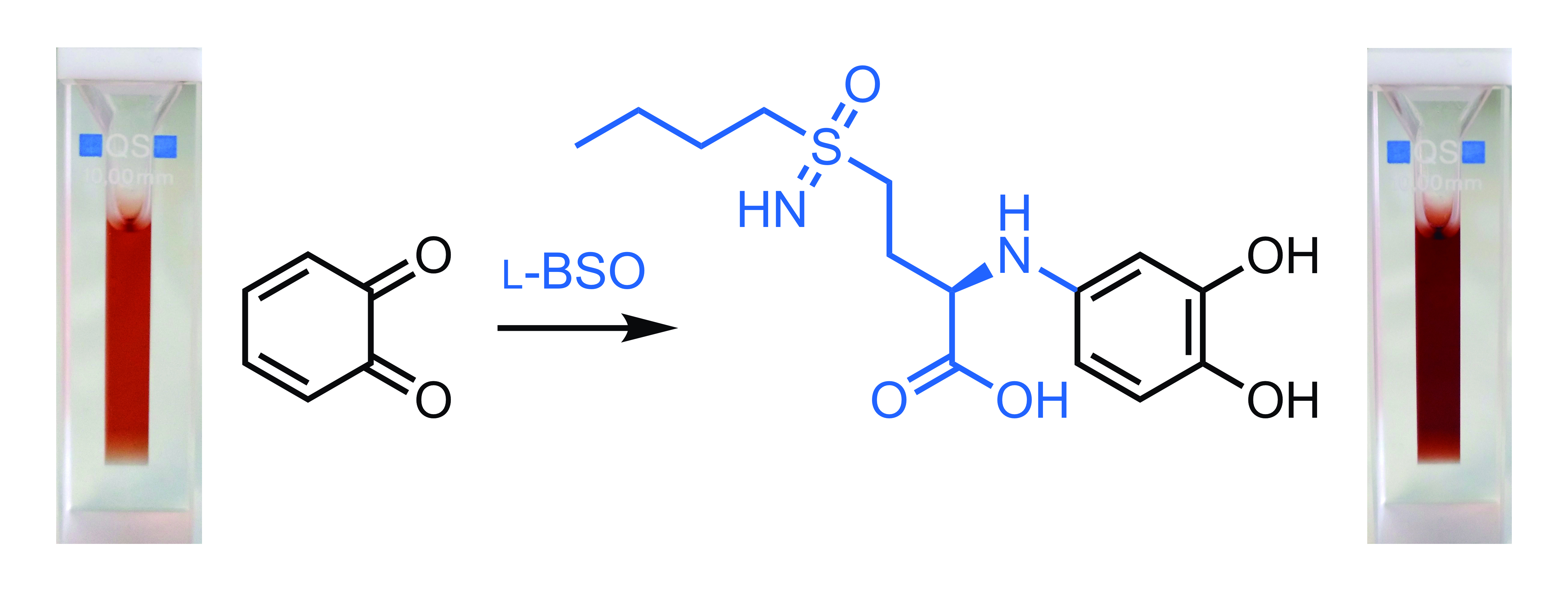
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
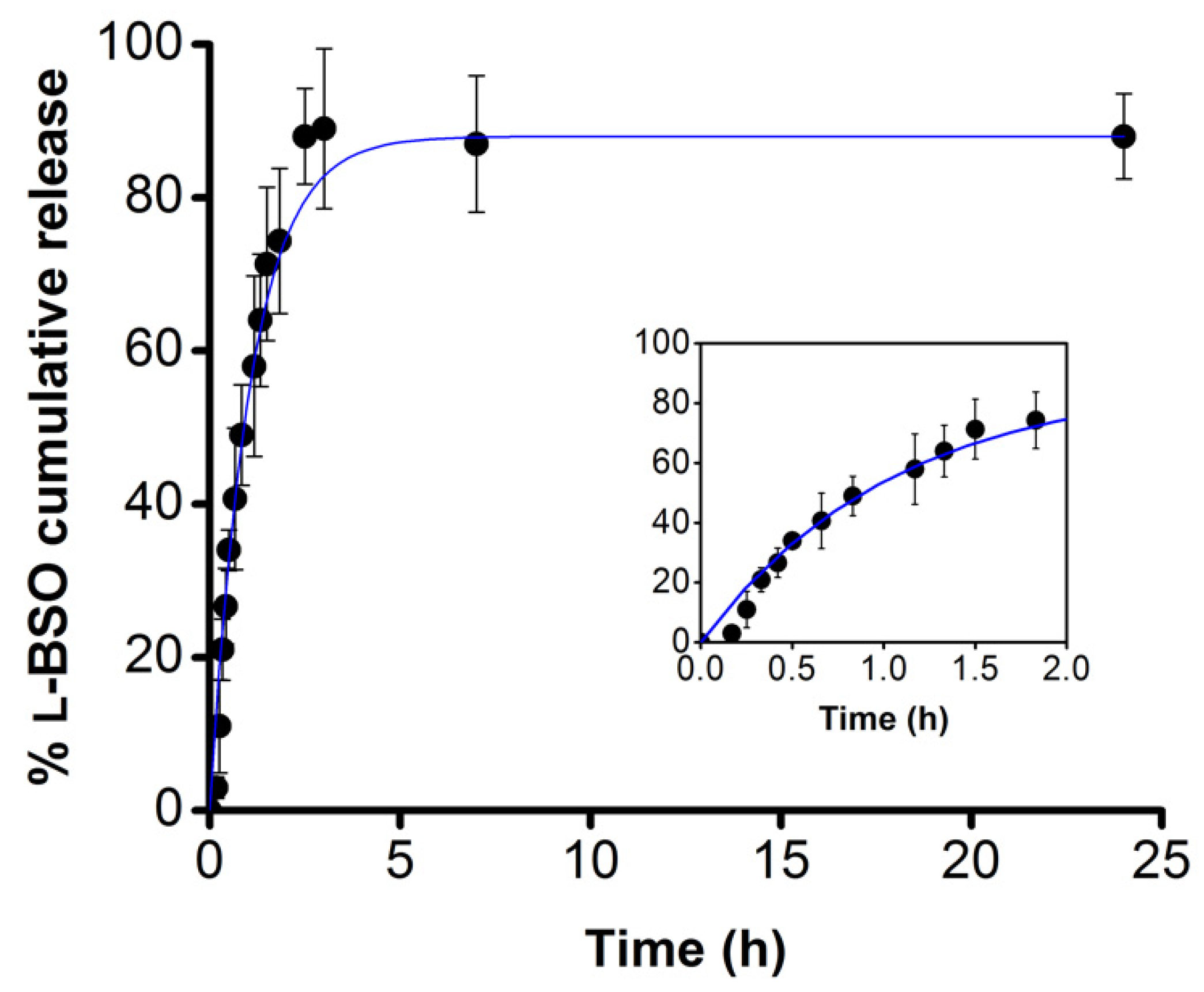
2.1. l-BSO Encapsulation and Release Studies
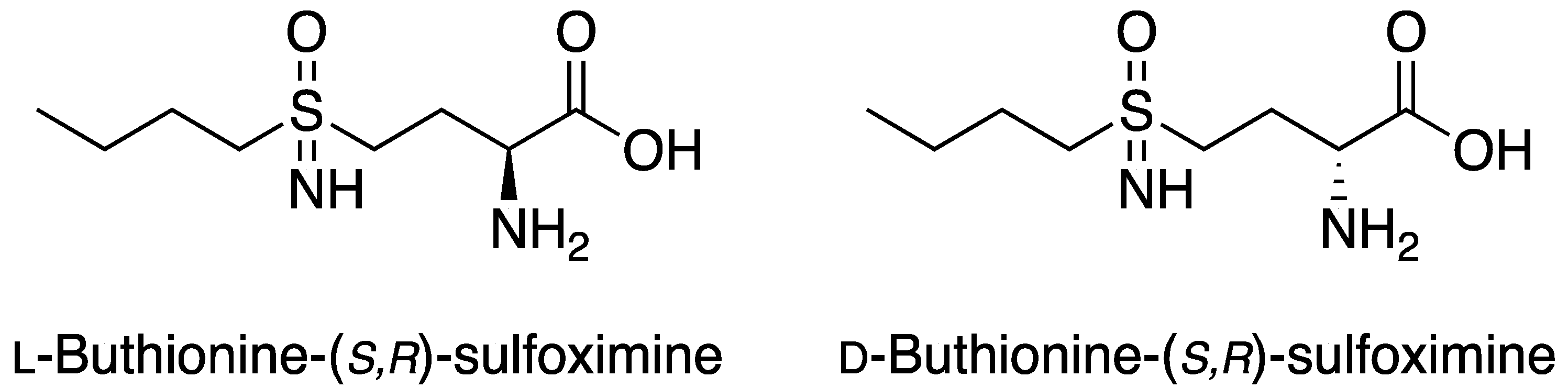
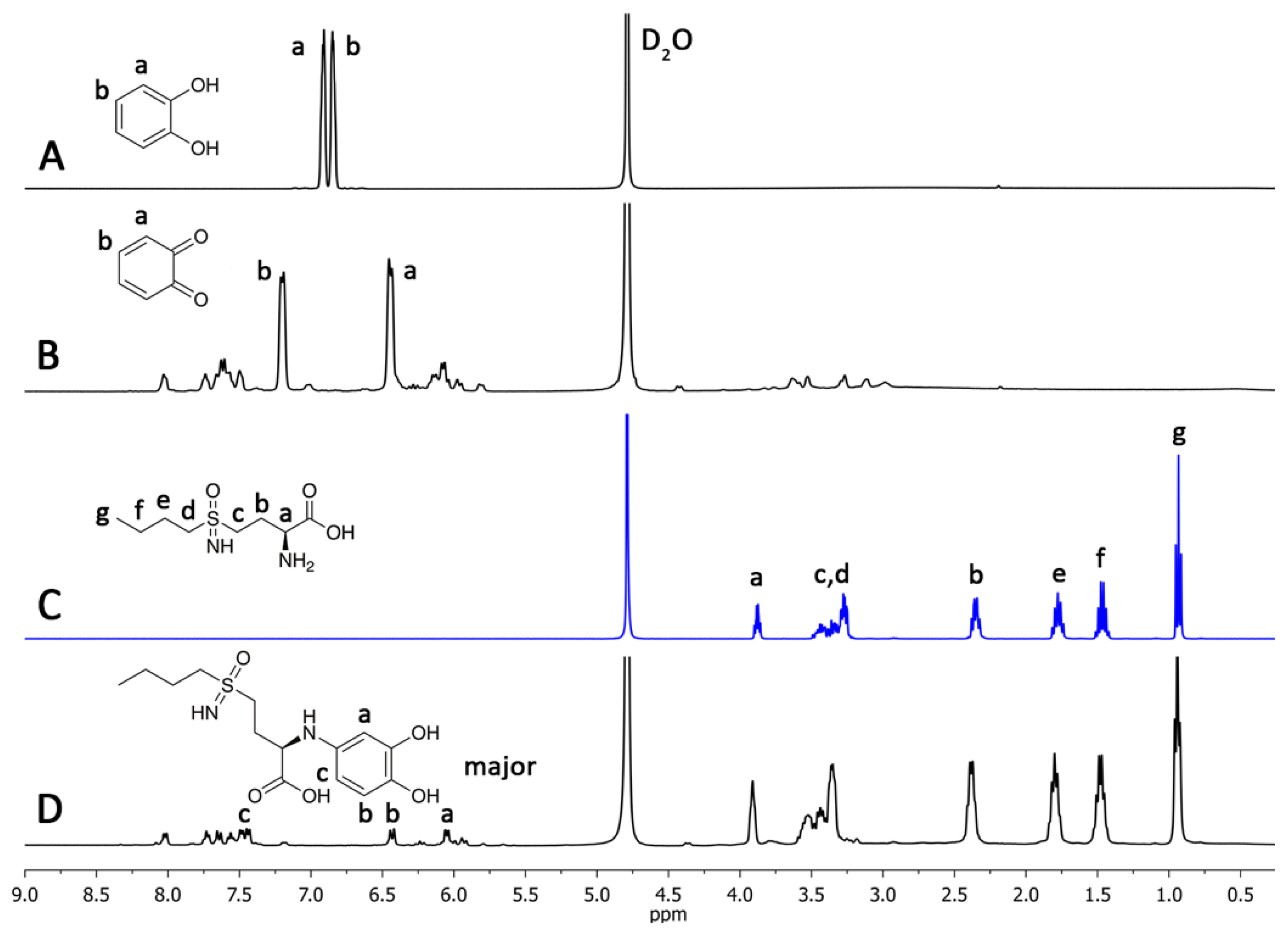
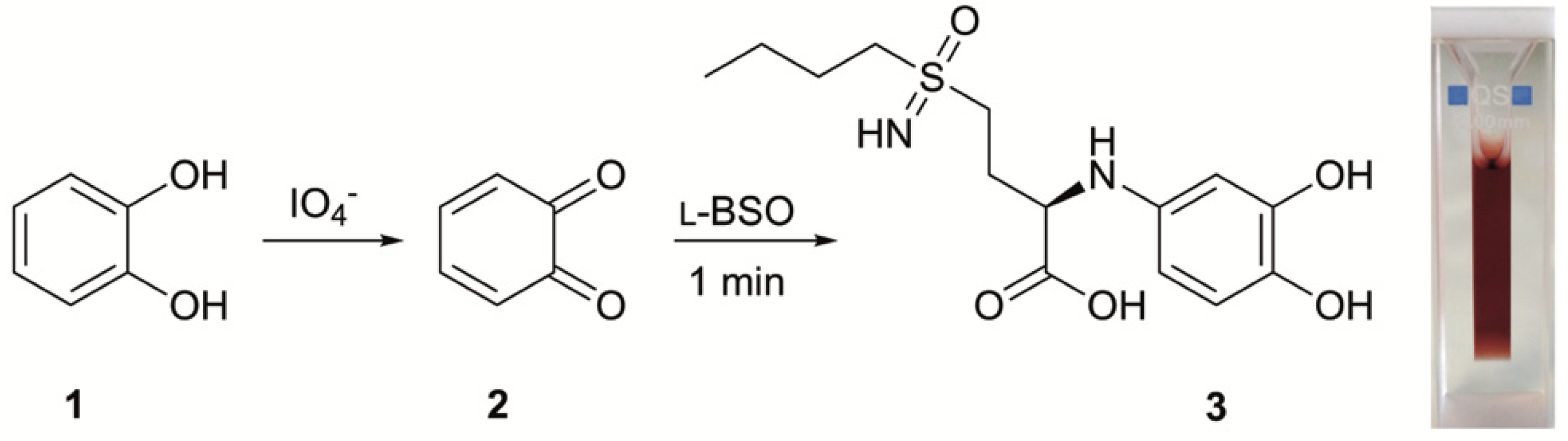
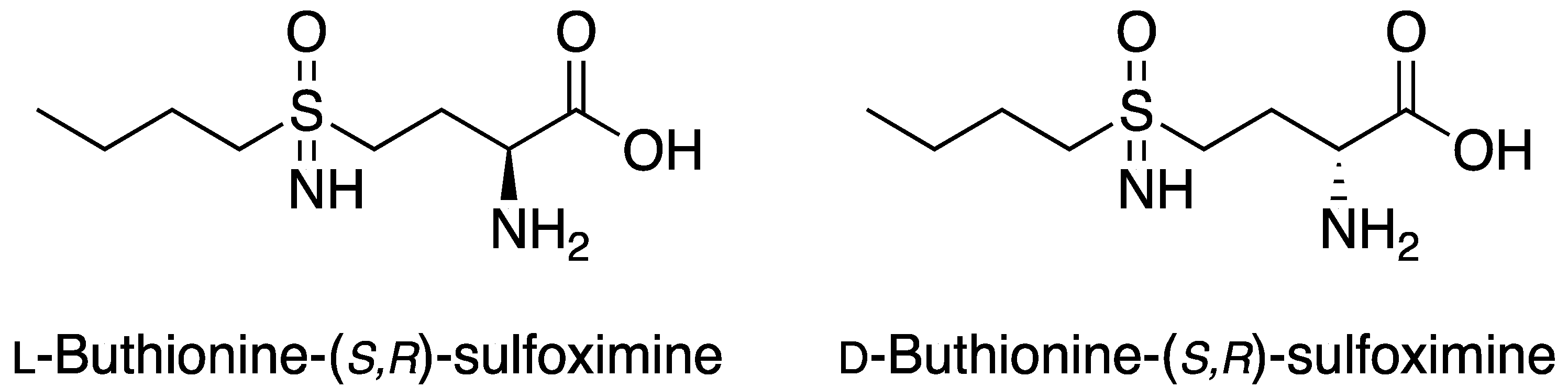
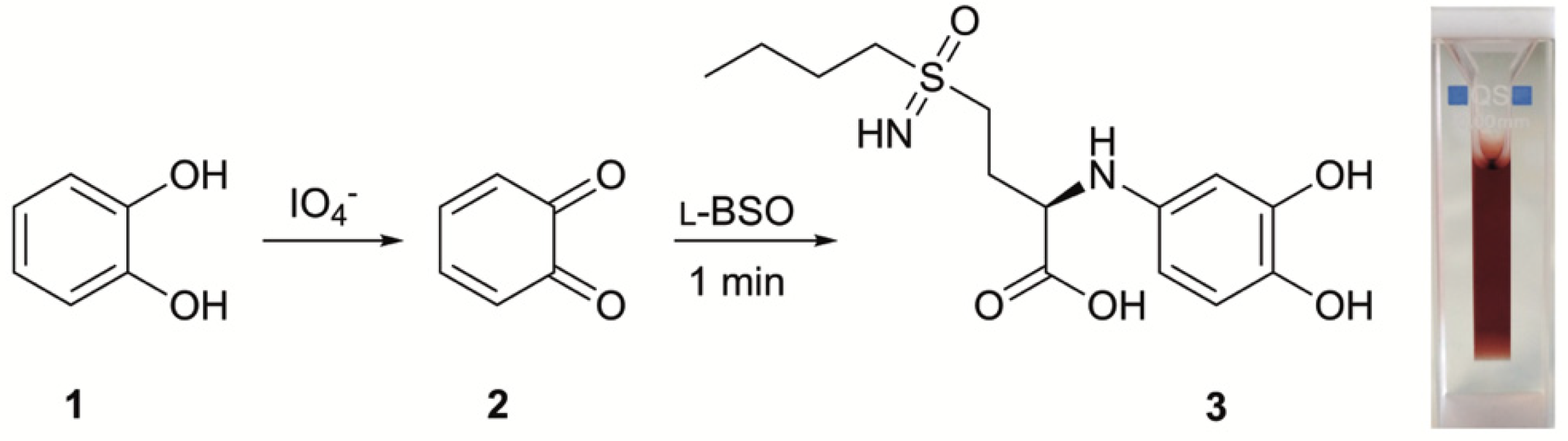
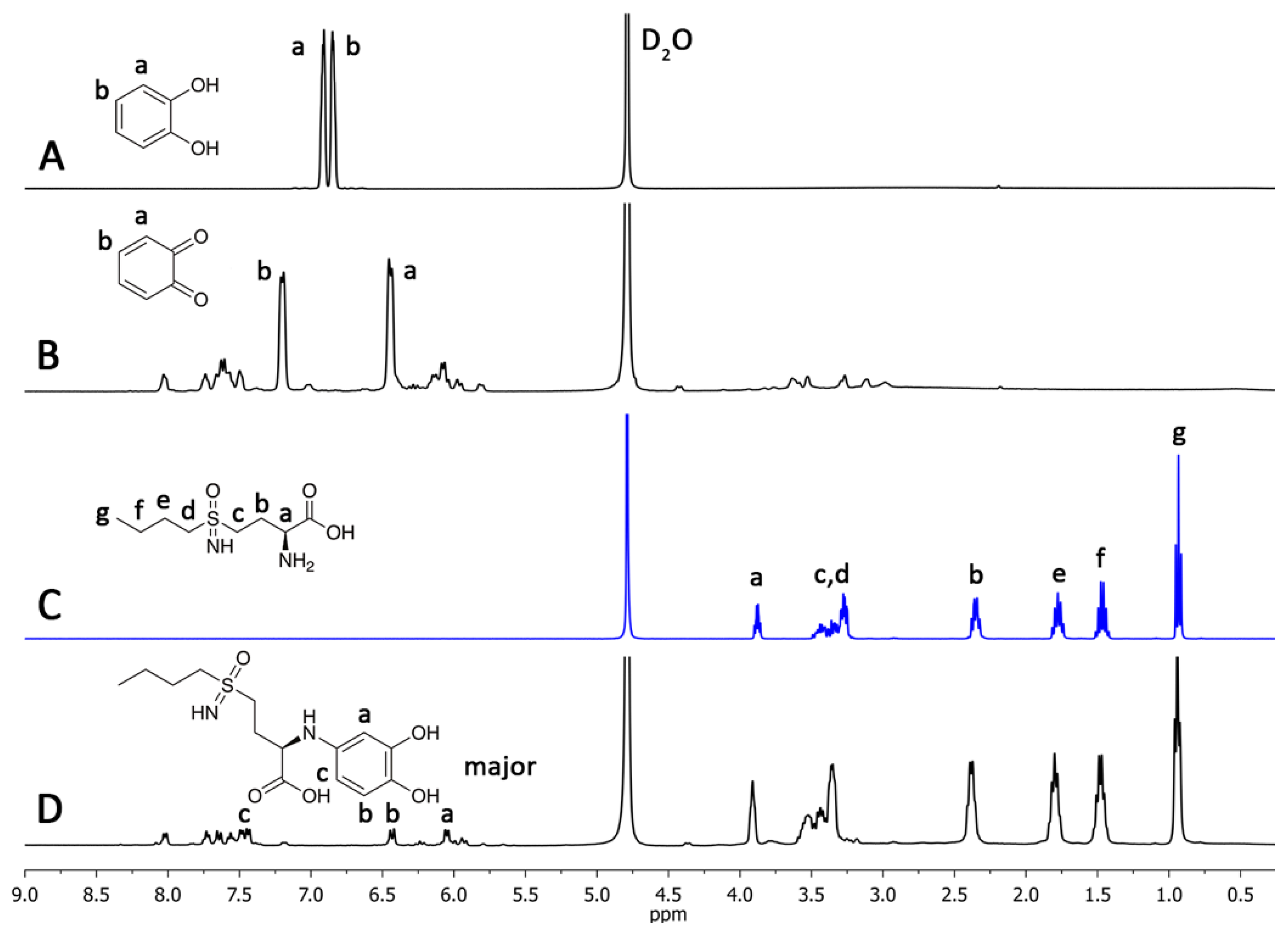
2.2. l-BSO Detection and Quantification
3. Materials and Methods
3.1. Reagents and Materials
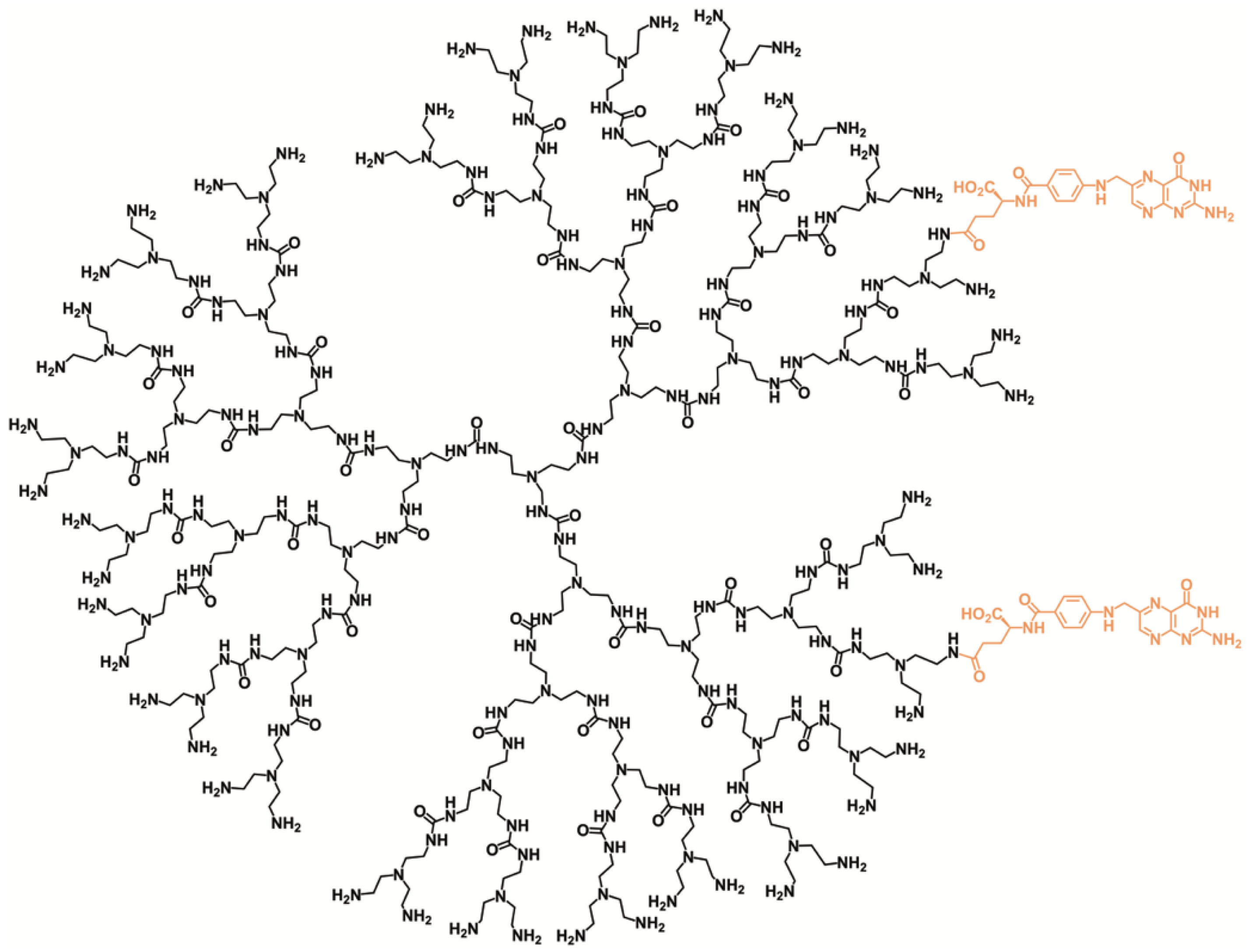
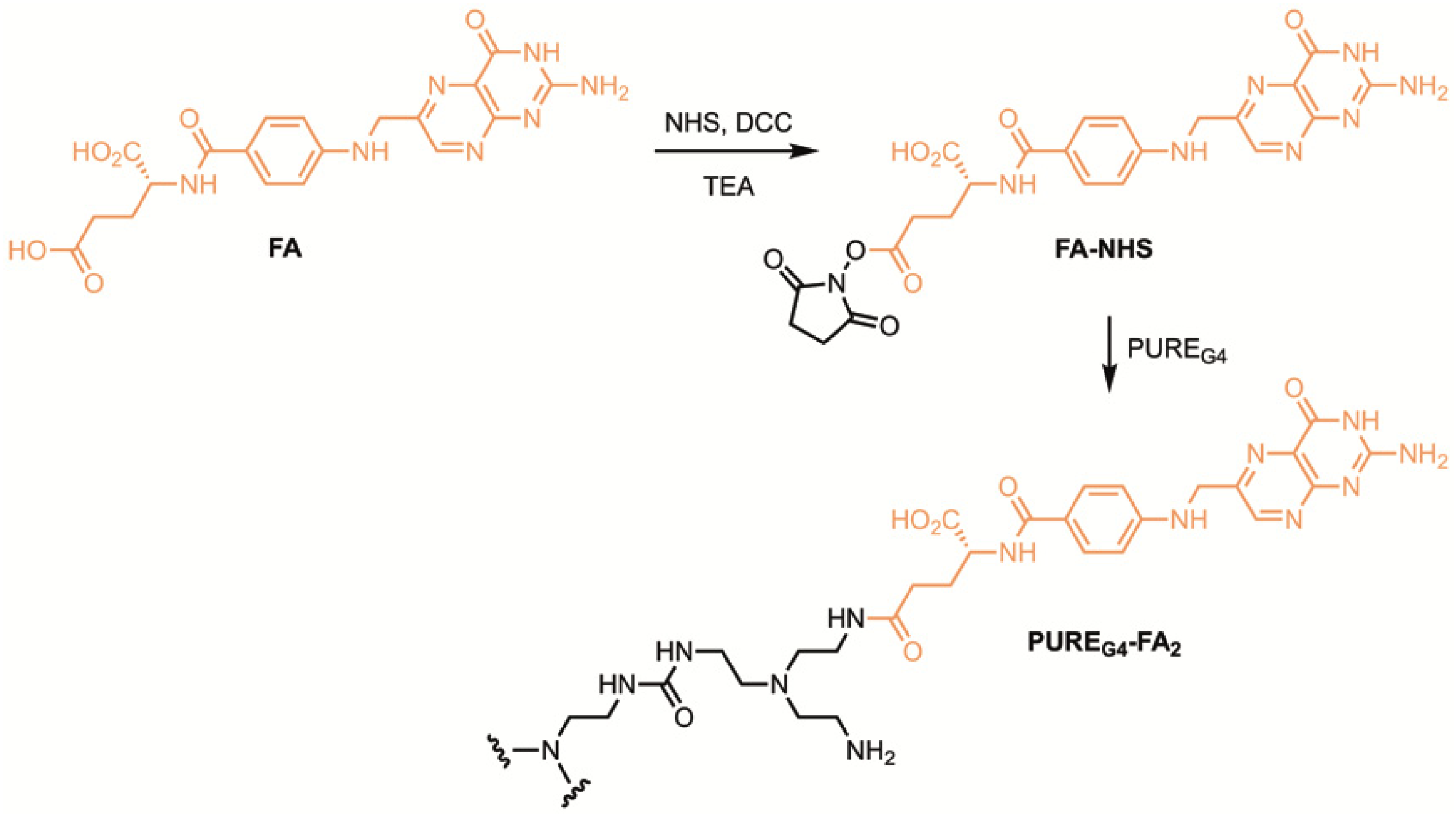
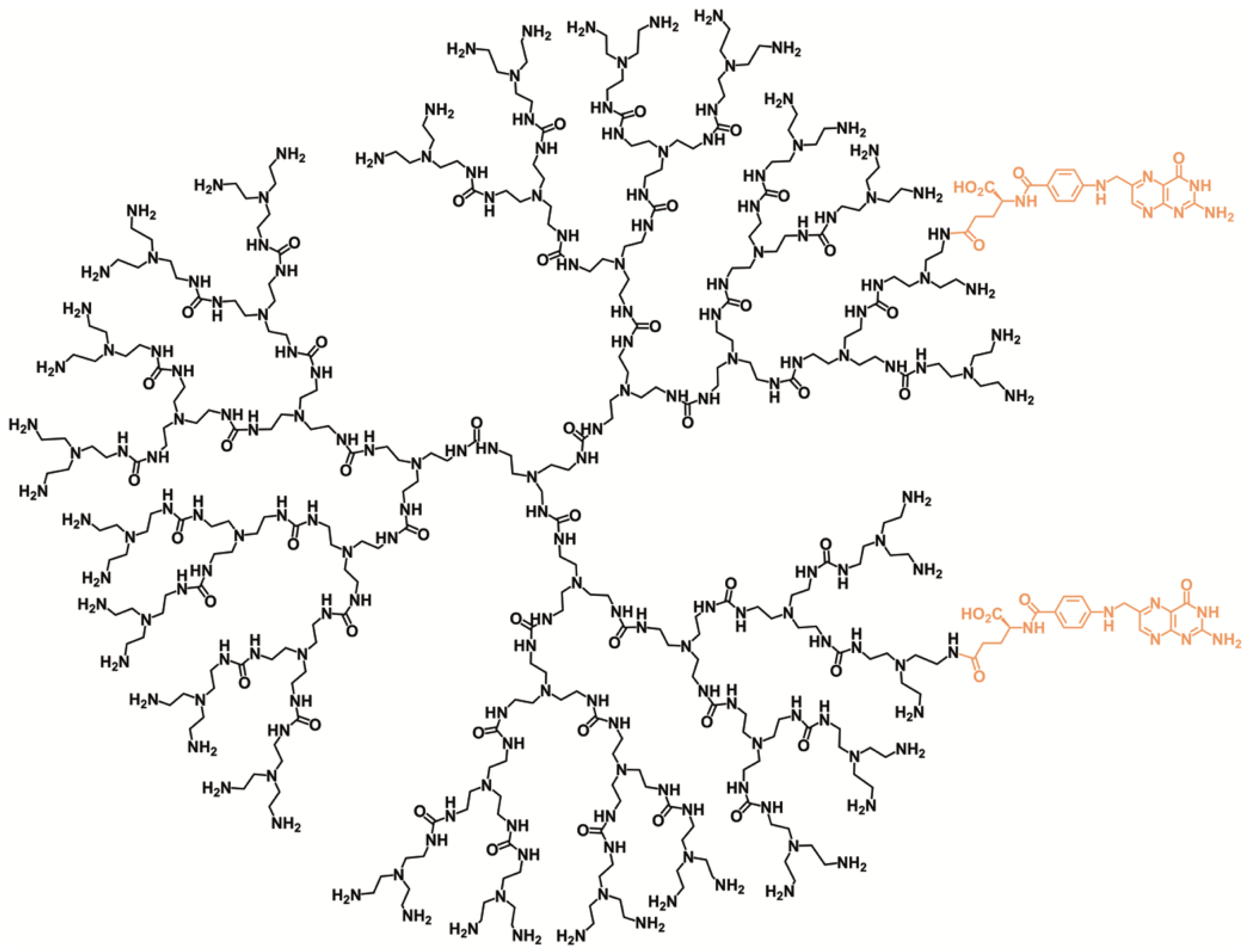
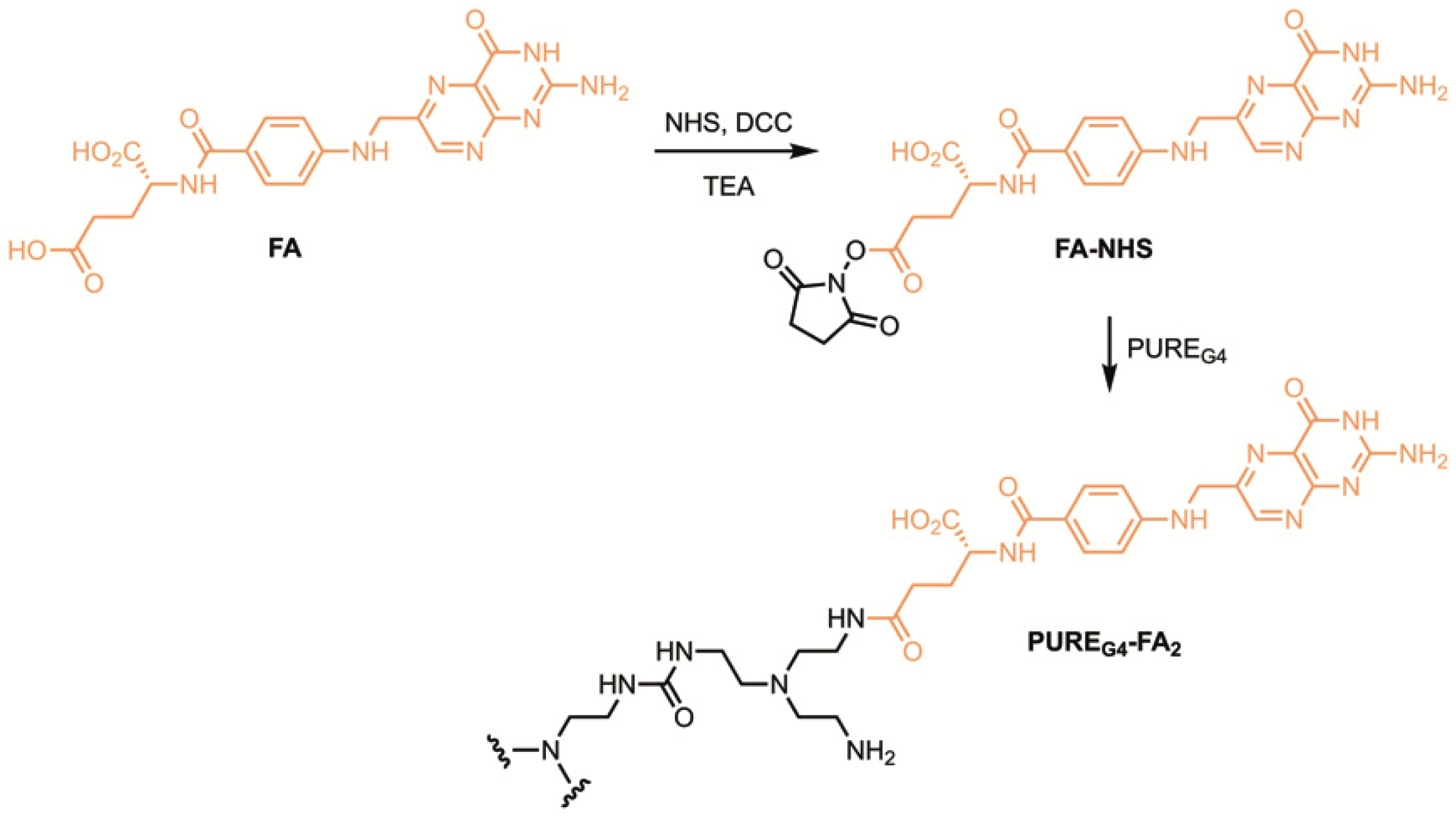
3.2. Synthesis of PUREG4-FA2
3.3. Encapsulation of l-BSO in PUREG4-FA2
3.4. l-BSO Release Profile
3.5. Quantification of BSO by UV-Vis Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Görög, S. Ultraviolet-Visible Spectrophotometry in Pharmaceutical Analysis; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Haddad, J.J. l-Buthionine-(S,R)-sulfoximine, an irreversible inhibitor of gamma-glutamylcysteine synthetase, augments LPS-mediated pro-inflammatory cytokine biosynthesis: Evidence for the implication of an IkappaB-alpha/NF-kappaB insensitive pathway. Eur Cytokine Netw. 2001, 12, 614–624. [Google Scholar] [PubMed]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar] [PubMed]
- Griffith, O.W. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem. 1982, 257, 13704–13712. [Google Scholar] [PubMed]
- Buglioni, L.; Bizet, V.; Bolm, C. Methionine and buthionine sulfoximines: Syntheses under mild and safe imidation/oxidation conditions. Adv. Synth. Catal. 2014, 356, 2209–2213. [Google Scholar] [CrossRef]
- Schnelldorfer, T.; Gansauge, S.; Gansauge, F.; Schlosser, S.; Beger, H.G.; Nussler, H.K. Glutathione depletion causes cell growth inhibition and enhanced apoptosis in pancreatic cancer cells. Cancer 2000, 89, 1440–1447. [Google Scholar] [CrossRef]
- Vanhoefer, U.; Cao, S.; Minderman, H.; Toth, K.; Skenderis, B.S.; Slovak, M.L.; Rustum, Y.M. D, l-Buthionine-(S,R)-sulfoximine potentiates in vivo the therapeutic efficacy of doxorubicin against multidrug resistance protein-expressing tumors. Clin. Cancer Res. 1996, 2, 1961–1968. [Google Scholar] [PubMed]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Gonçalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Félix, A.; Pereira, S.A.; Serpa, J. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumor Biol. 2016, 37, 4813–4829. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor energy metabolism and potential of 3-bromopyruvate as an inhibitor of aerobic glycolysis: Implications in tumor treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Flarakos, T.; Batist, G.; Wainer, I.W.; Lloyd, D.K. Quantitation of the diastereoisomers of l-buthionine-(R,S)-sulfoximine in human plasma: A validated assay by capillary electrophoresis. J. Chromatogr. B Biomed. Sci. Appl. 1995, 673, 123–131. [Google Scholar] [CrossRef]
- Duff, R.; Murrill, E. Determination of l-buthionine-(S,R)-sulfoximine in plasma by high-performance liquid chromatography with o-phthalaldehyde derivatization and fluorometric detection. J. Chromatogr. 1987, 385, 275–282. [Google Scholar] [CrossRef]
- Campbell, E.B.; Hayward, M.L.; Griffith, O.G. Analytical and preparative separation of the diastereomers of l-buthionine (SR)-sulfoximine, a potent inhibitor of glutathione biosynthesis. Anal. Biochem. 1991, 194, 268–277. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1923, 309, 97–519. [Google Scholar] [CrossRef] [PubMed]
- Pires, R.F.; Moro, A.; Lourenço, A.; Lima, J.C.; Casimiro, T.; Bonifácio, V.D.B. Molecular weight determination by luminescent chemoenzymatics. ChemistrySelect 2016, 1, 6818–6822. [Google Scholar] [CrossRef]
- Restani, R.B.; Conde, J.; Baptista, P.V.; Cidade, M.T.; Bragança, A.M.; Morgado, J.; Correia, I.J.; Aguiar-Ricardo, A.; Bonifácio, V.D.B. Polyurea dendrimer for efficient cytosolic siRNA delivery. RSC Adv. 2014, 4, 54872–54878. [Google Scholar] [CrossRef]
- Restani, R.B.; Conde, J.; Pires, R.F.; Martins, P.; Fernandes, A.R.; Baptista, P.V.; Bonifácio, V.D.B.; Aguiar-Ricardo, A. POxylated polyurea dendrimers: Smart core-shell vectors with IC50 lowering capacity. Macromol. Biosci. 2015, 15, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Restani, R.B.; Silva, A.S.; Pires, R.F.; Cabral, R.; Correia, I.J.; Casimiro, T.; Bonifácio, V.D.B.; Aguiar-Ricardo, A. Nano-in-micro POxylated polyurea dendrimers and chitosan dry powder formulations for pulmonary delivery. Part. Part. Syst. Charact. 2016, 33, 851–858. [Google Scholar] [CrossRef]
- Khalafi, L.; Rafiee, M.; Fathi, S. Effect of β-cyclodextrin on intra and intermolecular Michael addition of some catechol derivatives. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 118, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, D.; Rafiee, M.; Fotouhi, L. Mechanistic study of homogeneous reactions coupled with electrochemical oxidation of catechols. J. Iran Chem. Soc. 2009, 6, 448–476. [Google Scholar] [CrossRef]
- Obermeyer, A.C.; Jarman, J.B.; Francis, M.B. N-Terminal modification of proteins with o-aminophenols. J. Am. Chem. Soc. 2014, 136, 9572–9579. [Google Scholar] [CrossRef] [PubMed]
- Restani, R.B.; Morgado, P.I.; Ribeiro, M.P.; Correia, I.J.; Aguiar-Ricardo, A.; Bonifácio, V.D.B. Biocompatible polyurea dendrimers with pH-dependent fluorescence. Angew. Chem. Int. Ed. 2012, 51, 5162–5165. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Harris, J.M.; Bentley, M.D.; Fang, Z.; Viegas, T. Multifunctional Forms of Polyoxazoline Copolymers and Drug Compositions Comprising the Same. U.S. Patent 8,501,899, 23 January 2012. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mota, P.; Pires, R.F.; Serpa, J.; Bonifácio, V.D.B. l-Buthionine Sulfoximine Detection and Quantification in Polyurea Dendrimer Nanoformulations. Molecules 2019, 24, 3111. https://doi.org/10.3390/molecules24173111
Mota P, Pires RF, Serpa J, Bonifácio VDB. l-Buthionine Sulfoximine Detection and Quantification in Polyurea Dendrimer Nanoformulations. Molecules. 2019; 24(17):3111. https://doi.org/10.3390/molecules24173111
Chicago/Turabian StyleMota, Pedro, Rita F. Pires, Jacinta Serpa, and Vasco D. B. Bonifácio. 2019. "l-Buthionine Sulfoximine Detection and Quantification in Polyurea Dendrimer Nanoformulations" Molecules 24, no. 17: 3111. https://doi.org/10.3390/molecules24173111
APA StyleMota, P., Pires, R. F., Serpa, J., & Bonifácio, V. D. B. (2019). l-Buthionine Sulfoximine Detection and Quantification in Polyurea Dendrimer Nanoformulations. Molecules, 24(17), 3111. https://doi.org/10.3390/molecules24173111