
Very Recent Advances in Vinylogous Mukaiyama Aldol Reactions and Their Applications to Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
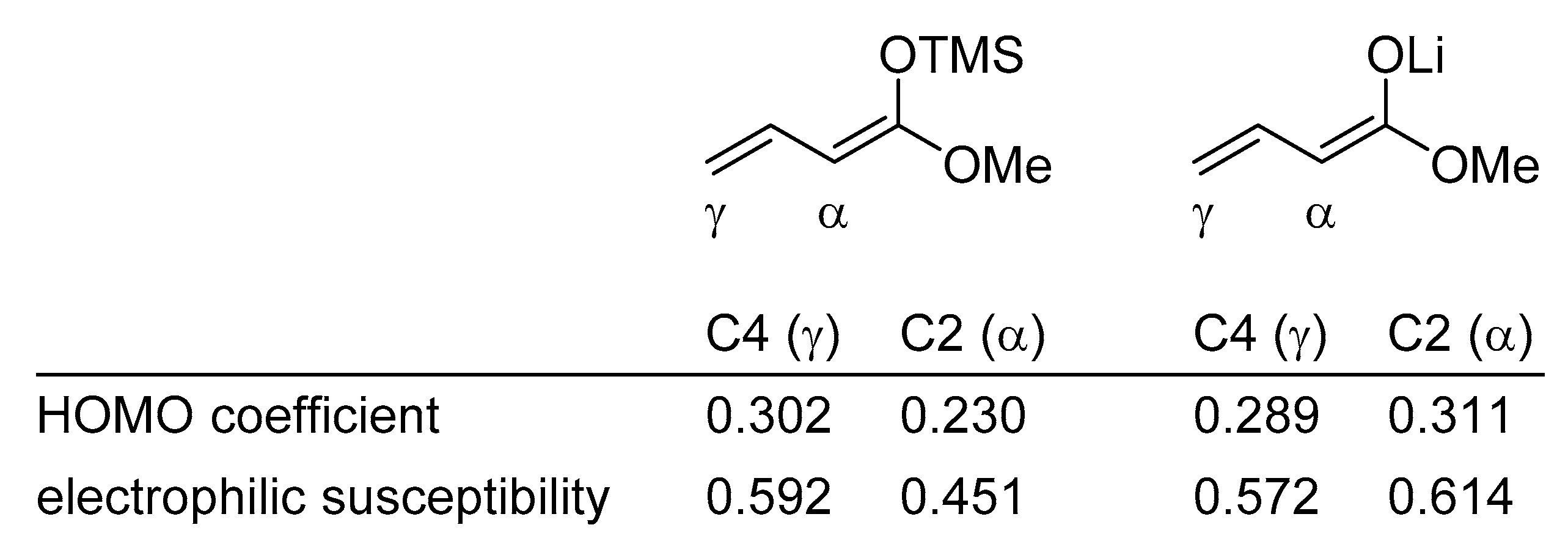{kind=link}
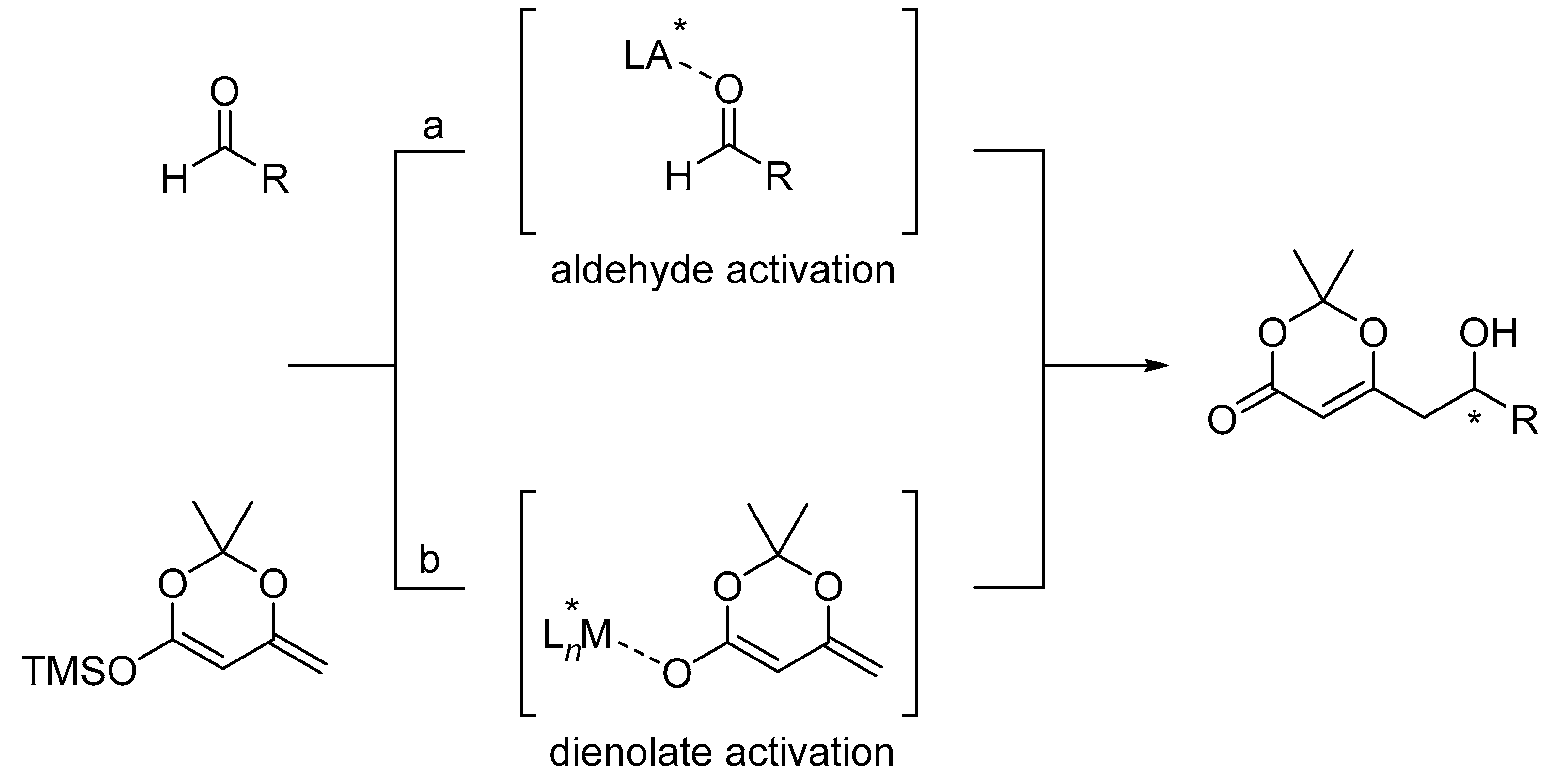{kind=link}
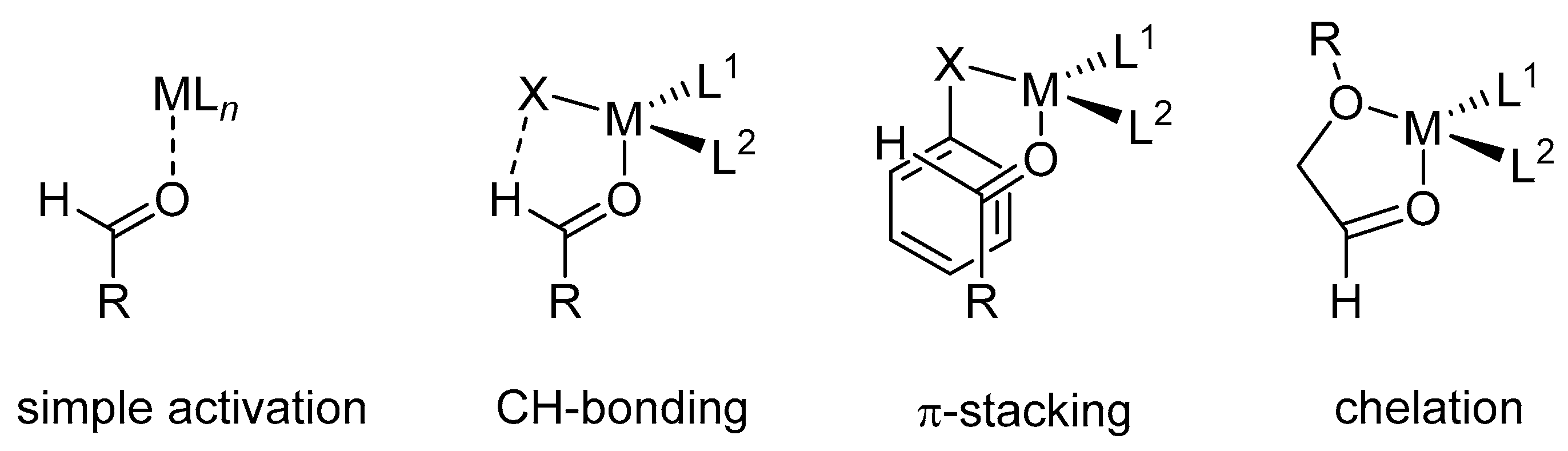{kind=link}
{kind=link}
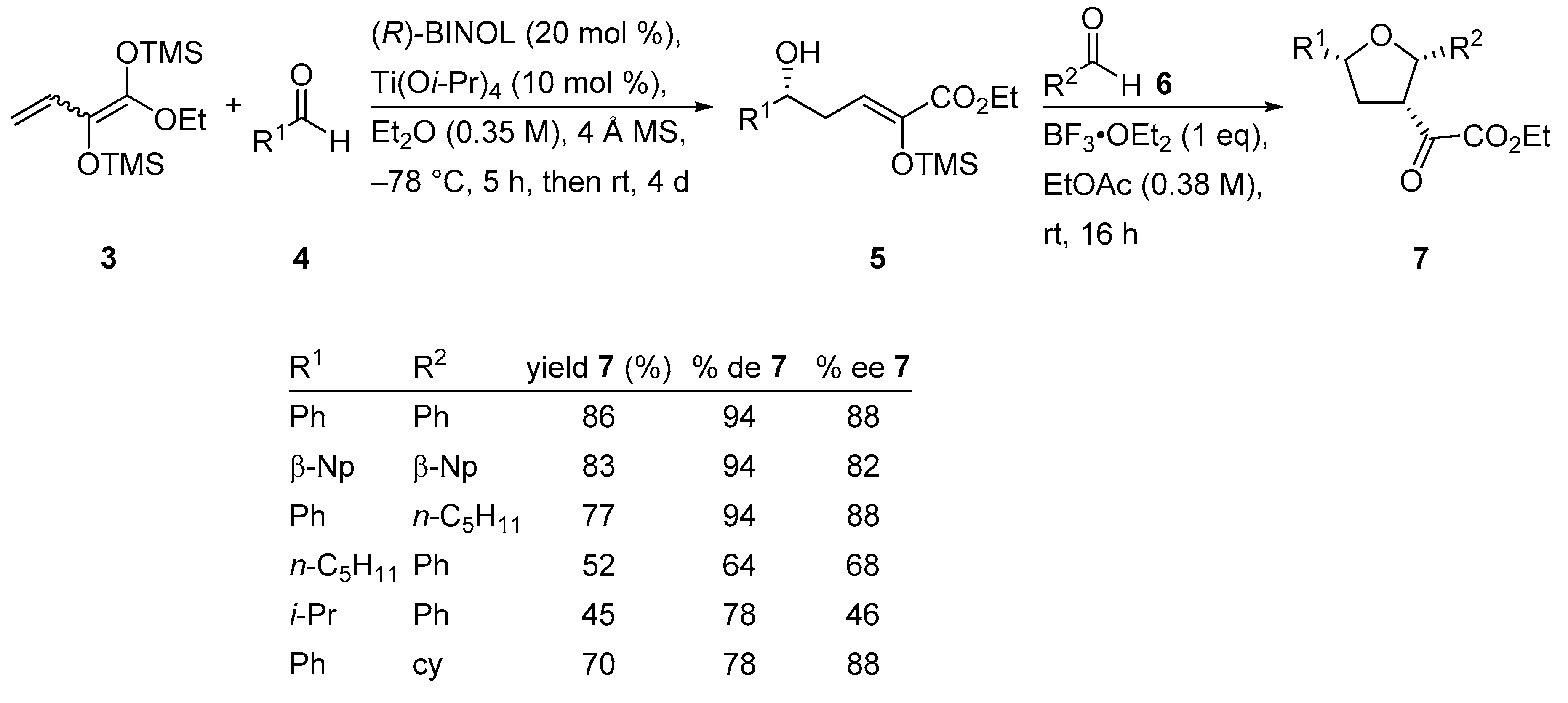{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
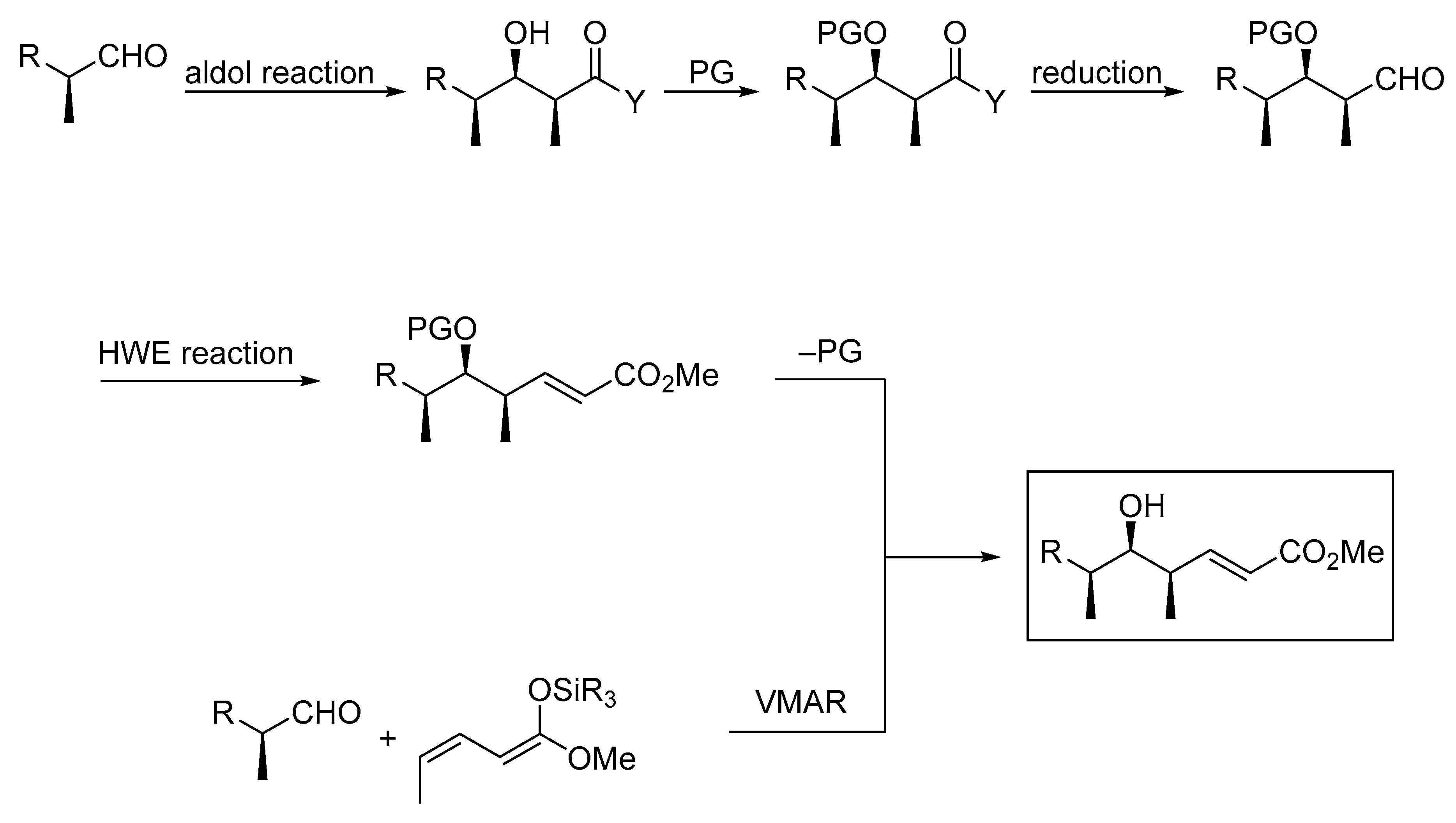
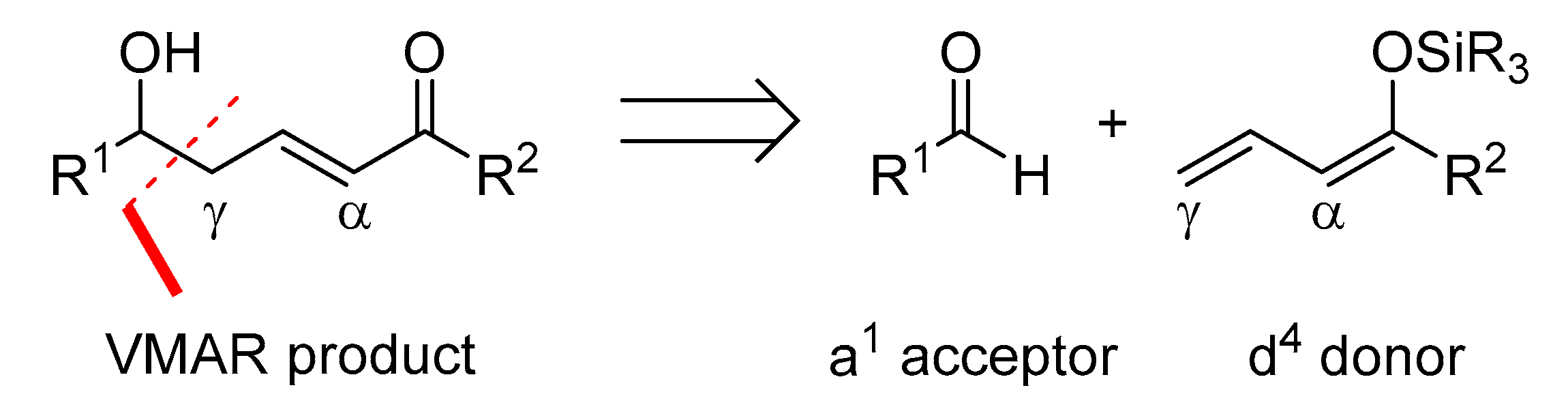
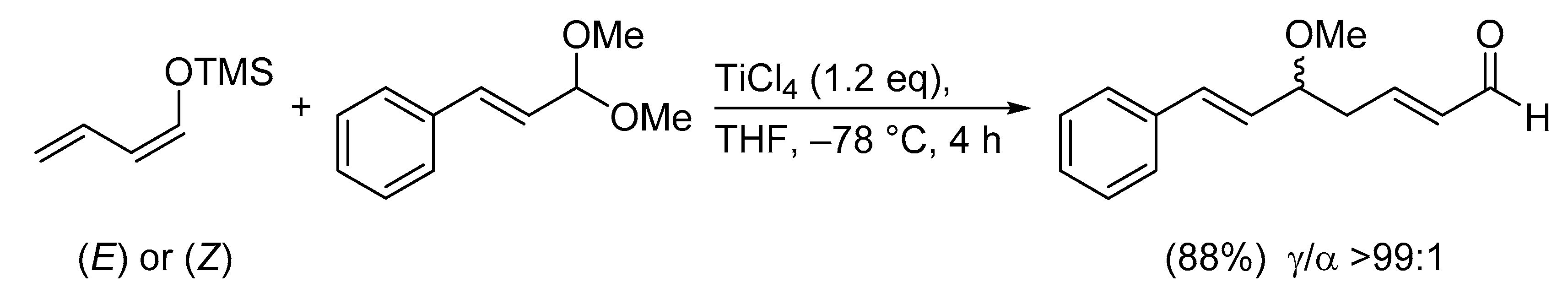
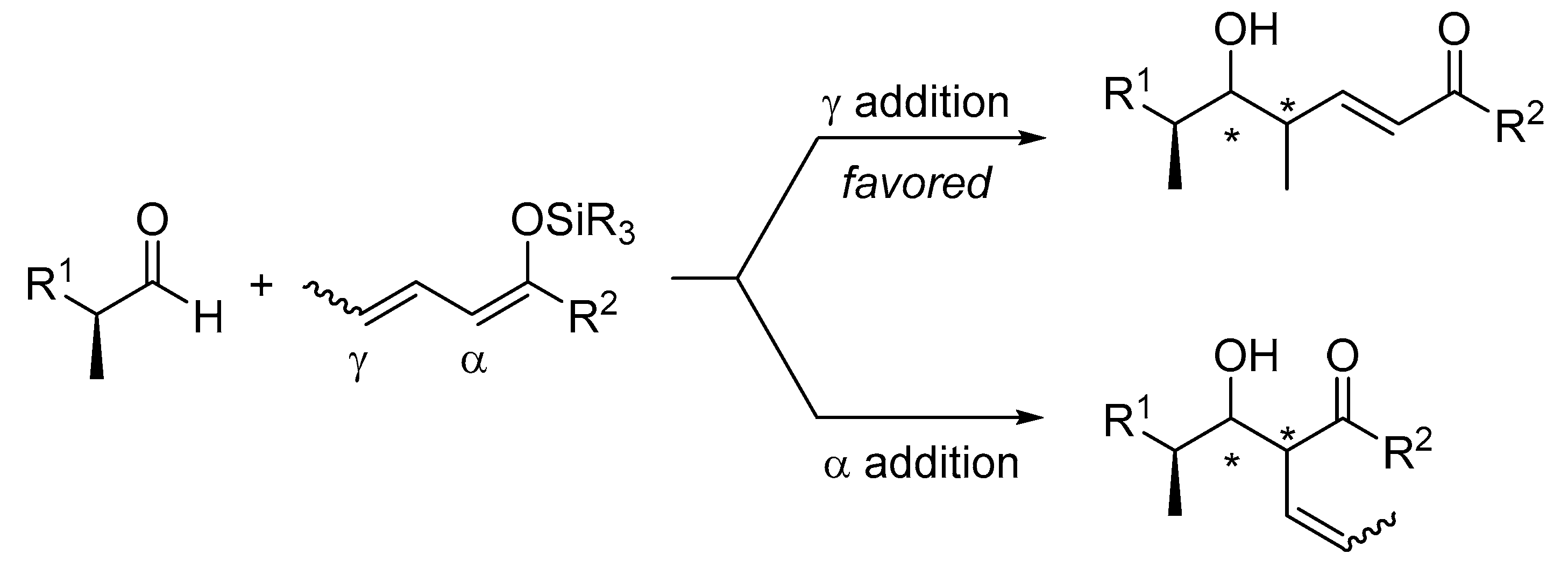
1. Introduction
2. Enantioselective VMA Reactions
2.1. Formal Synthesis of a CB2 Agonist Drug Candidate
2.2. Enantioselective Synthesis of 2,3,5-Trisubstituted Tetrahydrofurans
3. Diastereoselective VMA Reactions
3.1. Oxazaborolidinone Catalysis
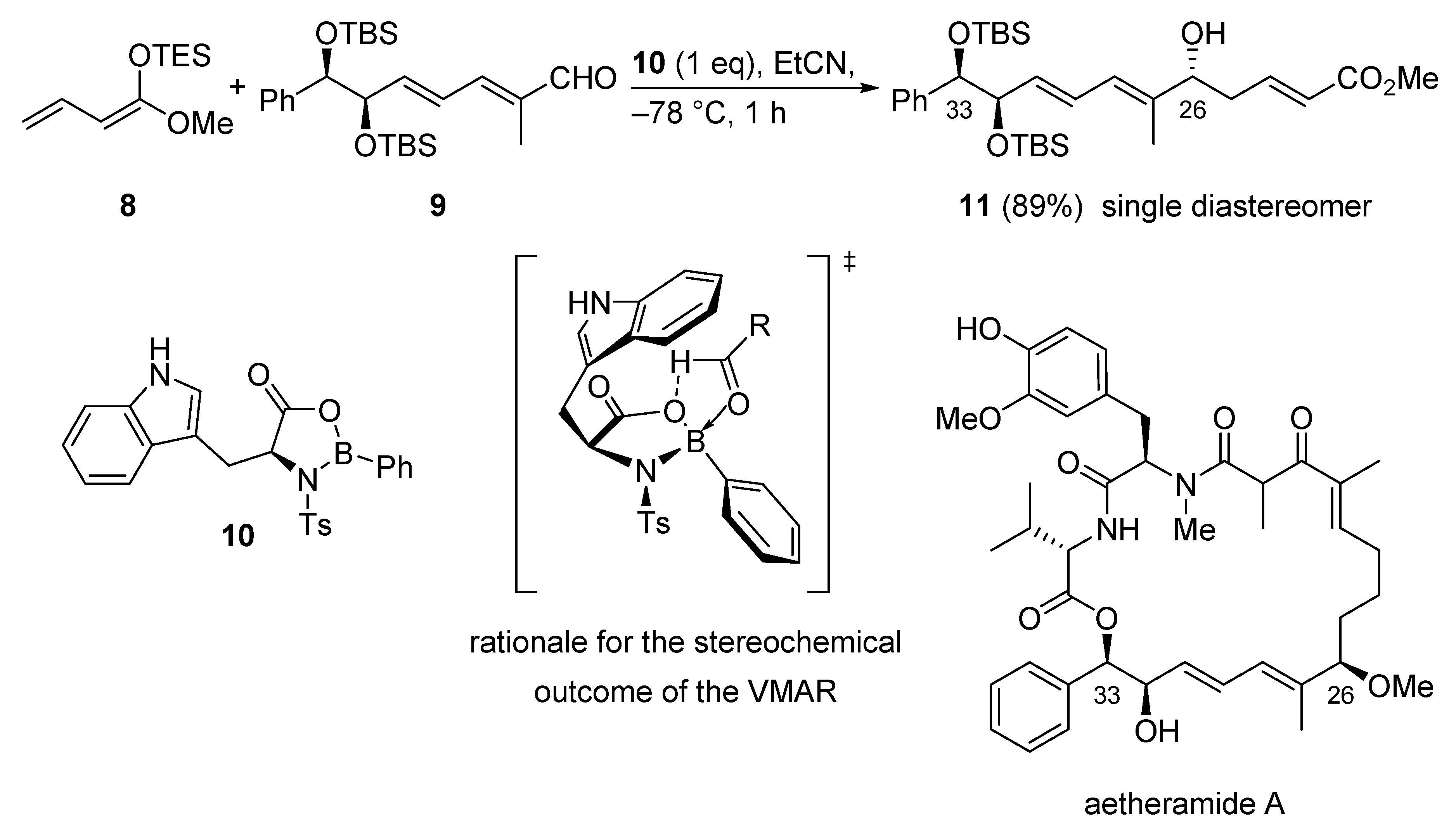
3.1.1. Aetheramide A
3.1.2. Nannocystin Ax
3.2. The Kobayashi Protocol
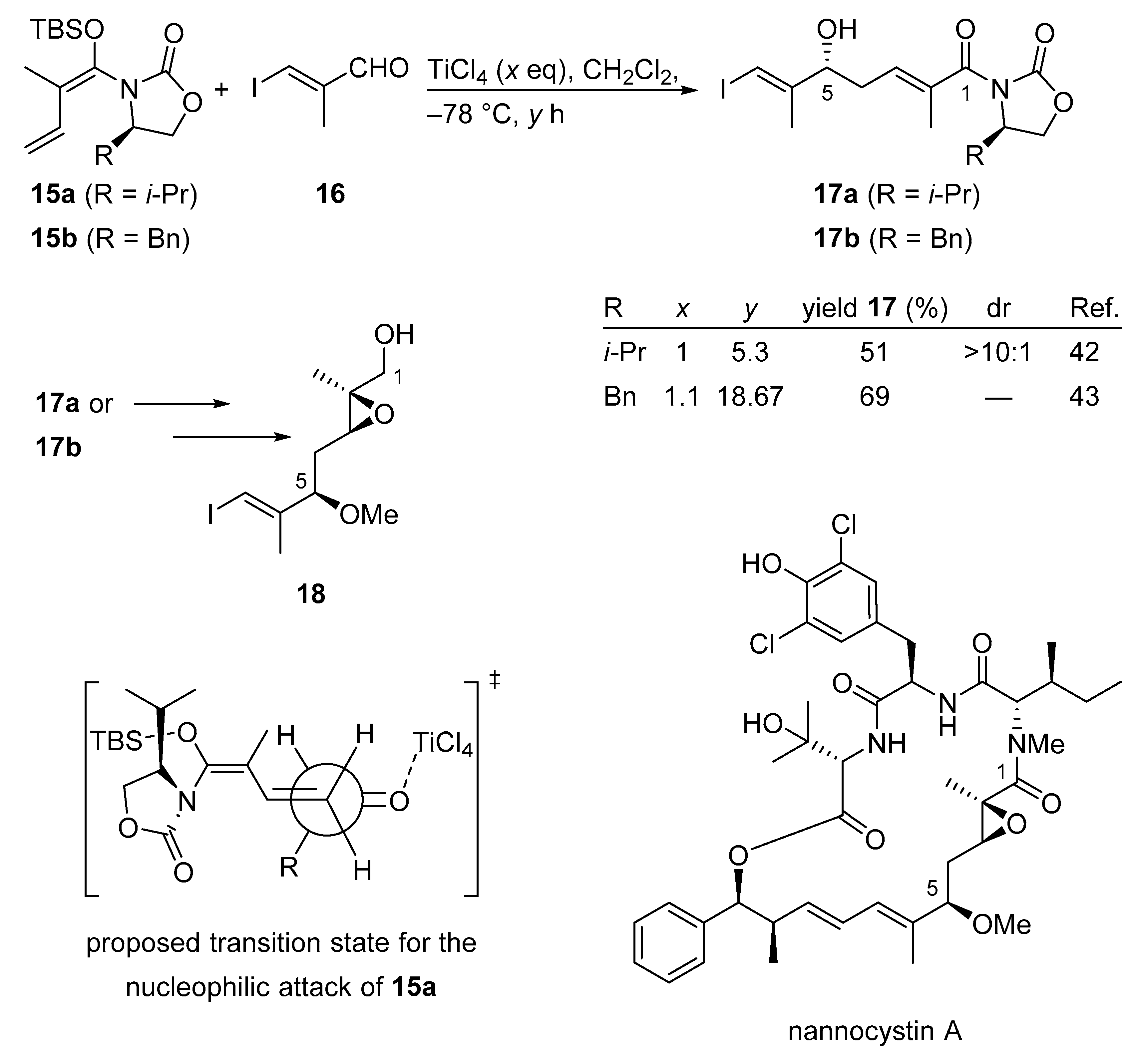
3.2.1. Nannocystin A
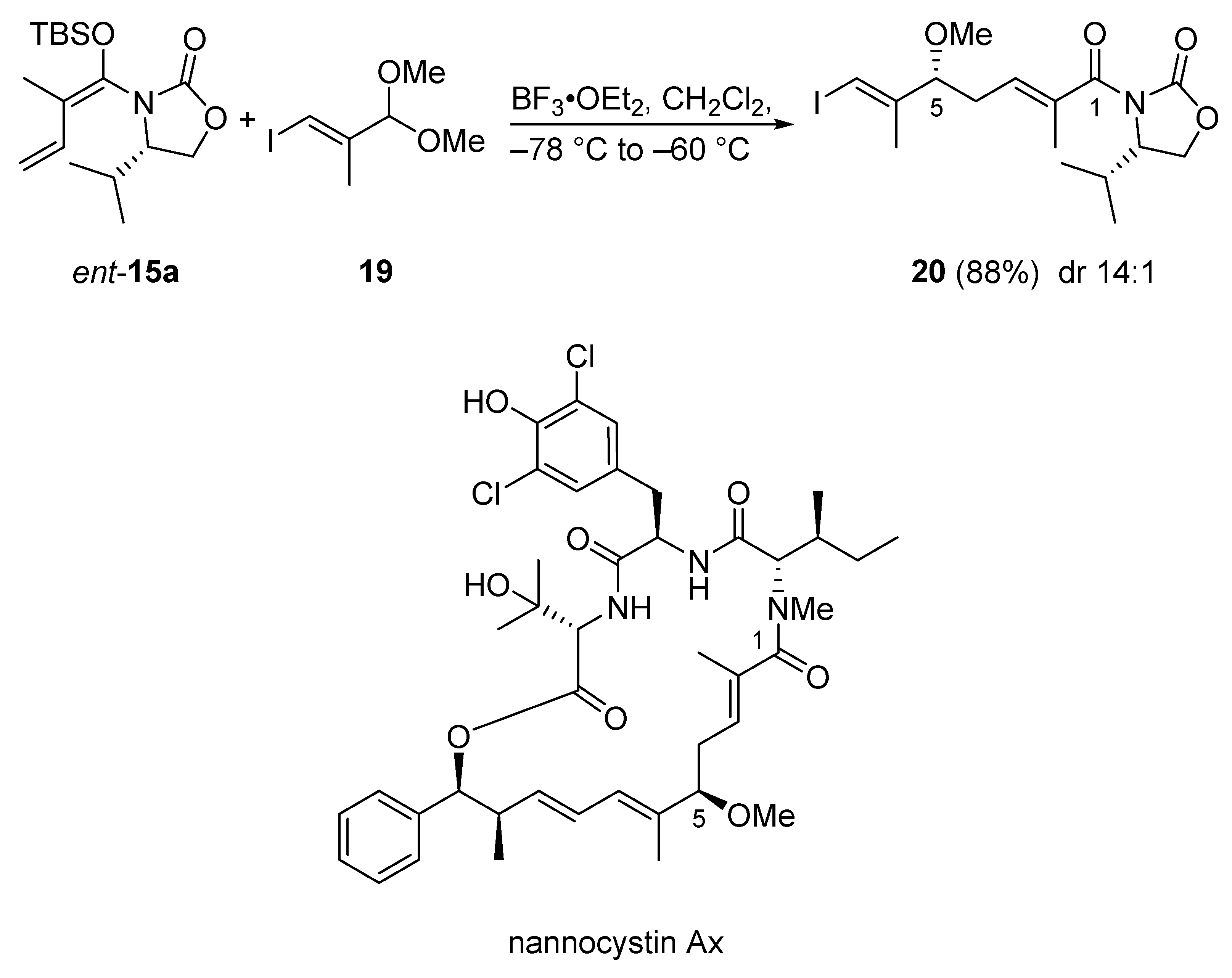
3.2.2. Nannocystin Ax
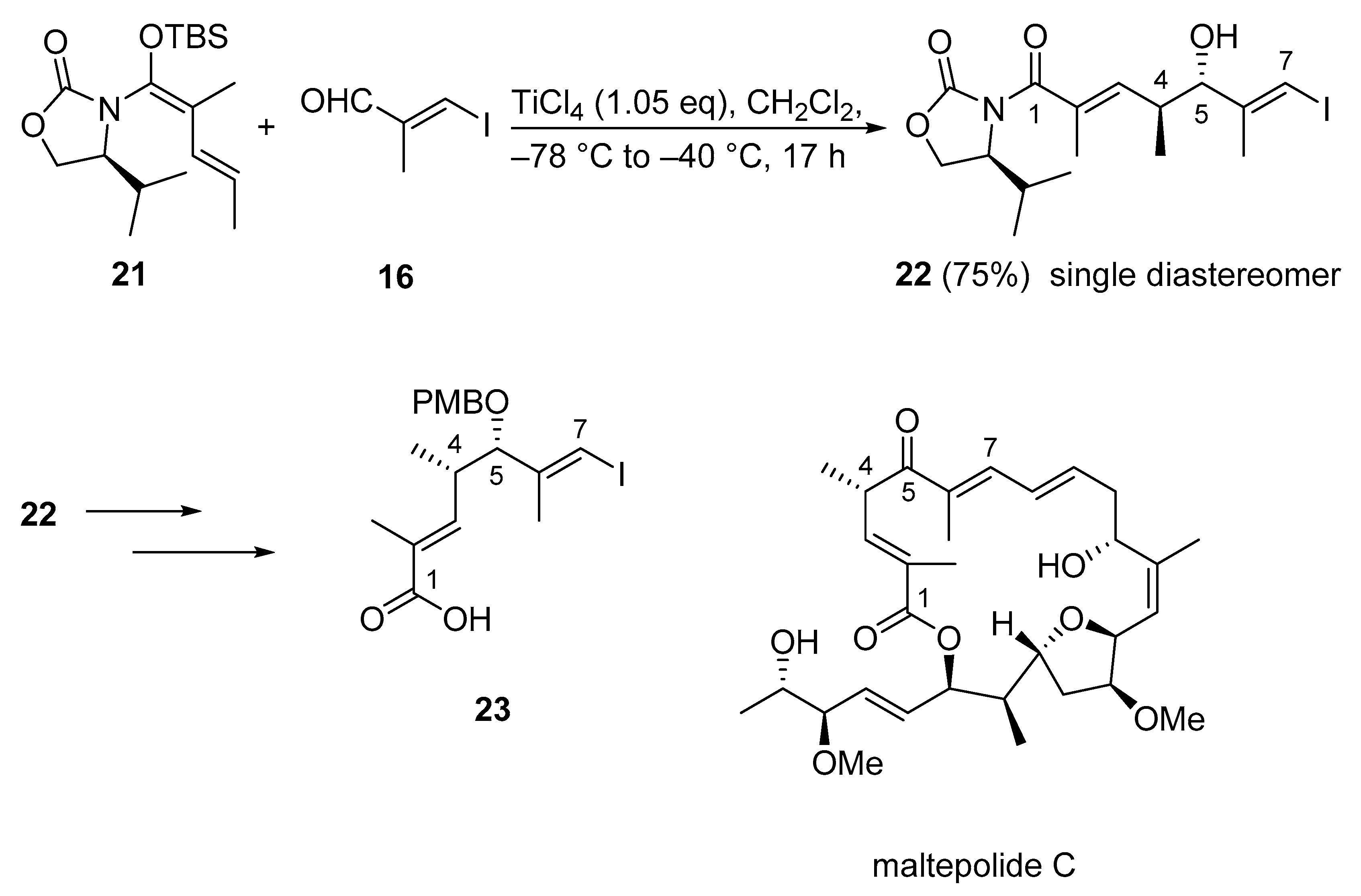
3.2.3. Maltepolide C
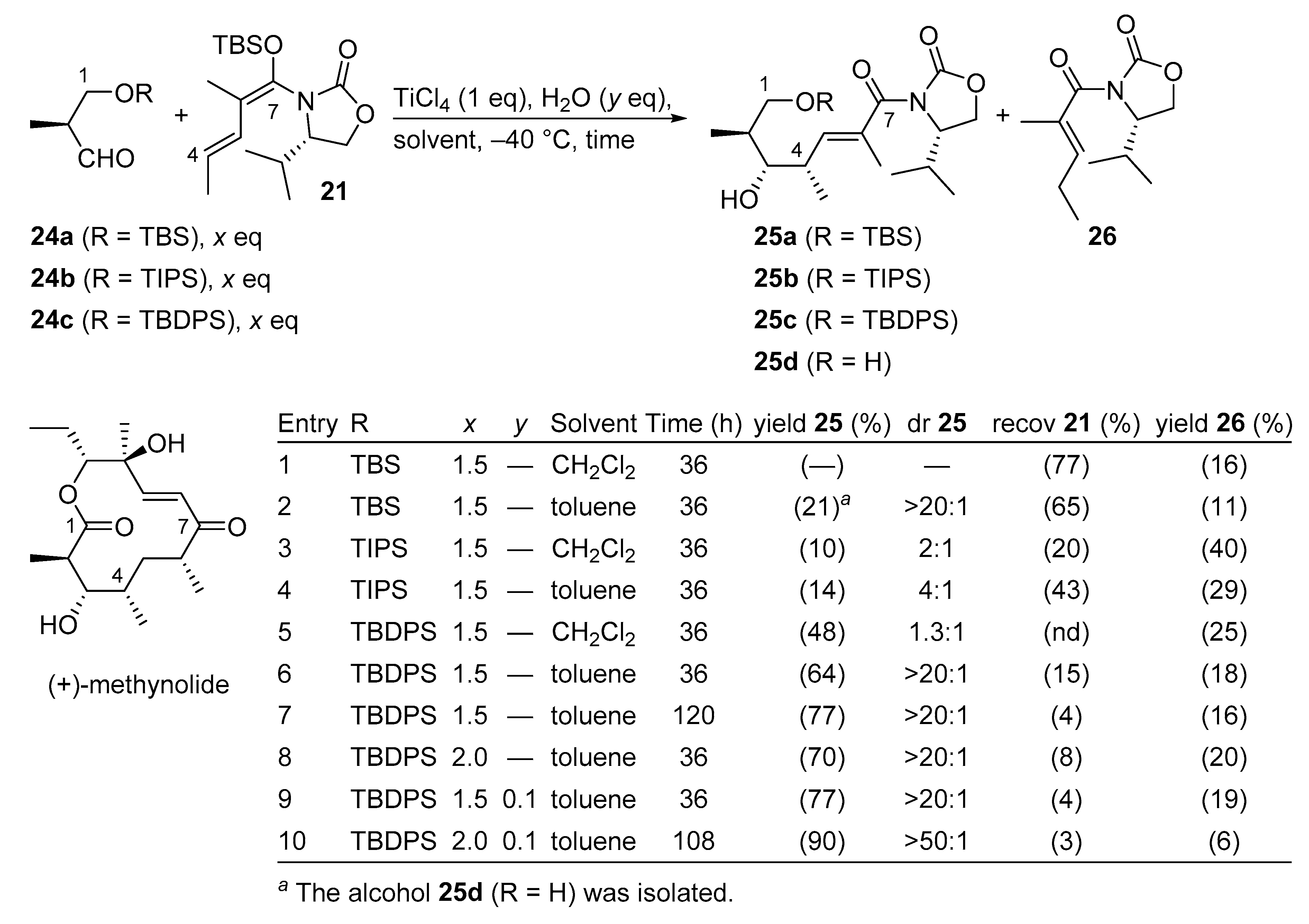
3.2.4. (+)-Methynolide
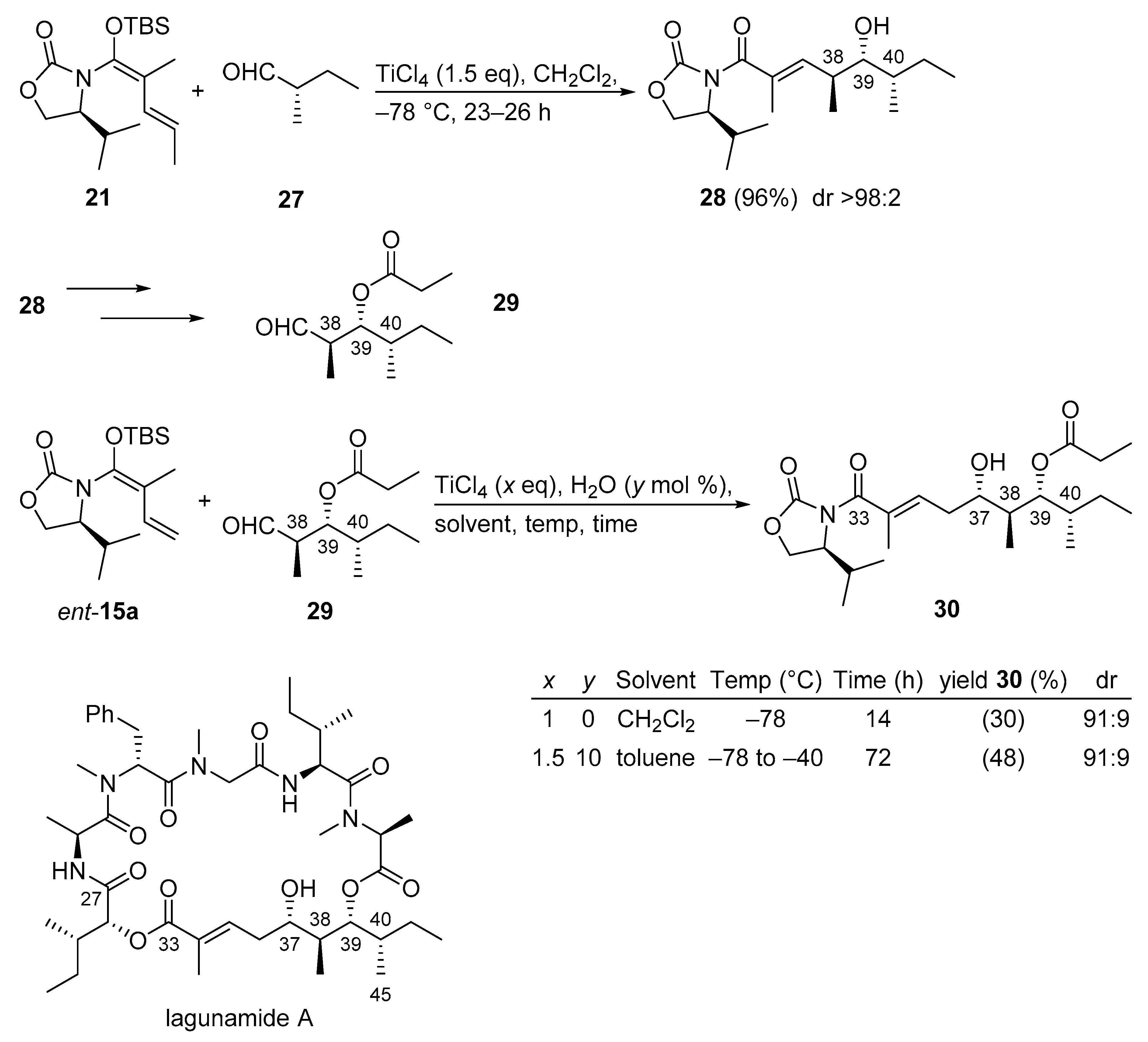
3.2.5. Lagunamide A
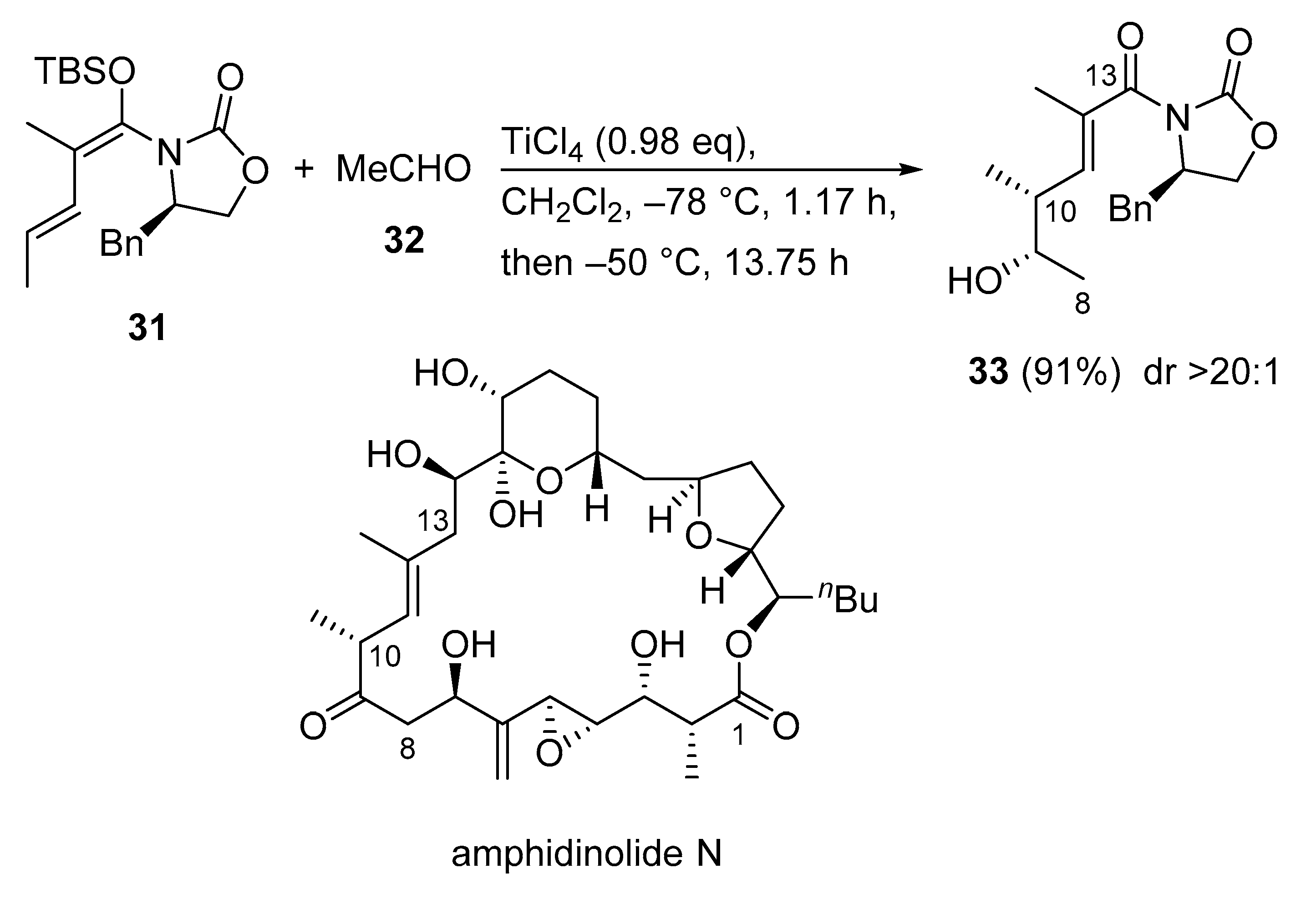
3.2.6. Amphidinolide N
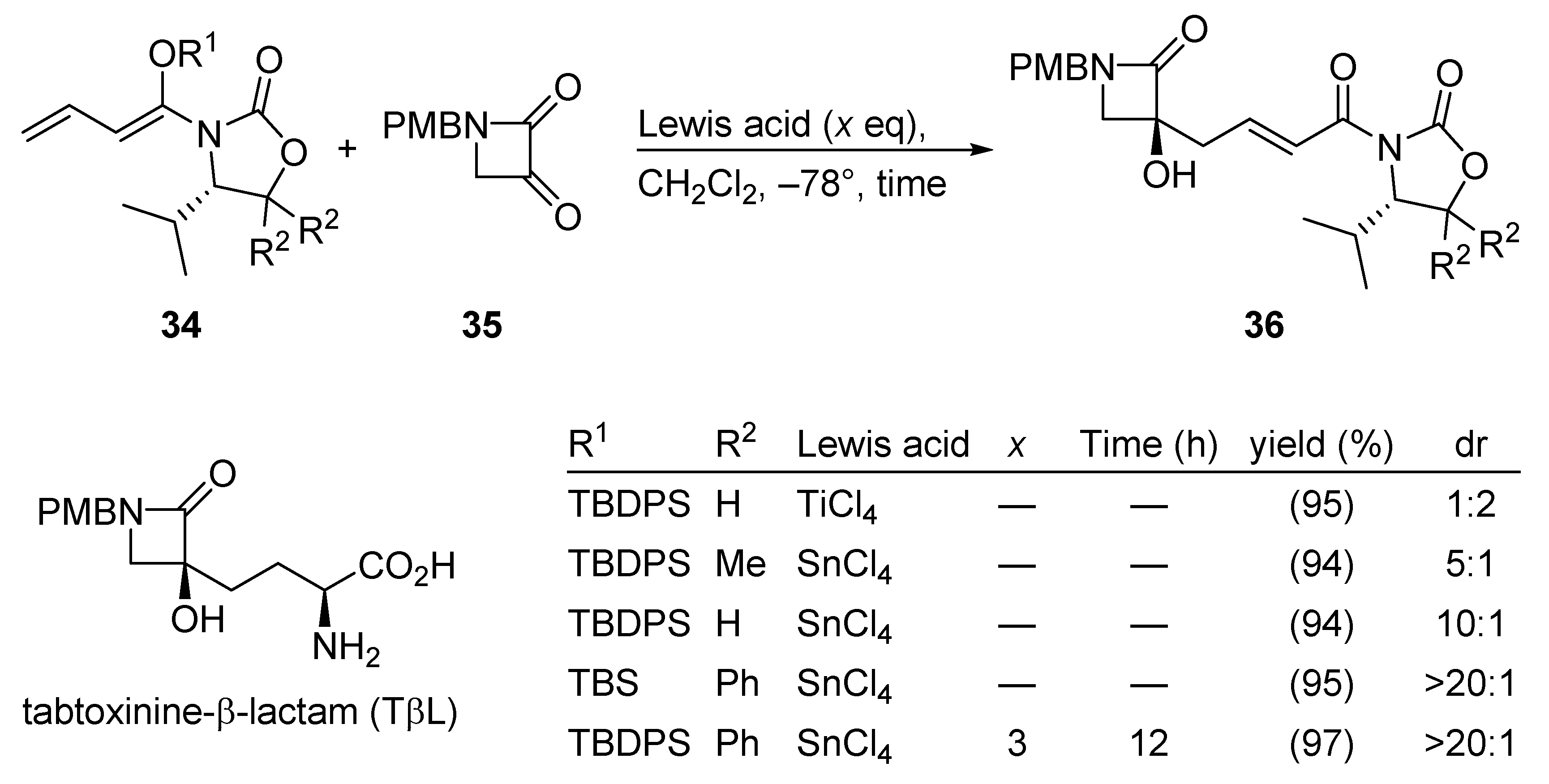
3.2.7. Tabtoxinine-β-Lactam (TβL)
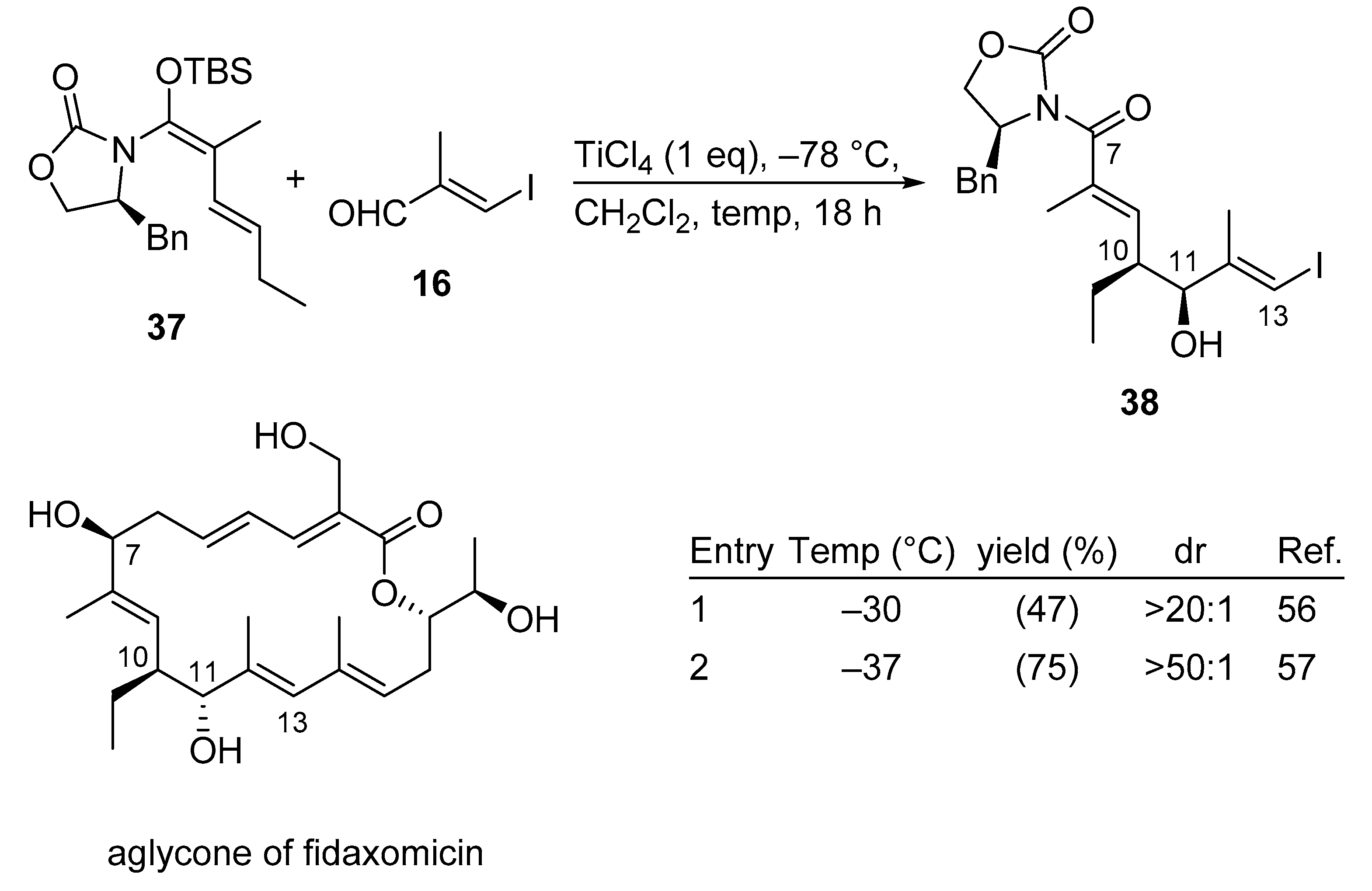
3.2.8. Fidaxomicin
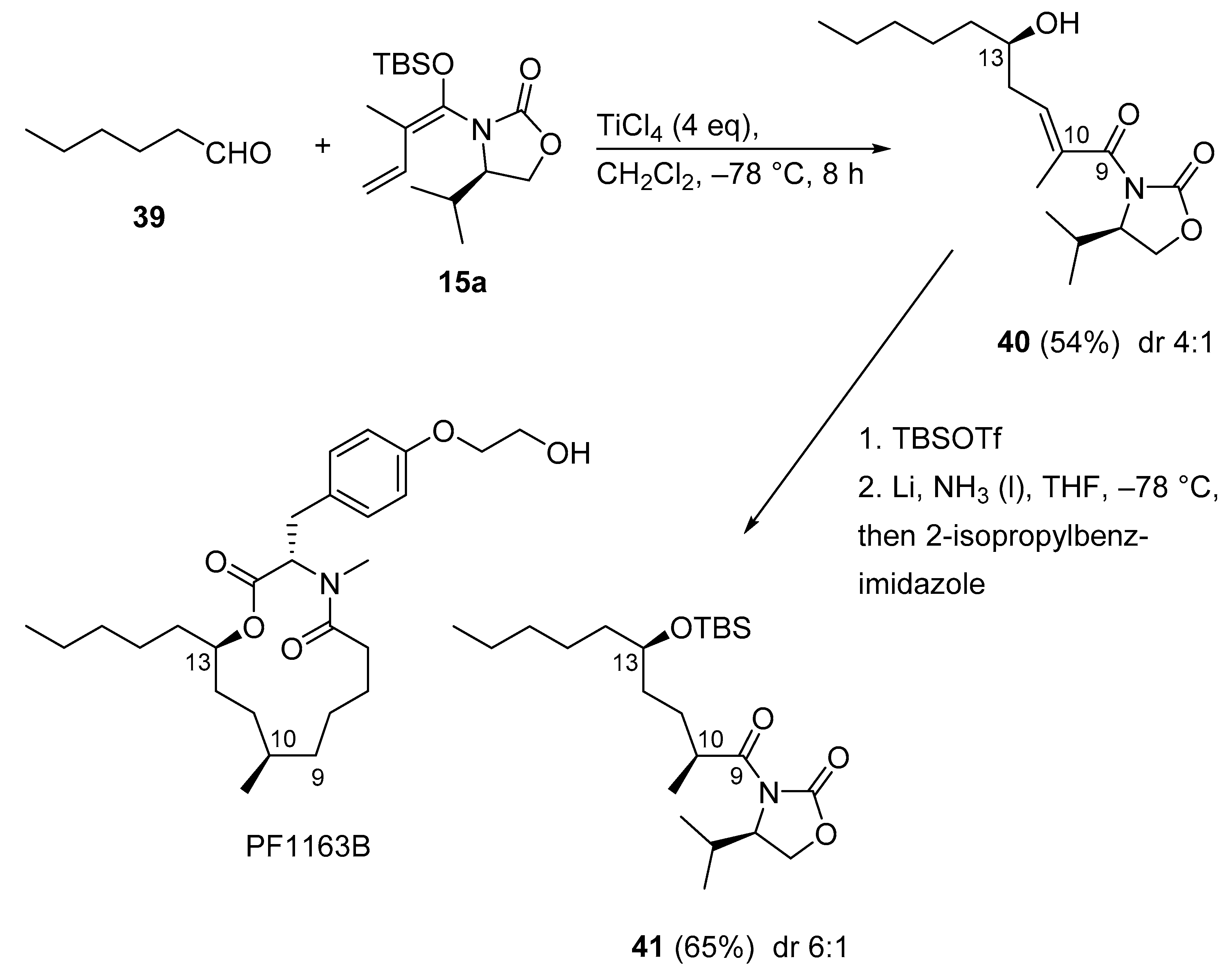
3.2.9. PF1163B
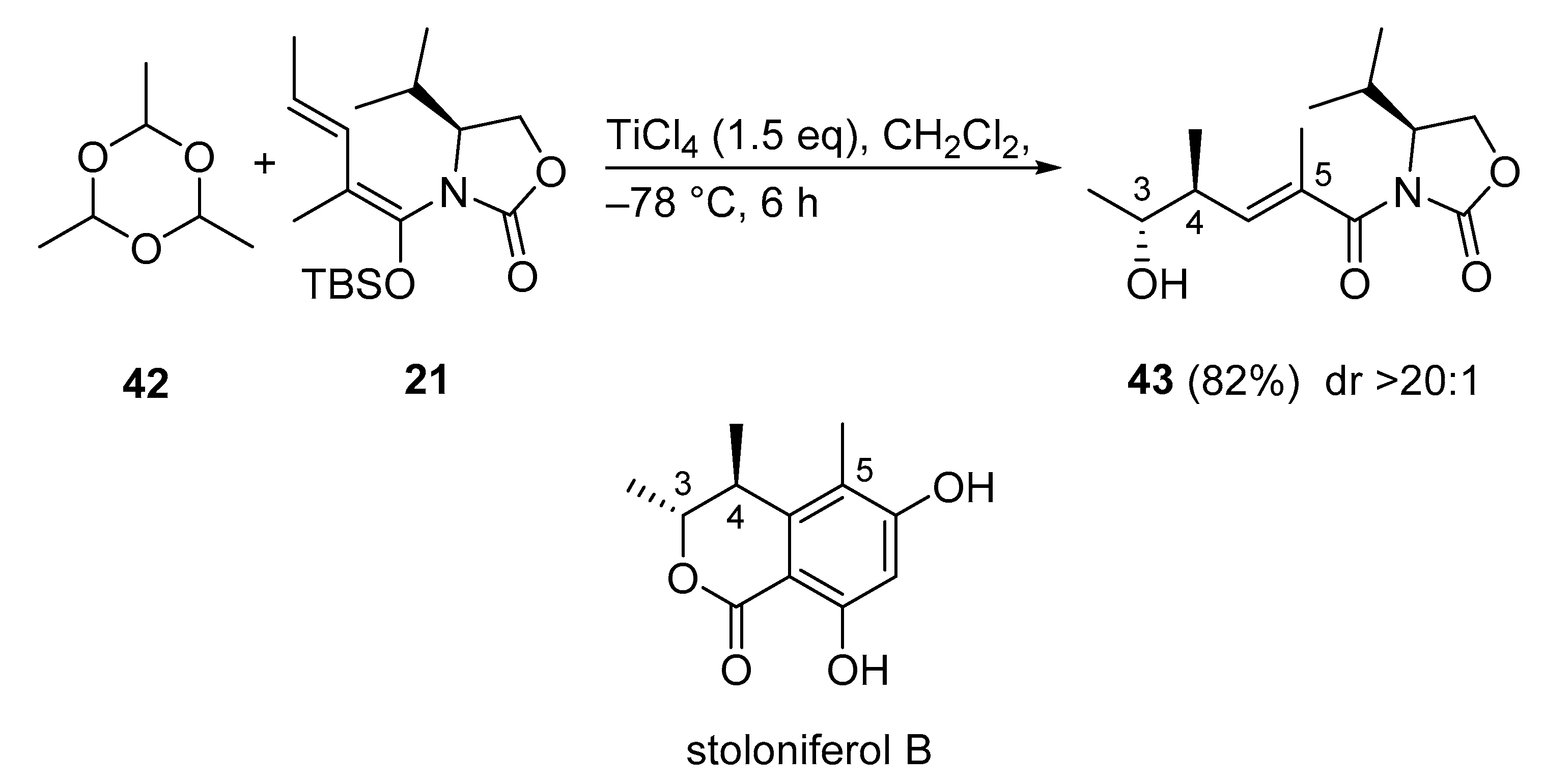
3.2.10. Stoloniferol B
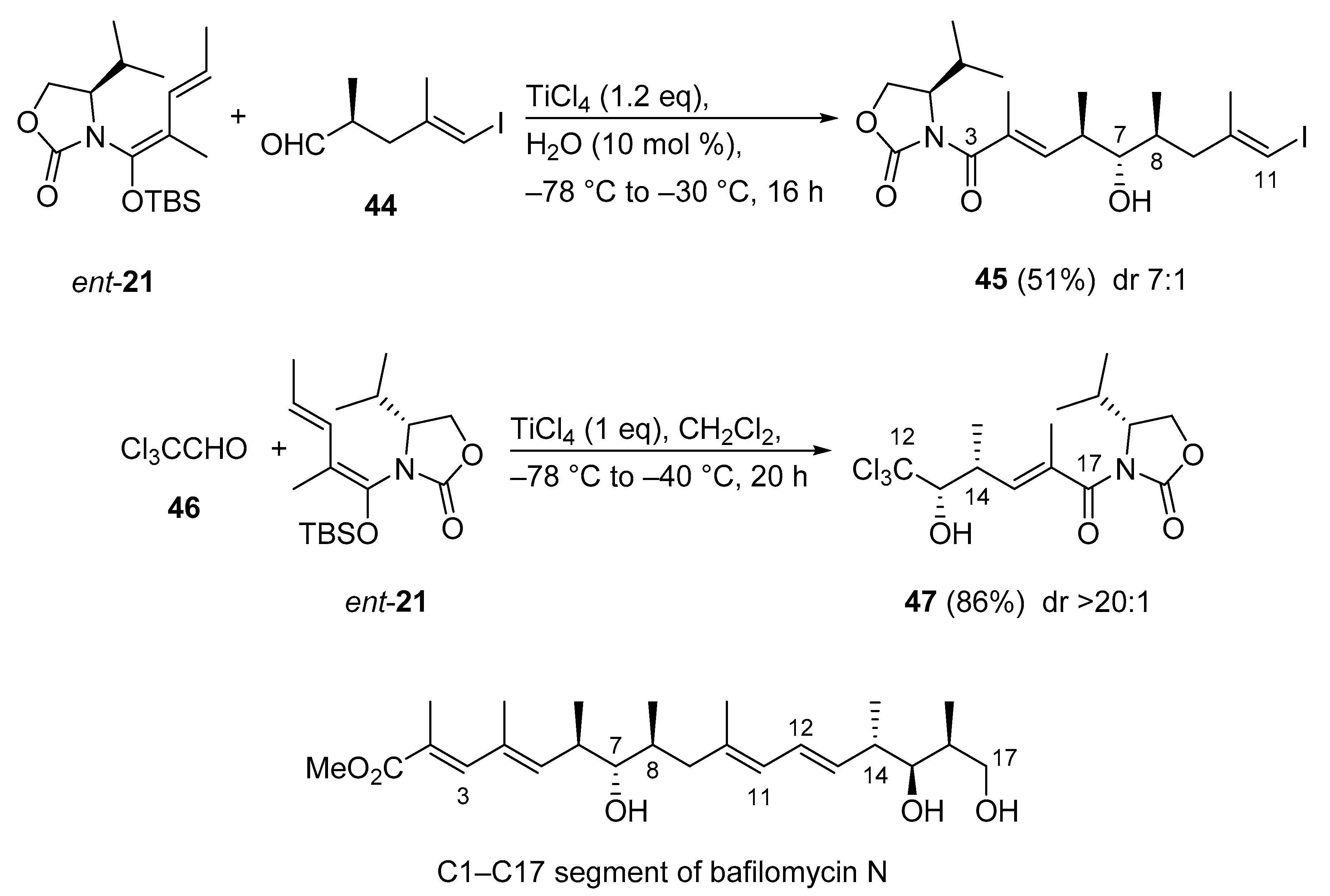
3.2.11. C1–C17 Segment of Bafilomycin N
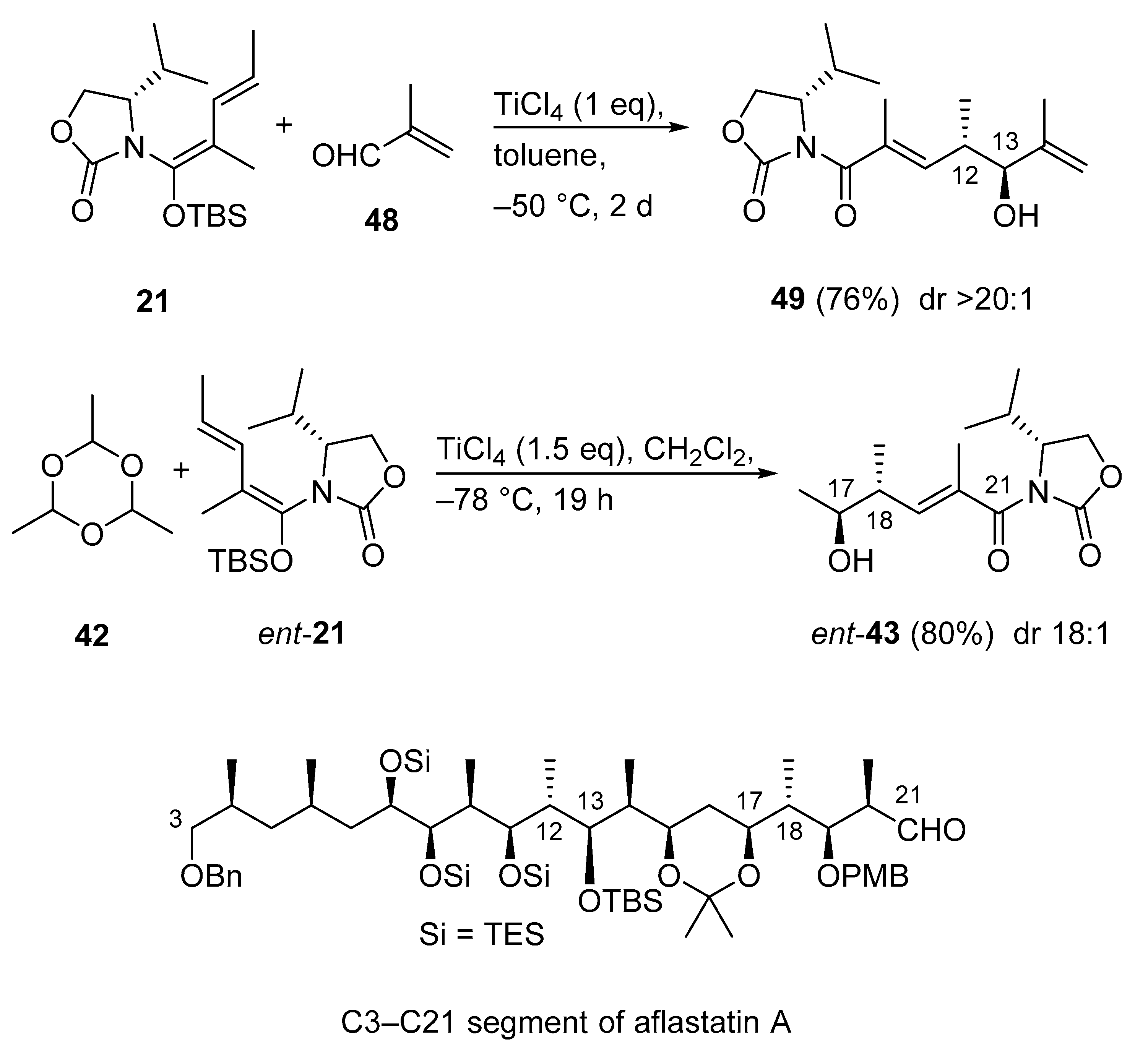
3.2.12. C3–C21 Segment of Aflastatin A
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cordes, M.H.; Kalesse, M. The Asymmetric Vinylogous Mukaiyama Aldol Reaction. Org. React. 2019, 98, 173–174. [Google Scholar]
- Kalesse, M. Recent Advances in Vinylogous Aldol Reactions and Their Applications in the Syntheses of Natural Products. Top. Curr. Chem. 2005, 244, 43–76. [Google Scholar]
- Denmark, S.E.; Heemstra, J.R., Jr.; Beutner, G.L. Catalytic, Enantioselective, Vinylogous Aldol Reactions. Angew. Chem. Int. Ed. 2005, 44, 4682–4698. [Google Scholar] [CrossRef] [PubMed]
- Soriente, A.; DeRosa, M.; Villano, R.; Scettri, A. Recent Advances in Asymmetric Aldol Reaction of Masked Acetoacetic Esters Promoted by Ti(IV) / BINOL: A New Methodology, Non-Linear Effects and Autoinduction. Curr. Org. Chem. 2004, 8, 993–1007. [Google Scholar] [CrossRef]
- Casiraghi, G.; Zanardi, F.; Appendino, G.; Rassu, G. The Vinylogous Aldol Reaction: A Valuable, Yet Understated Carbon−Carbon Bond-Forming Maneuver. Chem. Rev. 2000, 100, 1929–1972. [Google Scholar] [CrossRef] [PubMed]
- Fuson, R.C. The Principle of Vinylogy. Chem. Rev. 1935, 16, 1–27. [Google Scholar] [CrossRef]
- Casiraghi, G.; Battistini, L.; Curti, C.; Rassu, G.; Zanardi, F. The Vinylogous Aldol and Related Addition Reactions: Ten Years of Progress. Chem. Rev. 2011, 111, 3076–3154. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.B.J.; Ng, K.K.H.; Paterson, I. The Impact of the Mukaiyama Aldol Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2013, 52, 9097–9108. [Google Scholar] [CrossRef] [PubMed]
- Kalesse, M.; Cordes, M.; Symkenberg, G.; Lu, H.-H. The vinylogous Mukaiyama aldol reaction (VMAR) in natural product synthesis. Nat. Prod. Rep. 2014, 31, 563–594. [Google Scholar] [CrossRef] [PubMed]
- Frías, M.; Cieślik, W.; Fraile, A.; Rosado-Abón, A.; Garrido-Castro, A.F.; Yuste, F.; Alemán, J. Development and Application of Asymmetric Organocatalytic Mukaiyama and Vinylogous Mukaiyama-Type Reactions. Chem. Eur. J. 2018, 24, 10906–10933. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, S. Recent development of vinylogous Mukaiyama aldol reactions. Tetrahedron Lett. 2018, 59, 77–88. [Google Scholar] [CrossRef]
- Hosokawa, S. Remote Asymmetric Induction Reactions using a E,E-Vinylketene Silyl N,O-Acetal and the Wide Range Stereocontrol Strategy for the Synthesis of Polypropionates. Acc. Chem. Res. 2018, 51, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Mukaiyama, T.; Ishida, A. A Convenient Method for the Preparation of δ-alkoxy-α,β-unsaturated Aldehydes by Reaction of Acetals with 1-trimethylsiloxy-1,3-butadiene. Chem. Lett. 1975, 4, 319–322. [Google Scholar] [CrossRef]
- Ishida, A.; Mukaiyama, T. New Method for the Preparation of Polyenals from δ-alkoxy-α,β-unsaturated Aldehydes. Chem. Lett. 1975, 4, 1167–1170. [Google Scholar] [CrossRef]
- Ishida, A.; Mukaiyama, T. A New Method for the Preparation of δ-Alkoxy-α,β-unsaturated Aldehydes and Polyenals. Bull. Chem. Soc. Jpn. 1977, 50, 1161–1168. [Google Scholar] [CrossRef]
- Sato, M.; Sunami, S.; Sugita, Y.; Kaneko, C. Use of 1, 3-Dioxin-4-ones and Related Compounds in Synthesis. XLIV. Asymmetric Aldol Reaction of 4-Trimethylsiloxy-6-methylene-1, 3-dioxines: Use of Tartaric Acid-Derived (Acyloxy)borane Complex as the Catalyst. Chem. Pharm. Bull. 1994, 42, 839–845. [Google Scholar] [CrossRef]
- Sato, M.; Sunami, S.; Sugita, Y.; Kaneko, C. An Efficient Asymmetric Aldol Reaction of 4-Trimethylsiloxy-6-methylene-1,3-dioxines by Chiral Binaphthol-Titanium Complex Catalysis. Heterocycles 1995, 41, 1435–1444. [Google Scholar]
- Singer, R.A.; Carreira, E.M. Catalytic, Enantioselective Dienolate Additions to Aldehydes: Preparation of Optically Active Acetoacetate Aldol Adducts. J. Am. Chem. Soc. 1995, 117, 12360–12361. [Google Scholar] [CrossRef]
- Evans, D.A.; Kozlowski, M.C.; Murry, J.A.; Burgey, C.S.; Campos, K.R.; Connell, B.T.; Staples, R.J. C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of Catalytic Enantioselective Aldol Additions of Enolsilanes to (Benzyloxy)acetaldehyde. J. Am. Chem. Soc. 1999, 121, 669–685. [Google Scholar] [CrossRef]
- Evans, D.A.; Burgey, C.S.; Kozlowski, M.C.; Tregay, S.W. C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Scope and Mechanism of the Catalytic Enantioselective Aldol Additions of Enolsilanes to Pyruvate Esters. J. Am. Chem. Soc. 1999, 121, 686–699. [Google Scholar] [CrossRef]
- Denmark, S.E.; Beutner, G.L. Lewis Base Activation of Lewis Acids. Vinylogous Aldol Reactions. J. Am. Chem. Soc. 2003, 125, 7800–7801. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Beutner, G.L.; Wynn, T.; Eastgate, M.D. Lewis Base Activation of Lewis Acids: Catalytic, Enantioselective Addition of Silyl Ketene Acetals to Aldehydes. J. Am. Chem. Soc. 2005, 127, 3774–3789. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Heemstra, J.R., Jr. Lewis Base Activation of Lewis Acids: Catalytic, Enantioselective Vinylogous Aldol Addition Reactions. J. Org. Chem. 2007, 72, 5668–5688. [Google Scholar] [CrossRef]
- Kruger, J.; Carreira, E.M. Apparent Catalytic Generation of Chiral Metal Enolates: Enantioselective Dienolate Additions to Aldehydes Mediated by Tol-BINAP•Cu(II) Fluoride Complexes. J. Am. Chem. Soc. 1998, 120, 837–838. [Google Scholar] [CrossRef]
- Pagenkopf, B.L.; Krüger, J.; Stojanovic, A.; Carreira, E.M. Mechanistic Insights into Cu-Catalyzed Asymmetric Aldol Reactions: Chemical and Spectroscopic Evidence for a Metalloenolate Intermediate. Angew. Chem. Int. Ed. 1998, 37, 3124–3126. [Google Scholar] [CrossRef]
- Bluet, G.; Bazan-Tejeda, B.; Campagne, J.-M. Catalytic Asymmetric Access to α,β Unsaturated δ-Lactones through a Vinylogous Aldol Reaction: Application to the Total Synthesis of the Prelog-Djerassi Lactone. Org. Lett. 2001, 3, 3807–3810. [Google Scholar] [CrossRef]
- Corey, E.J.; Cywin, C.L.; Roper, T.D. Enantioselective Mukaiyama-aldol and aldol-dihydropyrone annulation reactions catalyzed by a tryptophan-derived oxazaborolidine. Tetrahedron Lett. 1992, 33, 6907–6910. [Google Scholar] [CrossRef]
- Brodmann, T.; Lorenz, M.; Schäckel, R.; Simsek, S.; Kalesse, M. Highly Stereoselective Aldol Reactions in the Total Syntheses of Complex Natural Products. Synlett 2009, 2, 174–192. [Google Scholar]
- Saito, S.; Shiozawa, M.; Ito, M.; Yamamoto, H. Conceptually New Directed Aldol Condensation Using Aluminum Tris(2,6-diphenylphenoxide). J. Am. Chem. Soc. 1998, 120, 813–814. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Nagata, C.; Shingu, H. Molecular Orbital Theory of Orientation in Aromatic, Heteroaromatic, and Other Conjugated Molecules. J. Chem. Phys. 1954, 22, 1433–1442. [Google Scholar] [CrossRef]
- Denmark, S.E.; Fan, Y. Catalytic, Enantioselective Aldol Additions to Ketones. J. Am. Chem. Soc. 2002, 124, 4233–4235. [Google Scholar] [CrossRef]
- Myers, A.G.; Widdowson, K.L. A silicon-directed aldol condensation. Synthesis of enantiomerically pure anti aldols. J. Am. Chem. Soc. 1990, 112, 9672–9674. [Google Scholar] [CrossRef]
- Denmark, S.E.; Griedel, B.D.; Coe, D.M.; Schnute, M.E. Chemistry of Enoxysilacyclobutanes: Highly Selective Uncatalyzed Aldol Additions. J. Am. Chem. Soc. 1994, 116, 7026–7043. [Google Scholar] [CrossRef]
- Myers, A.G.; Kephart, S.E.; Chen, H. Silicon-directed aldol reactions. Rate acceleration by small rings. J. Am. Chem. Soc. 1992, 114, 7922–7923. [Google Scholar] [CrossRef]
- Myers, A.G.; Widdowson, K.L.; Kukkola, P.J. Silicon-directed aldol condensation. Evidence for a pseudorotational mechanism. J. Am. Chem. Soc. 1992, 114, 2765–2767. [Google Scholar] [CrossRef]
- Denmark, S.E.; Lee, W. Investigations on Transition-State Geometry in the Lewis Acid- (Mukaiyama) and Fluoride-Promoted Aldol Reactions. J. Org. Chem. 1994, 59, 707–709. [Google Scholar] [CrossRef]
- Laina-Martín, V.; Humbrías-Martín, J.; Fernández-Salas, J.A.; Alemán, J. Asymmetric vinylogous Mukaiyama aldol reaction of isatins under bifunctional organocatalysis: Enantioselective synthesis of substituted 3-hydroxy-2-oxindoles. Chem. Commun. 2018, 54, 2781–2784. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, P.; Schneider, C. Stereoselective Synthesis of 2,3,5-Trisubstituted Tetrahydrofurans Initiated by a Titanium–BINOLate-Catalyzed Vinylogous Aldol Reaction. J. Org. Chem. 2019, 84, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Gerstmann, L.; Kalesse, M. Total Synthesis of Aetheramide A. Chem. Eur. J. 2016, 22, 11210. [Google Scholar] [CrossRef] [PubMed]
- Poock, C.; Kalesse, M. Total Synthesis of Nannocystin Ax. Org. Lett. 2017, 19, 4536–4539. [Google Scholar] [CrossRef]
- Shirokawa, S.-I.; Kamiyama, M.; Nakamura, T.; Okada, M.; Nakazaki, A.; Hosokawa, S.; Kobayashi, S. Remote Asymmetric Induction with Vinylketene Silyl N,O-Acetal. J. Am. Chem. Soc. 2004, 126, 13604–13605. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xu, X.; Yang, C.-H.; Tian, Y.; Chen, X.; Lian, L.; Pan, W.; Su, X.; Zhang, W.; Chen, Y. Total Synthesis of Nannocystin A. Org. Lett. 2016, 18, 5768–5770. [Google Scholar] [CrossRef]
- Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Concise Total Synthesis of Nannocystin A. Angew. Chem. Int. Ed. 2016, 55, 13263–13266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Liu, R.; Liu, B. Total synthesis of nannocystin Ax. Chem. Commun. 2017, 53, 5549–5552. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, H.; Mukaeda, Y.; Hosokawa, S. syn-Selective Kobayashi Aldol Reaction Using Acetals. Org. Lett. 2013, 15, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Hoecker, J.; Gademann, K. Enantioselective Total Syntheses and Absolute Configuration of JBIR-02 and Mer-A2026B. Org. Lett. 2013, 15, 670–673. [Google Scholar] [CrossRef]
- Rao, K.N.; Kanakaraju, M.; Kunwar, A.C.; Ghosh, S. Total Synthesis of the Proposed Structure of Maltepolide C. Org. Lett. 2016, 18, 4092–4095. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Fujimura, M.; Fujita, K.; Kobayashi, S. Total synthesis of (+)-methynolide using a Ti-mediated aldol reaction of a lactyl-bearing oxazolidin-2-one, and a vinylogous Mukaiyama aldol reaction. Tetrahedron 2017, 73, 3652–3659. [Google Scholar] [CrossRef]
- Jürjens, G.; Kirschning, A. Synthesis of a Cytotoxic Ansamycin Hybrid. Org. Lett. 2014, 16, 3000–3003. [Google Scholar] [CrossRef]
- Banasik, B.A.; Wang, L.; Kanner, A.; Bergdahl, B.M. Further insight into the asymmetric vinylogous Mukaiyama aldol reaction (VMAR); application to the synthesis of the C27–C45 segment of lagunamide A. Tetrahedron 2016, 72, 2481–2490. [Google Scholar] [CrossRef]
- Yamaoka, M.; Nakazaki, A.; Kobayashi, S. Rate enhancement by water in a TiCl4-mediated stereoselective vinylogous Mukaiyama aldol reaction. Tetrahedron Lett. 2010, 51, 287–289. [Google Scholar] [CrossRef]
- Hosokawa, S.; Matsushita, K.; Tokimatsu, S.; Toriumi, T.; Suzuki, Y.; Tatsuta, K. The first total synthesis and structural determination of epi-cochlioquinone A. Tetrahedron Lett. 2010, 51, 5532–5536. [Google Scholar] [CrossRef]
- Toyoshima, A.; Sasaki, M. Toward a total synthesis of amphidinolide N: Convergent synthesis of the C1–C13 segment. Tetrahedron Lett. 2016, 57, 3532–3534. [Google Scholar] [CrossRef]
- Nagasawa, T.; Kuwahara, S. Formal Total Synthesis of Lactimidomycin. Org. Lett. 2013, 15, 3002–3005. [Google Scholar] [CrossRef]
- Ejima, H.; Wakita, F.; Imamura, R.; Kato, T.; Hosokawa, S. Stereoselective Synthesis of Tabtoxinine-β-lactam by Using the Vinylogous Mukaiyama Aldol Reaction with Acetate-Type Vinylketene Silyl N,O-Acetal and α-Keto-β-lactam. Org. Lett. 2017, 19, 2530–2532. [Google Scholar] [CrossRef]
- Miyatake-Ondozabal, H.; Kaufmann, E.; Gademann, K. Total Synthesis of the Protected Aglycon of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). Angew. Chem. Int. Ed. 2015, 54, 1933–1936. [Google Scholar] [CrossRef]
- Hattori, H.; Kaufmann, E.; Miyatake-Ondozabal, H.; Berg, R.; Gademann, K. Total Synthesis of Tiacumicin A. Total Synthesis, Relay Synthesis, and Degradation Studies of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). J. Org. Chem. 2018, 83, 7180–7205. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Hosokawa, S. Total Synthesis of PF1163B. Synlett 2019, 30, 709–712. [Google Scholar]
- Mukaeda, Y.; Kato, T.; Hosokawa, S. Syn-Selective Kobayashi Aldol Reaction Using the E,E-Vinylketene Silyl N,O-Acetal. Org. Lett. 2012, 14, 5298–5301. [Google Scholar] [CrossRef]
- Ohashi, T.; Hosokawa, S. Total Syntheses of Stoloniferol B and Penicitol A, and Structural Revision of Fusaraisochromanone. Org. Lett. 2018, 20, 3021–3024. [Google Scholar] [CrossRef]
- Sato, H.; Hosokawa, S. Synthesis of the C1–C17 Segment of Bafilomycin N. Synlett 2019, 30, 577–580. [Google Scholar]
- Shinoyama, M.; Shirokawa, S.-I.; Nakazaki, A.; Kobayashi, S. A Switch of Facial Selectivities Using α-Heteroatom-Substituted Aldehydes in the Vinylogous Mukaiyama Aldol Reaction. Org. Lett. 2009, 11, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, S.; Hosokawa, S. Synthesis of C3–C21 Segment of Aflastatin A Using Remote Asymmetric Induction Reactions. Org. Lett. 2019, 21, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Matsui, R.; Seto, K.; Sato, Y.; Suzuki, T.; Nakazaki, A.; Kobayashi, S. Convergent Total Synthesis of (+)-TMC-151C by a Vinylogous Mukaiyama Aldol Reaction and Ring-Closing Metathesis. Angew. Chem. Int. Ed. 2011, 50, 680–683. [Google Scholar] [CrossRef] [PubMed]























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordes, M.; Kalesse, M. Very Recent Advances in Vinylogous Mukaiyama Aldol Reactions and Their Applications to Synthesis. Molecules 2019, 24, 3040. https://doi.org/10.3390/molecules24173040
Cordes M, Kalesse M. Very Recent Advances in Vinylogous Mukaiyama Aldol Reactions and Their Applications to Synthesis. Molecules. 2019; 24(17):3040. https://doi.org/10.3390/molecules24173040
Chicago/Turabian StyleCordes, Martin, and Markus Kalesse. 2019. "Very Recent Advances in Vinylogous Mukaiyama Aldol Reactions and Their Applications to Synthesis" Molecules 24, no. 17: 3040. https://doi.org/10.3390/molecules24173040
APA StyleCordes, M., & Kalesse, M. (2019). Very Recent Advances in Vinylogous Mukaiyama Aldol Reactions and Their Applications to Synthesis. Molecules, 24(17), 3040. https://doi.org/10.3390/molecules24173040