Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
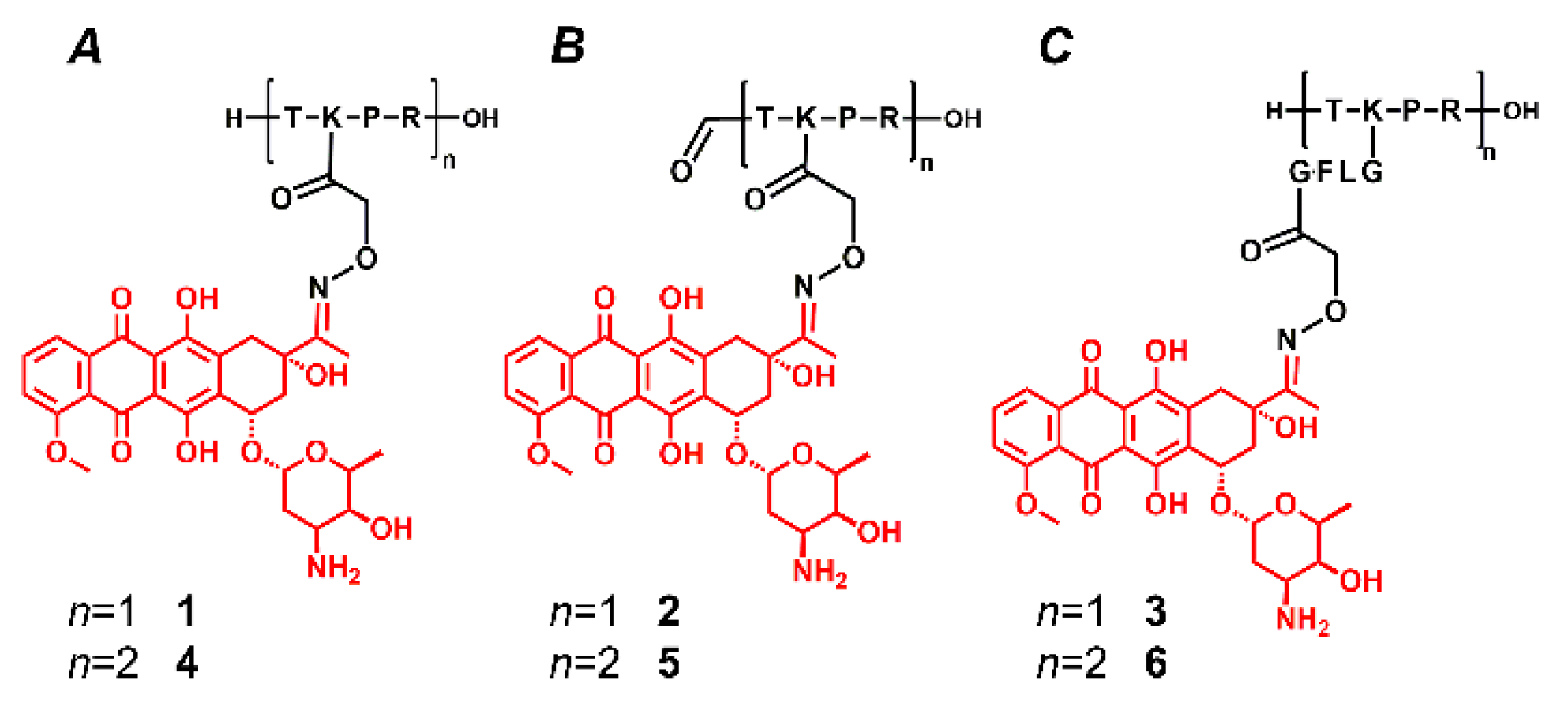
2.1. Synthesis of the Conjugates
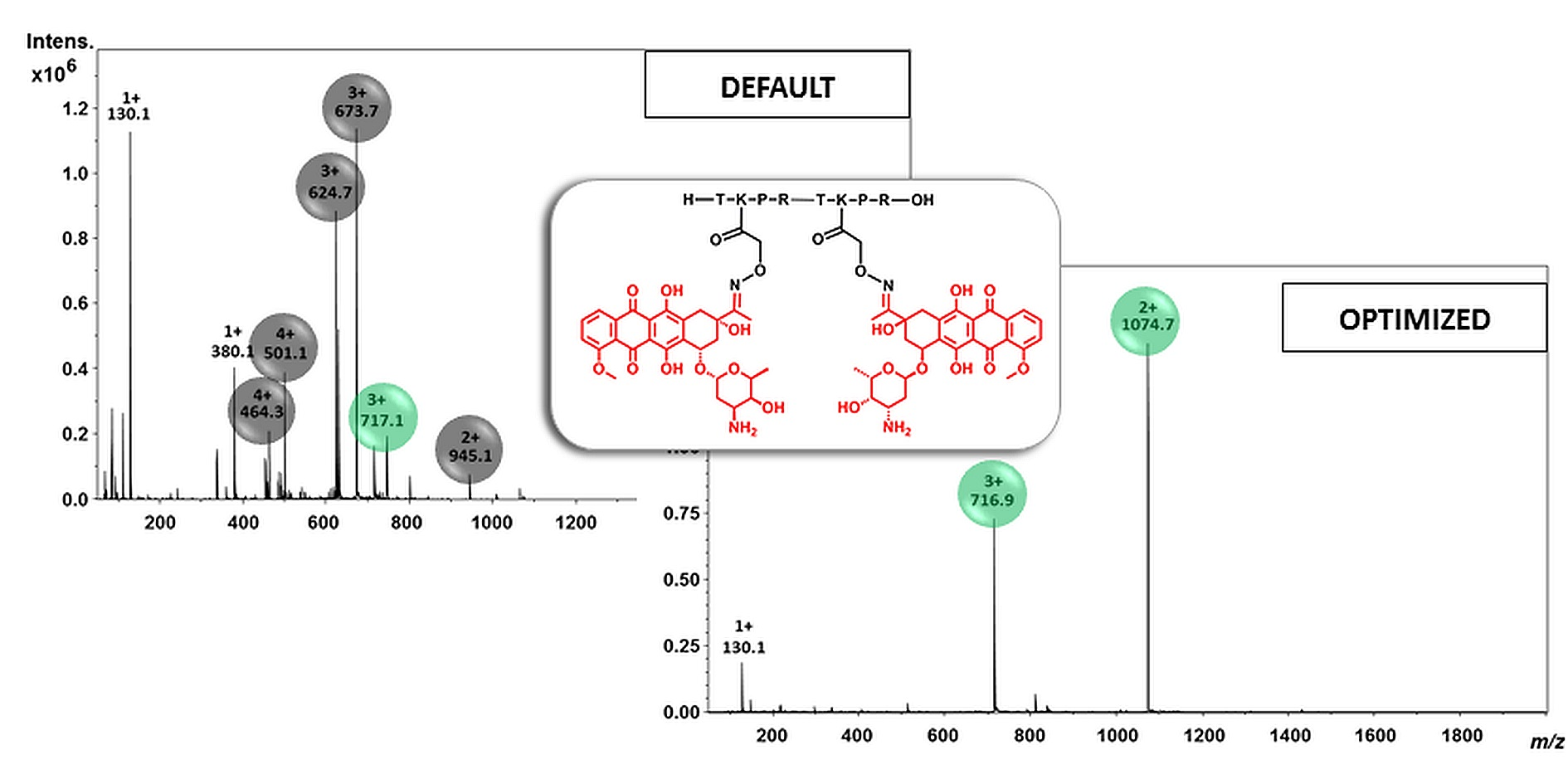
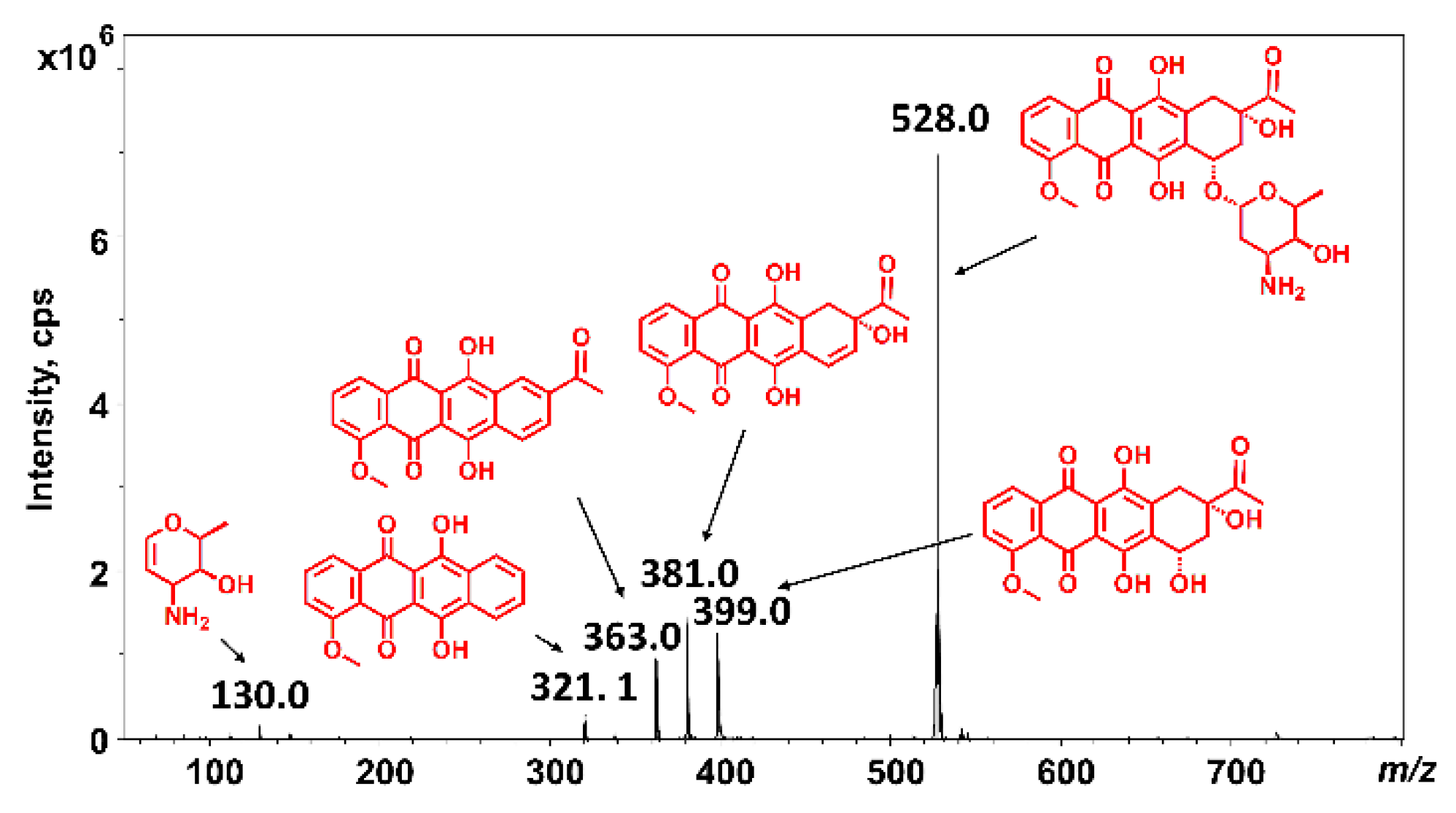
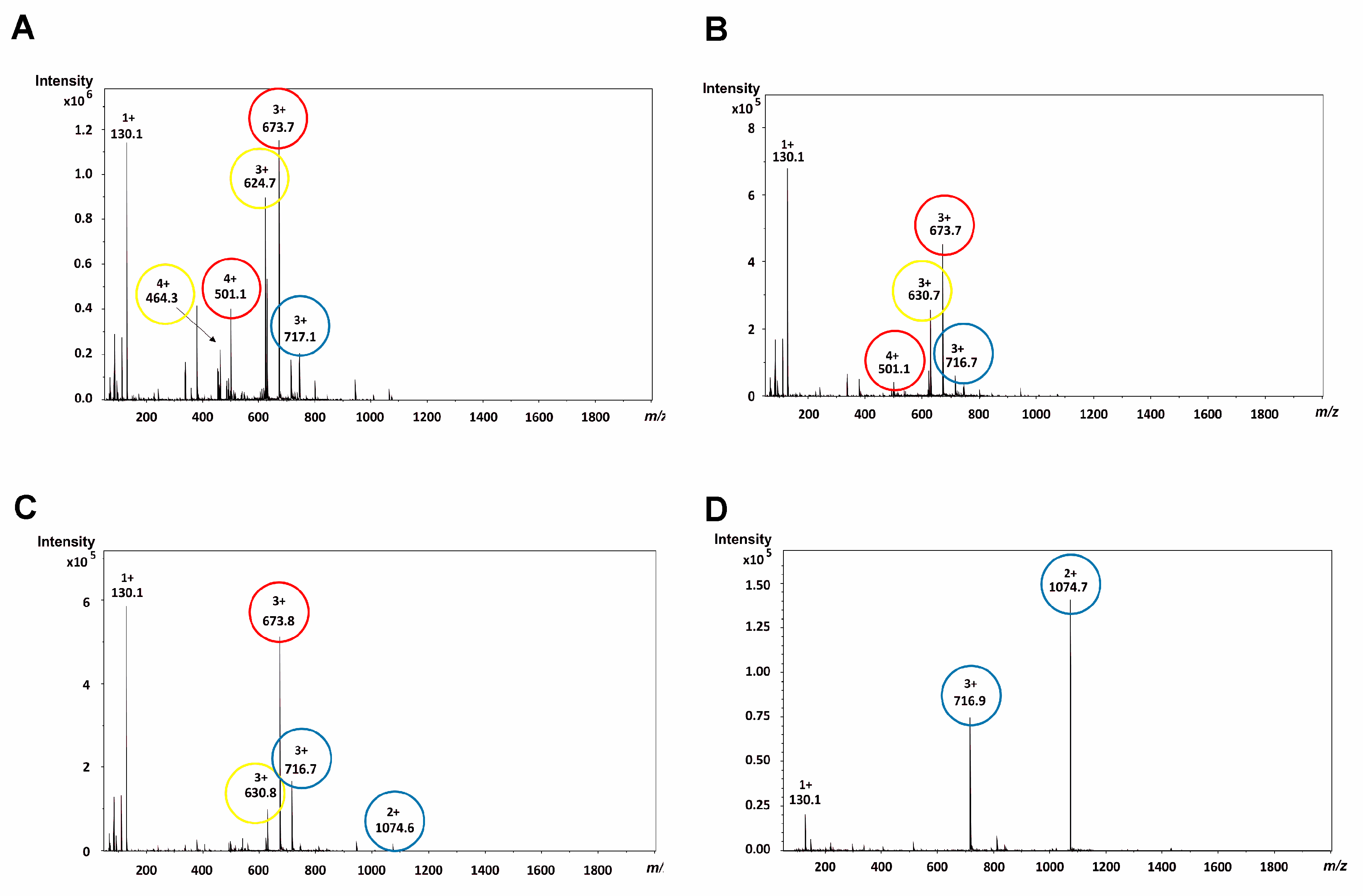
2.2. Mass Spectrometric Analysis under the Commonly Used Conditions
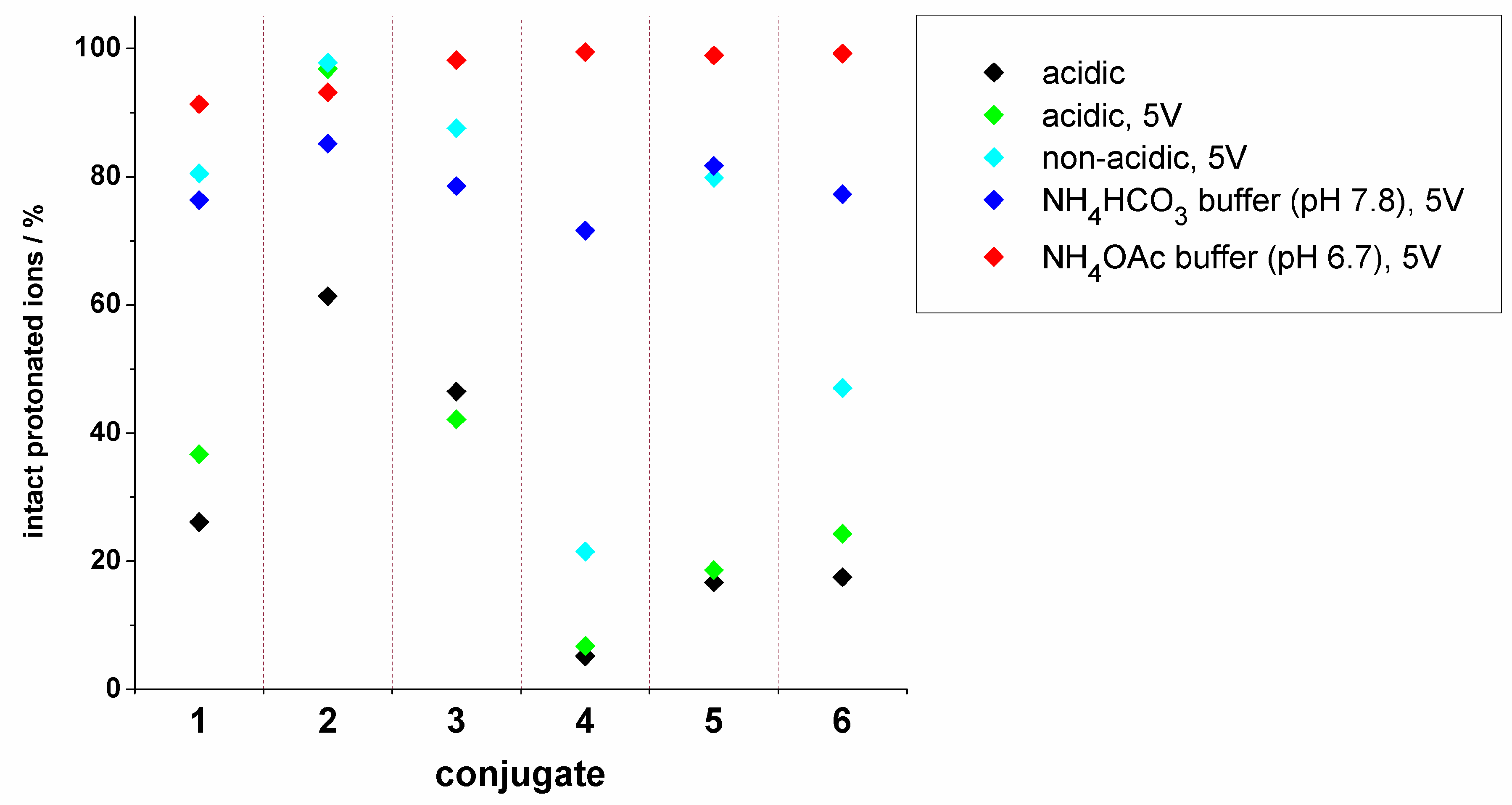
2.3. Mass Spectrometric Analysis under the Changed Conditions
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Preparation of Daunomycin-Peptide Bioconjugates
4.3. Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- DiMarco, A.; Soldati, M.; Fioretti, A.; Dasdia, T. Activity of Daunomycin, a new antitumor antibiotic on normal and neoplastic cells grown in vitro. Cancer Chemother. Rep. 1964, 38, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Enyedi, K.N.; Tóth, S.; Szakács, G.; Mező, G. NGR-peptide-drug conjugates with dual targeting properties. PLoS ONE 2017, 12, e0178632. [Google Scholar] [CrossRef] [PubMed]
- Kapuvári, B.; Hegedüs, R.; Schulcz, Á.; Manea, M.; Tóvári, J.; Gacs, A.; Vincze, B.; Mező, G. Improved in vivo antitumor effect of a daunorubicin-GnRH-III bioconjugate modified by apoptosis inducing agent butyric acid on colorectal carcinoma bearing mice. Investig. New Drugs 2016, 34, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Lelle, M.; Kaloyanova, S.; Freidel, C.; Theodoropoulou, M.; Musheev, M.; Niehrs, C.; Stalla, G.; Peneva, K. Octreotide-mediated tumor-targeted drug delivery via a cleavable doxorubicin–peptide conjugate. Mol. Pharm. 2015, 12, 4290–4300. [Google Scholar] [CrossRef] [PubMed]
- Mező, G.; Manea, M. Receptor-mediated tumor targeting based on peptide hormones. Expert Opin. Drug Deliv. 2010, 7, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Najjar, V.A.; Nishioka, K. “Tuftsin”: A natural phagocytosis stimulating peptide. Nature 1970, 228, 672–673. [Google Scholar] [CrossRef] [PubMed]
- Mező, G.; Láng, O.; Jakab, A.; Bai, K.B.; Szabó, I.; Schlosser, G.; Láng, J.; Kőhidai, L.; Hudecz, F. Synthesis of oligotuftsin-based branched oligopeptide conjugates for chemotactic drug targeting. J. Peptide Sci. 2006, 12, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Bai, K.B.; Láng, O.; Orbán, E.; Szabó, R.; Kőhidai, L.; Hudecz, F.; Mező, G. Design, synthesis, and In vitro activity of novel drug delivery systems containing tuftsin derivatives and methotrexate. Bioconjug. Chem. 2008, 19, 2260–2269. [Google Scholar] [CrossRef]
- Mező, G.; Szekerke, M.; Sármay, G.; Gergely, J. Synthesis and functional studies of tuftsin analogs containing isopeptide bond. Peptides 1990, 11, 405–415. [Google Scholar] [CrossRef]
- Kukowska-Kaszuba, M.; Dzierzbicka, K.; Serocki, M.; Skladanowski, A. Solid phase synthesis and biological activity of tuftsin conjugates. J. Med. Chem. 2011, 54, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Sleno, L.; Campagna-Slater, V.; Volmer, D.A. Dissociation reactions of protonated anthracycline antibiotics following electrospray ionization-tandem mass spectrometry. Int. J. Mass Spectrom. 2006, 255–256, 130–138. [Google Scholar] [CrossRef]
- Ryppa, C.; Mann-Steinberg, H.; Fichtner, I.; Weber, H.; Satchi-Fainaro, R.; Biniossek, M.L.; Kratz, F. In Vitro and in vivo evaluation of doxorubicin conjugates with the divalent peptide E-[c(RGDfK)2] that targets integrin αvβ3. Bioconjug. Chem. 2008, 19, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Schreier, V.N.; Pethő, L.; Orbán, E.; Marquardt, A.; Petre, B.A.; Mező, G.; Manea, M. Protein expression profile of HT-29 human colon cancer cells after treatment with a cytotoxic daunorubicin-GnRH-III derivative bioconjugate. PLoS ONE 2014, 9, e94041. [Google Scholar] [CrossRef] [PubMed]
- Orbán, E.; Mező, G.; Schlage, P.; Csík, G.; Kulić, Ž.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin–GnRH-III bioconjugates and DNA-binding properties of daunorubicin–amino acid metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Mező, G.; Szabó, I.; Kertész, I.; Hegedüs, R.; Orbán, E.; Leurs, U.; Bősze, S.; Halmos, G.; Manea, M. Efficient synthesis of an (aminooxy) acetylated-somatostatin derivative using (aminooxy) acetic acid as a ‘carbonyl capture’ reagent. J. Pept. Sci. 2011, 17, 39–46. [Google Scholar] [CrossRef]
- Krauss, U.; Kratz, F.; Beck-Sickinger, A.G. Novel daunorubicin-carrier peptide conjugates derived from human calcitonin segments. J. Mol. Recognit. 2003, 16, 280–287. [Google Scholar] [CrossRef]
- Kaushik, D.; Bansal, G. Four new degradation products of doxorubicin: An application of forced degradation study and hyphenated chromatographic techniques. J. Pharm. Anal. 2015, 5, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Schlage, P.; Mező, G.; Orbán, E.; Bősze, S.; Manea, M. Anthracycline-GnRH derivative bioconjugates with different linkages: Synthesis, In vitro drug release and cytostatic effect. J. Control. Release 2011, 156, 170–178. [Google Scholar] [CrossRef]
- Rejmanová, P.; Kopeček, J.; Pohl, J.; Baudyš, M.; Kostka, V. Polymers containing enzymatically degradable bonds 8. Degradation of oligopeptide sequences in N-(2-hydroxypropyl)methacrylamide copolymers by bovine spleen cathepsin B. Makromol. Chem. 1983, 184, 2009–2020. [Google Scholar] [CrossRef]
- Wysocki, V.H.; Tsaprailis, G.; Smith, L.L.; Breci, L.A. Mobile and localized protons: A framework for understanding peptide dissociation. J. Mass Spectrom. 2000, 35, 1399–1406. [Google Scholar] [CrossRef]
- Boyd, R.; Somogyi, Á. The mobile proton hypothesis in fragmentation of protonated peptides: A perspective. J. Am. Soc. Mass Spectrom. 2010, 21, 1275–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iavarone, A.T.; Williams, E.R. Supercharging in electrospray ionization: Effects on signal and charge. Int. J. Mass Spectrom. 2002, 219, 63–72. [Google Scholar] [CrossRef]
- Sterling, H.J.; Williams, E.R. Origin of supercharging in electrospray ionization of noncovalent complexes from aqueous solution. J. Am. Soc. Mass Spectrom. 2009, 20, 1933–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miladinović, S.A.; Fornelli, L.; Lu, Y.; Piech, K.M.; Girault, H.H.; Tsybin, Y.O. In-spray supercharging of peptides and proteins in electrospray ionization mass spectrometry. Anal. Chem. 2012, 84, 4647–4651. [Google Scholar] [CrossRef] [PubMed]
- Going, C.C.; Xia, Z.; Williams, E.R. New supercharging reagents produce highly charged protein ions in native mass spectrometry. Analyst 2015, 140, 7184–7194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenaidee, M.A.; Donald, W.A. Extremely supercharged proteins in mass spectrometry: Profiling the pH of electrospray generated droplets, narrowing charge state distributions, and increasing ion fragmentation. Analyst 2015, 140, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Nshanian, M.; Lakshmanan, R.; Chen, H.; Ogorzalek Loo, R.R.; Loo, J.A. Enhancing sensitivity of liquid chromatography/mass spectrometry of peptides and proteins using supercharging agents. Int. J. Mass Spectrom. 2018, 427, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.L.; McLuckey, S.A. Ion/ion reactions in the gas phase: Proton transfer reactions involving multiply-charged proteins. J. Am. Chem. Soc. 1996, 118, 7390–7397. [Google Scholar] [CrossRef]
- Scalf, M.; Westphall, M.S.; Smith, L.M. Charge reduction electrospray mass spectrometry. Anal. Chem. 2000, 72, 52–60. [Google Scholar] [CrossRef]
- Stutzman, J.R.; Crowe, M.C.; Alexander, J.N.; Bell, B.M.; Dunkle, M.N. Coupling charge reduction mass spectrometry to liquid chromatography for complex mixture analysis. Anal. Chem. 2016, 88, 4130–4139. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, A.; Prentice, B.M.; Huang, T.-Y.; McLuckey, S.A. Electrospray droplet exposure to gaseous acids for the manipulation of protein charge state distributions. Anal. Chem. 2010, 82, 7422–7429. [Google Scholar] [CrossRef] [PubMed]
- Catalina, M.I.; van den Heuvel, R.H.H.; van Duijn, E.; Heck, A.J.R. Decharging of globular proteins and protein complexes in electrospray. Chem. Eur. J. 2005, 11, 960–968. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pethő, L.; Mező, G.; Schlosser, G. Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates. Molecules 2019, 24, 2981. https://doi.org/10.3390/molecules24162981
Pethő L, Mező G, Schlosser G. Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates. Molecules. 2019; 24(16):2981. https://doi.org/10.3390/molecules24162981
Chicago/Turabian StylePethő, Lilla, Gábor Mező, and Gitta Schlosser. 2019. "Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates" Molecules 24, no. 16: 2981. https://doi.org/10.3390/molecules24162981
APA StylePethő, L., Mező, G., & Schlosser, G. (2019). Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates. Molecules, 24(16), 2981. https://doi.org/10.3390/molecules24162981