
A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives




Abstract
:
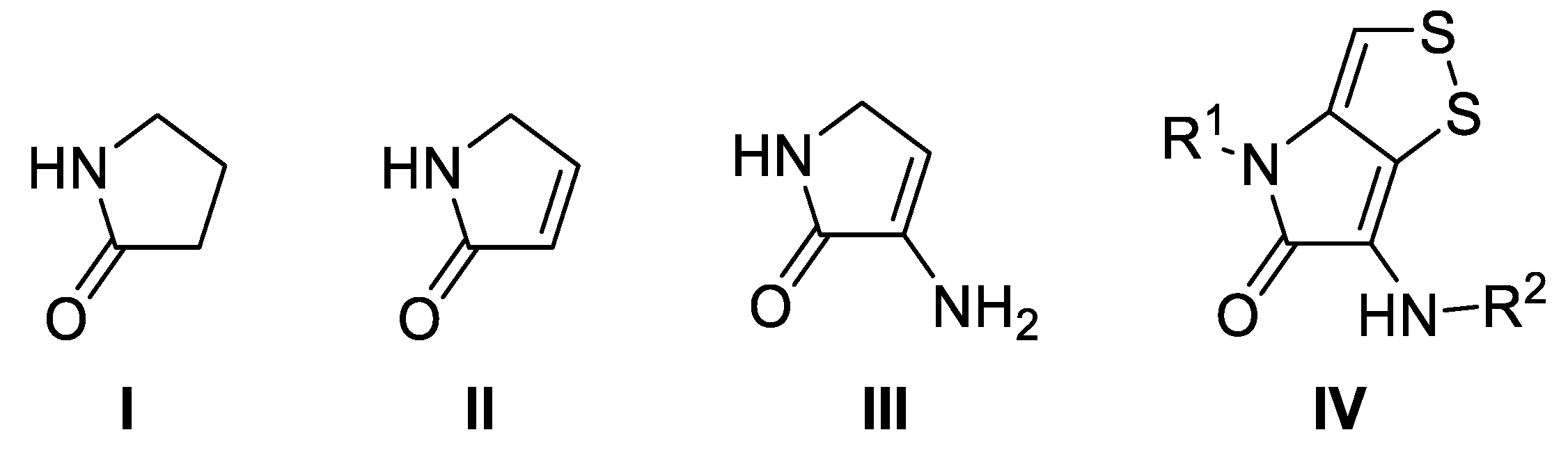
1. Introduction
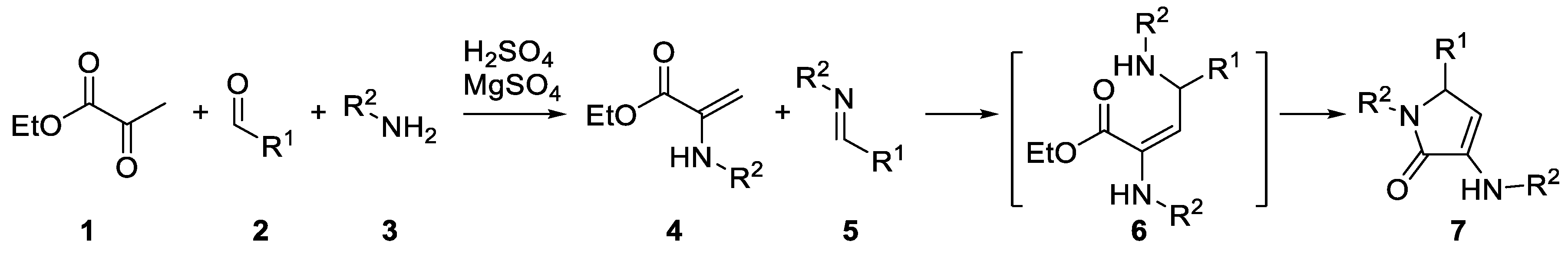
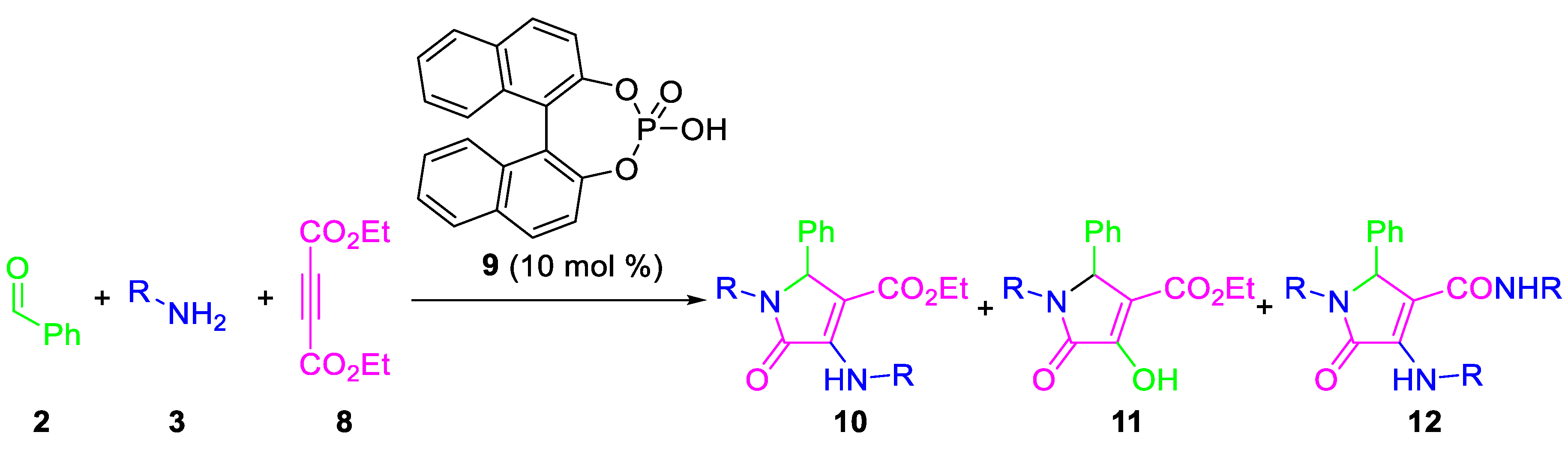
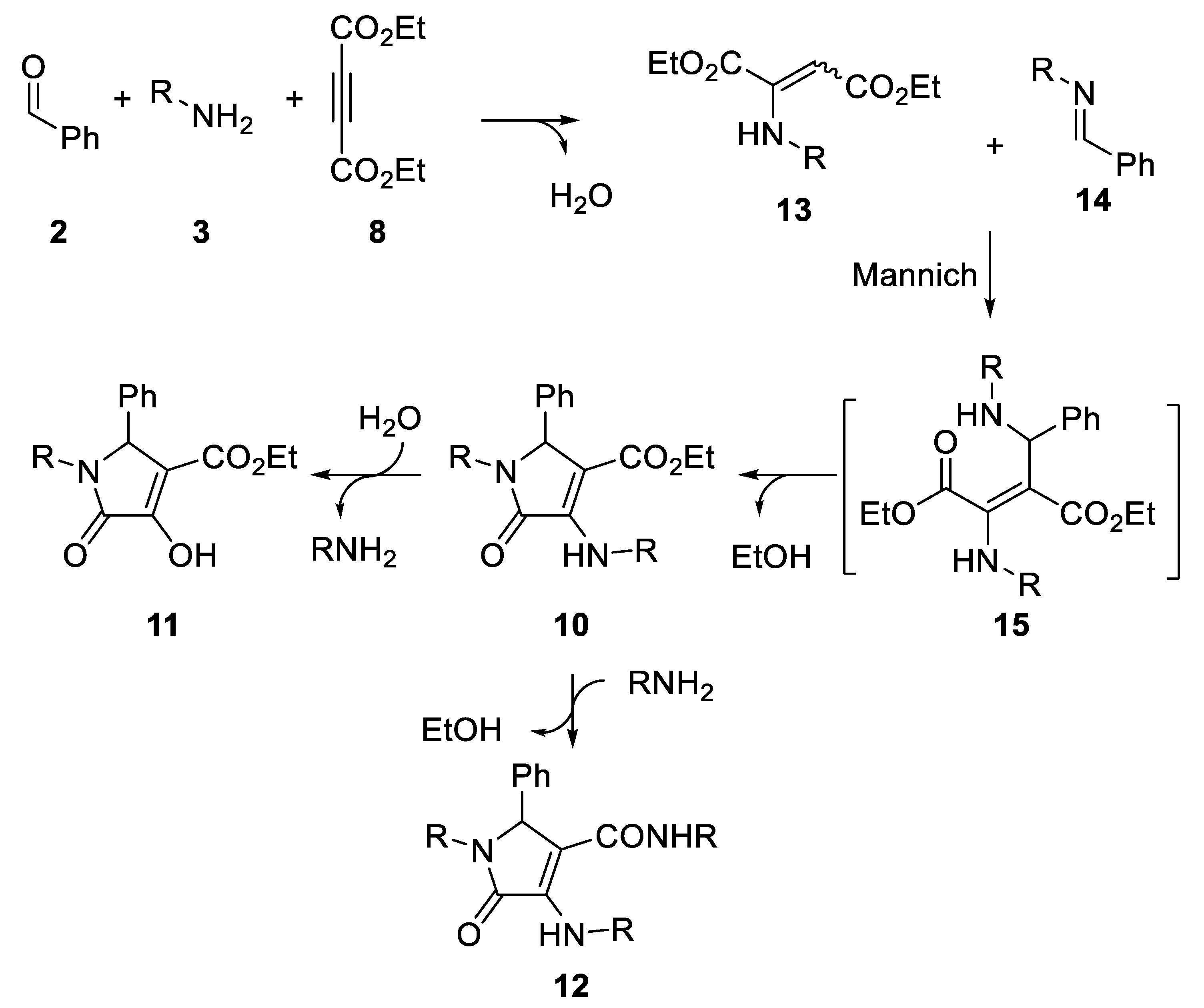
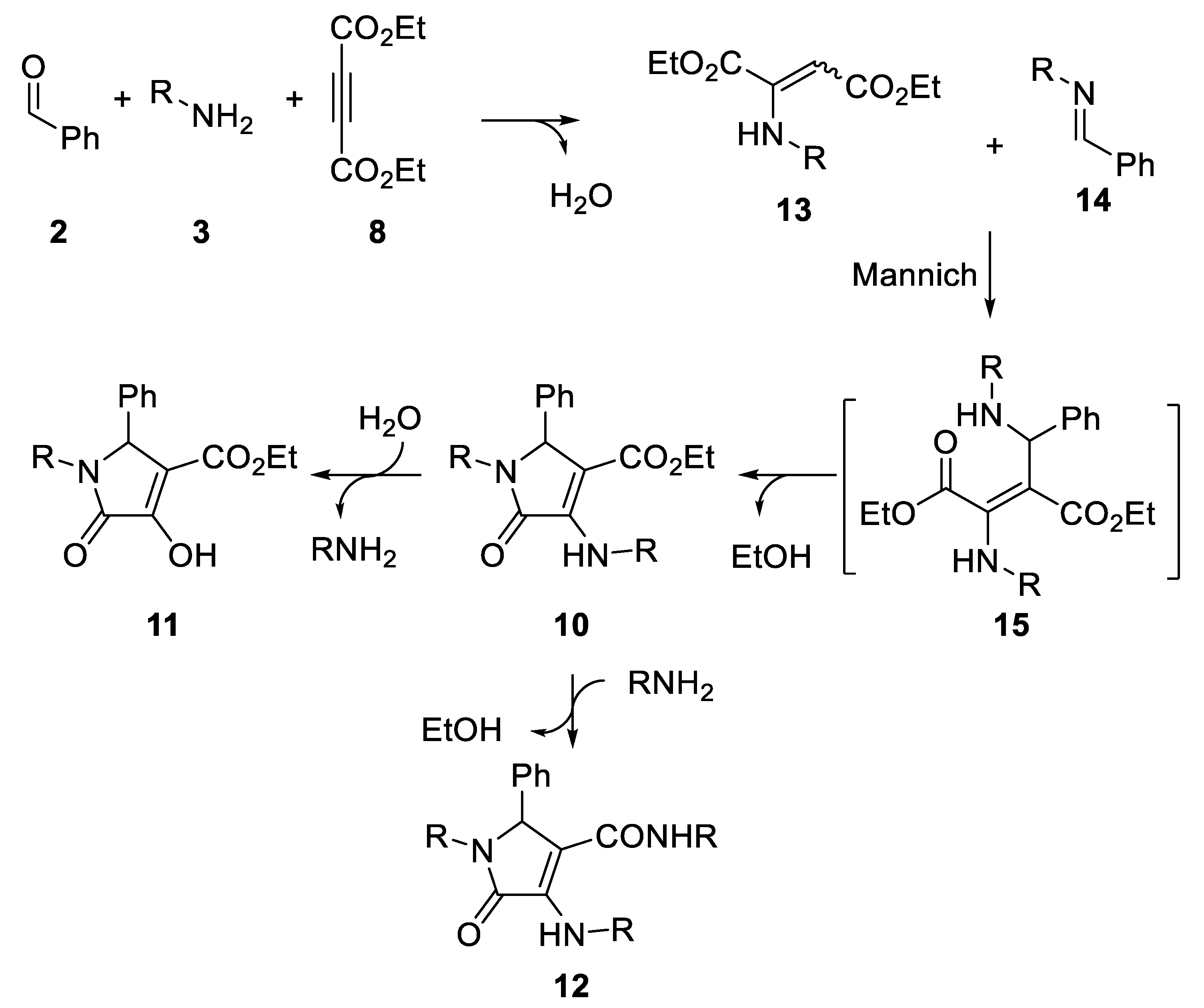
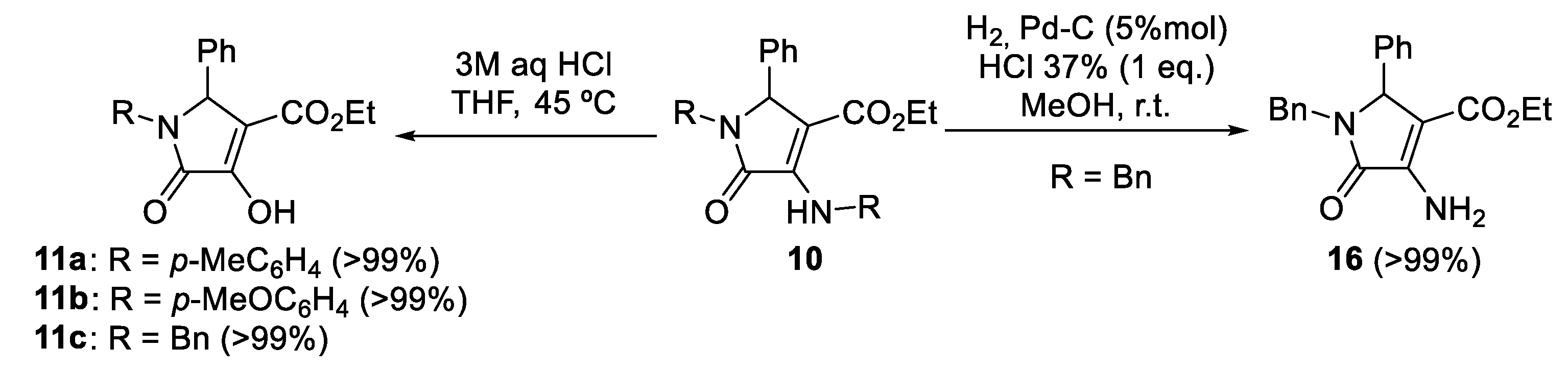
2. Results
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, J.; Wang, Q.; Wang, M.-X. (Eds.) Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germnay, 2015. [Google Scholar]
- Müller, T.J. J in Science of Synthesis, Multicomponent Reactions, Vol 1 and 2; Thieme: Stutgart, Germnay, 2014. [Google Scholar]
- Knapp, J.M.; Kurth, M.J.; Shaw, J.T.; Younai, A. Diversity-Oriented Synthesis; Trabocchi, A., Ed.; Willey: New York, NY, USA, 2013; Volume 41, pp. 29–57. [Google Scholar]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- de Moliner, F.; Kielland, N.; Lavilla, R.; Vendrell, M. Modern Synthetic Avenues for the Preparation of Functional Fluorophores. Angew. Chem. Int. Ed. 2017, 56, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G.; Rybak, T.; Verdelet, T. Multicomponent Hetero-[4 + 2] Cycloaddition/Allylboration Reaction: From Natural Product Synthesis to Drug Discovery. Acc. Chem. Res. 2016, 49, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
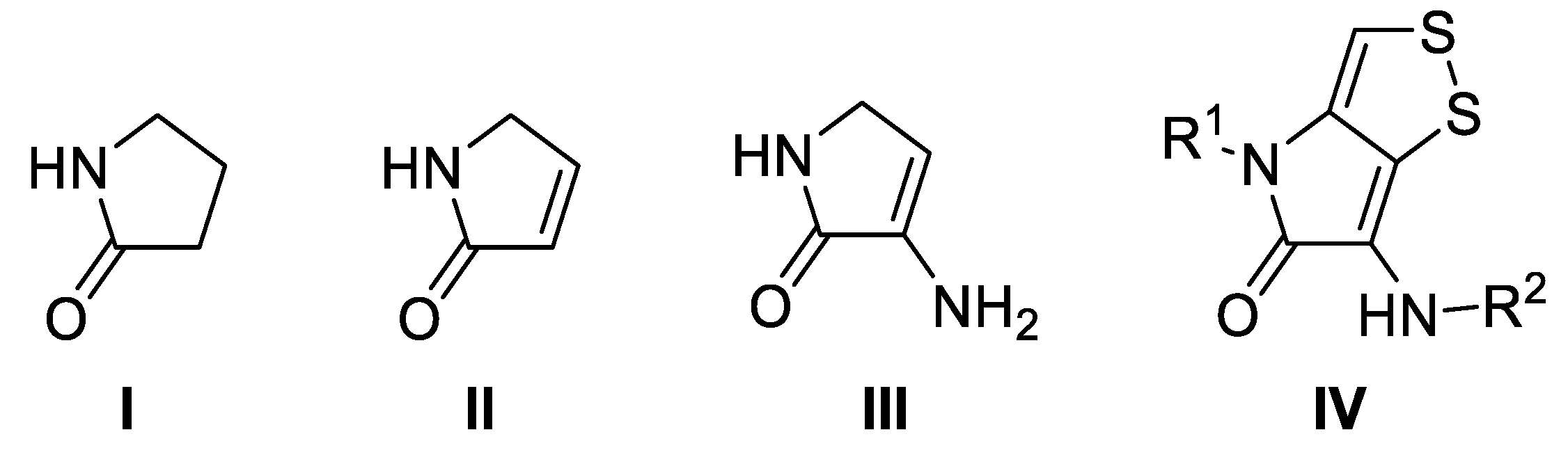
- Caruano, J.; Muccioli, G.G.; Robiette, R. Biologically active γ-lactams: Synthesis and natural sources. Org. Biomol. Chem. 2016, 14, 10134–10156. [Google Scholar] [CrossRef] [PubMed]
- Martinez de Marigorta, E.; de los Santos, J.; Ochoa de Retana, A.M.; Vicario, J.; Palacios, F. Multicomponent Reactions in the Synthesis of γ –Lactams. Synthesis 2018, 50, 4539–4554. [Google Scholar]
- Martinez de Marigorta, E.; de los Santos, J.; Ochoa de Retana, A.M.; Vicario, J.; Palacios, F. Multicomponent reactions (MCRs): A useful access to the synthesis of benzo-fused γ-lactams. Beilstein J. Org. Chem. 2019, 150, 1065–1085. [Google Scholar] [CrossRef] [PubMed]
- Kirpotina, L.N.; Schepetkin, I.A.; Khlebnikov, A.I.; Ruban, O.I.; Ge, Y.; Ye, R.D.; Kominsky, D.J.; Quinn, M.T. 4-Aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-ones as N-formyl peptide receptor 1 (FPR1) antagonists. Biochem. Pharmacol. 2017, 142, 120–132. [Google Scholar] [CrossRef]
- Ma, K.; Wang, P.; Fu, W.; Wan, X.; Zhou, L.; Chu, Y.; Ye, D. Rational design of 2-pyrrolinones as inhibitors of HIV-1 integrase. Bioorg. Med. Chem. Lett. 2011, 21, 6724–6727. [Google Scholar] [CrossRef]
- Zhuang, C.; Miao, Z.; Zhu, L.; Dong, G.; Guo, Z.; Wang, S.; Zhang, Y.; Wu, Y.; Yao, J.; Sheng, C.; et al. Discovery, Synthesis, and Biological Evaluation of Orally Active Pyrrolidone Derivatives as Novel Inhibitors of p53–MDM2 Protein–Protein Interaction. J. Med. Chem. 2012, 55, 9630–9642. [Google Scholar] [CrossRef]
- Peifer, C.; Selig, R.; Kinkel, K.; Ott, D.; Totzke, F.; Schaechtele, C.; Heidenreich, R.; Roecken, M.; Schollmeyer, D.; Laufer, S. Design, Synthesis, and Biological Evaluation of Novel 3-Aryl-4-(1H-indole-3yl)-1,5-dihydro-2H-pyrrole-2-ones as Vascular Endothelial Growth Factor Receptor (VEGF-R) Inhibitors. J. Med. Chem. 2008, 51, 3814–3824. [Google Scholar] [CrossRef]
- Liu, Q.-J.; Wang, L.; Kang, Q.-K.; Zhang, X.P.; Tang, Y. Cy-SaBOX/Copper(II)-Catalyzed Highly Diastereo- and Enantioselective Synthesis of Bicyclic N,O Acetals. Angew. Chem. Int. Ed. 2016, 55, 9220–9223. [Google Scholar] [CrossRef] [PubMed]
- Bures, J.; Armstrong, A.; Blackmond, D. Explaining Anomalies in Enamine Catalysis: “Downstream Species as a New Paradigm for Stereocontrol G. Acc. Chem. Res. 2016, 49, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, A.I.; Waigh, R.D.; Drummond, A.J.; Pringle, B.; McGroarty, I.; Skellern, G.G.; Suckling, C.J. Distamycin Analogues with Enhanced Lipophilicity: Synthesis and Antimicrobial Activity. J. Med. Chem. 2004, 47, 2133–2156. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Fang, F.; Li, Y. Synthesis and anti-biofilm activities of dihydro-pyrrol-2-one derivatives on Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 2015, 25, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, L.; Chen, Z.; Zheng, S.; Shu, H.; Li, J.; Jiang, H.; Liu, S. A novel class of small-molecule caspase-3 inhibitors prepared by multicomponent reactions. Eur. J. Med. Chem. 2012, 54, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Gein, V.L.; Popov, A.V.; Kolla, V.E.; Popova, N.A.; Potemkin, K.D. Synthesis and biological activity of 1,5-diaryl-3-arylamino-4-carboxymethyl-2,5-dihydro-2-pyrrolones and 1,5-diaryl-4-carboxymethyltetrahydropyrrole-2, 3-diones. Pharm. Chem. J. 1993, 27, 343–346. [Google Scholar] [CrossRef]
- Gein, V.L.; Popov, A.V.; Kolla, V.E.; Popova, N.A. Synthesis and biological activity of 1,5-diaryl-3-alkylamino-4-carboxymethyl-2,5-dihydropyrrol-2-ones and 1,5-diaryl-4-carboxymethyl-tetrahydropyrrol-2,3-diones. Pharmazie 1993, 8, 107–109. [Google Scholar]
- Li, B.; Wever, W.J.; Walsh, C.T.; Bowers, A.A. Dithiolopyrrolones: Biosynthesis, synthesis, and activity of a unique class of disulfide-containing antibiotics. Nat. Prod. Rep. 2014, 31, 905–923. [Google Scholar] [CrossRef] [PubMed]
- Rigby, J.H.; Hughes, R.C.; Heeg, M.J.J. Endo-Selective Cyclization Pathways in the Intramolecular Heck Reaction. Am. Chem. Soc. 1995, 117, 7834–7835. [Google Scholar] [CrossRef]
- Lewis, J.R. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 1994, 11, 329–332. [Google Scholar] [CrossRef]
- Palacios, F.; Vicario, J.; Aparicio, D. An efficient synthesis of achiral and chiral cyclic dehydro-α-amino acid derivatives through nucleophilic addition of Amines to β,γ-unsaturated α-keto esters. Eur. J. Org. Chem. 2006, 2843–2850. [Google Scholar] [CrossRef]
- Shaterian, H.R.; Ranjbar, M. Uncatalyzed synthesis of 3-amino-1,5-dihydro-2H-pyrrol-2-ones. Res. Chem. Intermed. 2014, 40, 2059–2074. [Google Scholar] [CrossRef]
- Niknam, K.; Mojikhalifeh, S. Synthesis of new 1,5-diaryl-3-(arylamino)-1H-pyrrol-2(5H)-ones under catalyst-free and solvent-free conditions. Mol. Diver. 2014, 18, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Quian, J.; Yi, W.; Cai, C. Recyclable fluorous organocatalysts promoted three-component reactions of pyruvate, aldehyde and amine at room temperature. Tetrahedron Lett. 2013, 54, 7100–7102. [Google Scholar] [CrossRef]
- Li, X.; Deng, H.; Luo, S.; Cheng, J.-P. Organocatalytic Three-Component Reactions of Pyruvate, Aldehyde and Aniline by Hydrogen-Bonding Catalysts. Eur. J. Org. Chem. 2008, 4350–4356. [Google Scholar] [CrossRef]
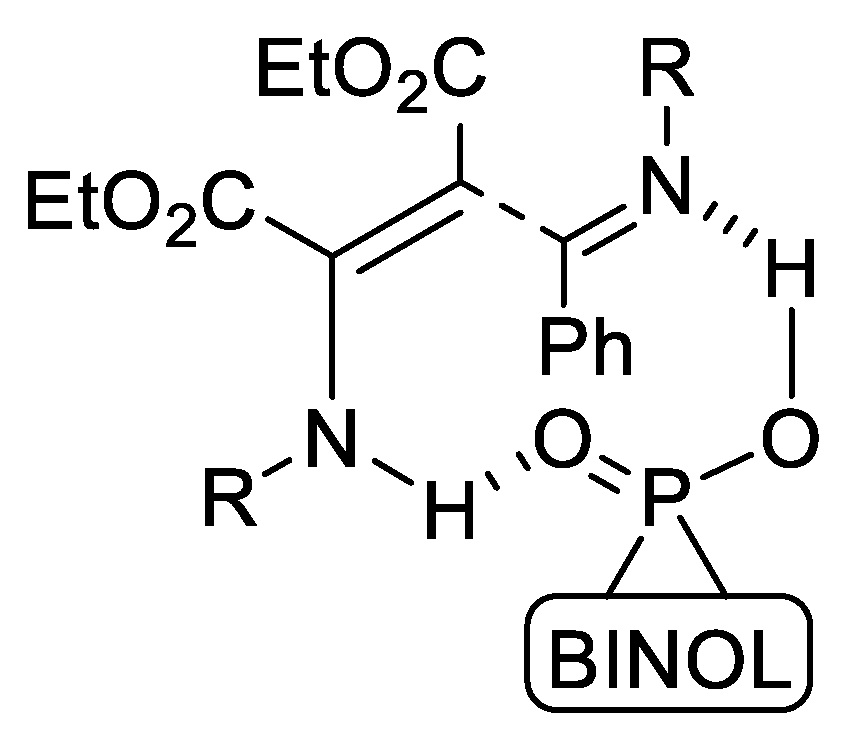
- del Corte, X.; Maestro, A.; Vicario, J.; Martinez de Marigorta, E.; Palacios, F. Brönsted-Acid-Catalyzed Asymmetric Three-Component Reaction of Amines, Aldehydes, and Pyruvate Derivatives. Enantioselective Synthesis of Highly Functionalized γ-Lactam Derivatives. Org. Lett. 2018, 20, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, J.; Yan, C.-G. Synthesis of functionalized 2-pyrrolidinones via domino reactions of arylamines, ethyl glyoxylate and acetylenedicarboxylates. Tetrahedron 2013, 69, 589–594. [Google Scholar] [CrossRef]
- Khan, A.T.; Gosh, A.; Khan, M.M. One-pot four-component domino reaction for the synthesis of substituted dihydro-2-oxypyrrole catalyzed by molecular iodine. Tetrahedron Lett. 2012, 53, 2622–2626. [Google Scholar] [CrossRef]
- Saha, M.; Das, A.R. Access of Diverse 2-Pyrrolidinone, 3,4,5-Substituted Furanone and 2-Oxo-dihydropyrroles Applying Graphene Oxide Nanosheet: Unraveling of Solvent Selectivity. ChemistrySelect 2017, 2, 10249–10260. [Google Scholar] [CrossRef]
- Palacios, F.; Aparicio, D.; García, J.; Vicario, J.; Ezpeleta, J.M. Regioselective alkylation reactions of enamines derived from phosphane oxides - Synthesis of phosphorus substituted enamino esters, δ-amino-phosphonates, pyridone derivatives and pyrroles. Eur. J. Org. Chem. 2001, 3357–3365. [Google Scholar] [CrossRef]
- Palacios, F.; Aparicio, D.; Vicario, J. Synthesis of quinolinylphosphane oxides and -phosphonates from N-arylimines derived from phosphane oxides and phosphonates. Eur. J. Org. Chem. 2002, 4131–4136. [Google Scholar]
- Palacios, F.; Vicario, J.; Aparicio, D. Aza-Diels-Alder reaction of α,β-unsaturated sulfinylimines derived from α-amino acids with enolethers and enamines. Tetrahedron Lett. 2007, 48, 6747–6750. [Google Scholar] [CrossRef]
- Palacios, F.; Vicario, J.; Aparicio, D. A diastereoselective aza-Diels-Alder reaction of N-aryl-1-azadienes derived from α-amino acids with enamines. Tetrahedron Lett. 2011, 52, 4109–4111. [Google Scholar]
- Palacios, F.; Vicario, J.; Aparicio, D. Efficient synthesis of 1-azadienes derived from α-aminoesters. Regioselective preparation of α-dehydroamino acids, vinylglycines, and α-amino acids. J. Org. Chem. 2006, 71, 7690–7696. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J.; Ortiz, P.; Ezpeleta, J.M.; Palacios, F. Asymmetric Synthesis of Functionalized Tetrasubstituted α-Aminophosphonates through Enantioselective Aza-Henry Reaction of Phosphorylated Ketimines. J. Org. Chem. 2015, 80, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J.; Ezpeleta, J.M.; Palacios, F. Asymmetric Cyanation of α-Ketiminophosphonates Catalyzed by Cinchona Alkaloids: Enantioselective Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives from Trisubstituted α-Aminophosphonates. Adv. Synth. & Catal. 2012, 354, 2641–2647. [Google Scholar]
- Maestro, A.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Enantioselective alpha-Aminophosphonate Functionalization of Indole Ring through an Organocatalyzed Friedel-Crafts Reaction. J. Org. Chem. 2019, 84, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Metten, B.; Kostermans, M.; Van Baelen, G.; Smet, M.; Dehaen, W. Synthesis of 5-aryl-2-oxopyrrole derivatives as synthons for highly substituted pyrroles. Tetrahedron 2006, 62, 6018. [Google Scholar] [CrossRef]
- Winstein-Holnes values: ABn = 1.68 kcal.mol-1 and APh = 2.80 kcal.mol-1. In Stereochemistry of Organic Compounds; Eliel, E.L.; Wilen, S.H.; Mander, L.N. (Eds.) Wiley: New York, NY, USA, 1994. [Google Scholar]
- Ávila, E.P.; Justo, R.M.S.; Gonçalves, V.P.; Pereira, A.A.; Diniz, R.; Amarante, G.W. Chiral Brønsted Acid-Catalyzed Stereoselective Mannich-Type Reaction of Azlactones with Aldimines. J. Org. Chem. 2015, 80, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Shaoa, Y.-D.; Chengb, D.-J. Catalytic Asymmetric 1,2-Difunctionalization of Indolenines with α-(Benzothiazol-2-ylsulfonyl) Carbonyl Compounds. Adv. Synth. Catal. 2017, 359, 2549–2556. [Google Scholar] [CrossRef]
- Huang, Q.; Cheng, Y.; Yuan, H.; Chang, X.; Li, P.; Li, W. Organocatalytic enantioselective Mannich-type addition of 5H-thiazol-4-ones to isatin-derived imines: Access to 3-substituted 3-amino-2-oxindoles featured by vicinal sulfur-containing tetrasubstituted stereocenters. Org. Chem. Front. 2018, 5, 3226–3230. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2010. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program. PLATON J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
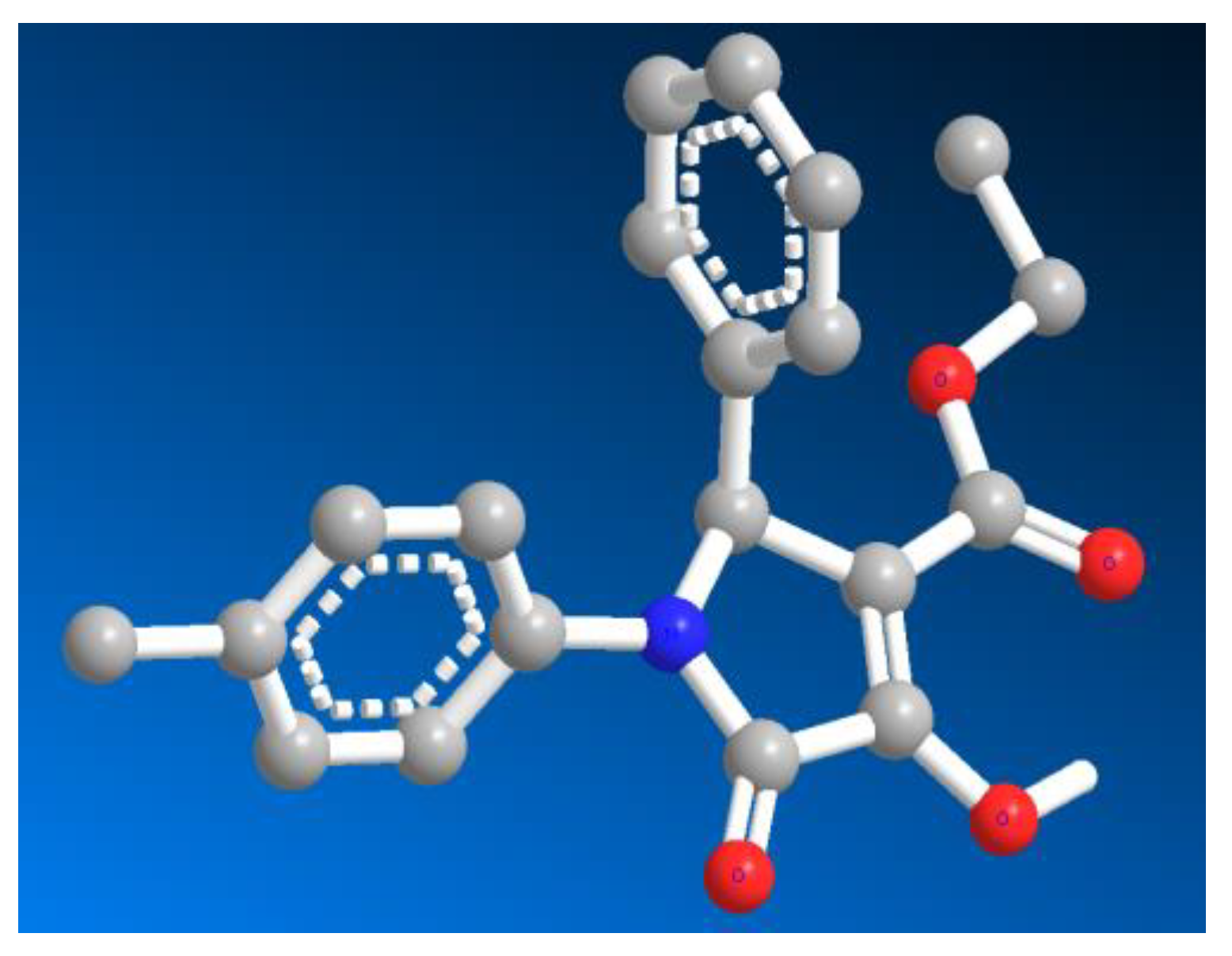
Sample Availability: Samples of the compounds 10, 11, 12, and 16 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
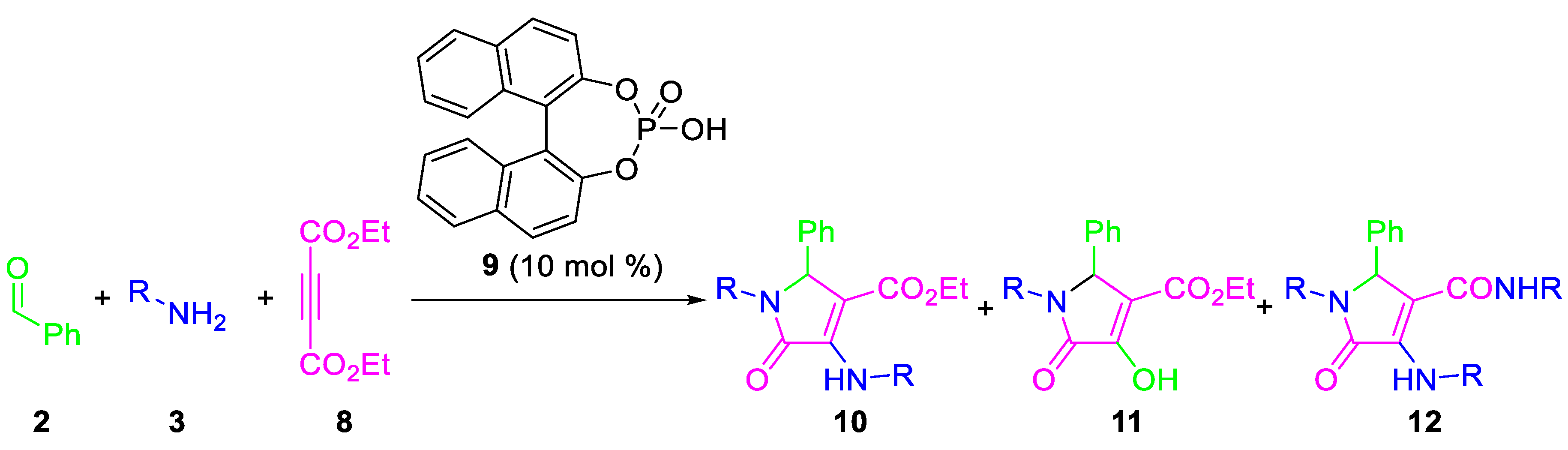
| Entry | R | 2/3/8 | Solvent | T (°C) | Yield (%) 1 | 10/11/12 2 |
|---|---|---|---|---|---|---|
| 1 | p-MeC6H4 | 1/2/1 | CH2Cl2 | 40 | 0 | n.d. |
| 2 | p-MeC6H4 | 1/2/1 | THF | 65 | 0 | n.d. |
| 3 | p-MeC6H4 | 1/2/1 | DME | 85 | 0 | n.d. |
| 4 | p-MeC6H4 | 1/2/1 | MTBE | 55 | 72 | 40/60/0 |
| 5 | p-MeC6H4 | 1/2/3 | MTBE | 55 | 0 | n.d. |
| 6 | p-MeC6H4 | 1/2/1 | Dioxane | 101 | 81 | 80/0/20 |
| 7 | p-MeC6H4 | 1/2/1 | Toluene | 110 | 77 | 95/0/5 |
| 8 | p-MeOC6H4 | 1/2/1 | Toluene | 110 | 76 | 70/0/30 |
| 9 | p-MeOC6H4 | 1/4/1 | Toluene | 110 | 76 | 70/0/30 |
| 10 | Bn | 1/2/1 | Toluene | 110 | 58 | 100/0/0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Corte, X.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives. Molecules 2019, 24, 2951. https://doi.org/10.3390/molecules24162951
del Corte X, Martinez de Marigorta E, Palacios F, Vicario J. A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives. Molecules. 2019; 24(16):2951. https://doi.org/10.3390/molecules24162951
Chicago/Turabian Styledel Corte, Xabier, Edorta Martinez de Marigorta, Francisco Palacios, and Javier Vicario. 2019. "A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives" Molecules 24, no. 16: 2951. https://doi.org/10.3390/molecules24162951
APA Styledel Corte, X., Martinez de Marigorta, E., Palacios, F., & Vicario, J. (2019). A Brønsted Acid-Catalyzed Multicomponent Reaction for the Synthesis of Highly Functionalized γ-Lactam Derivatives. Molecules, 24(16), 2951. https://doi.org/10.3390/molecules24162951